1. Introduction
Mesenchymal stem cells (MSCs) are multipotent cells present in different adult tissues that possess the capacity to differentiate into various specialized cell lineages including osteoblasts, adipocytes, and chondrocytes. Due to this ability, in the last few years, there has been an increasing interest in using MSCs-based approaches to improve bone repair and regeneration [
1]. In particular, the use of MSCs-based therapies would benefit the treatment of critical size bone defects resulting from direct trauma or from the removal of large bone areas through surgical procedures in patients with osteosarcoma, osteonecrosis, or other pathologies. Due to the known drawbacks of autologous and allogeneic bone graft, bone tissue engineering has emerged as an interesting alternative. One of the main obstacles for the development of a successful MSCs-based therapy is to obtain an efficient osteogenic differentiation of the transplanted cells. Different MSCs modifications have been designed to achieve this point [
2,
3,
4,
5].
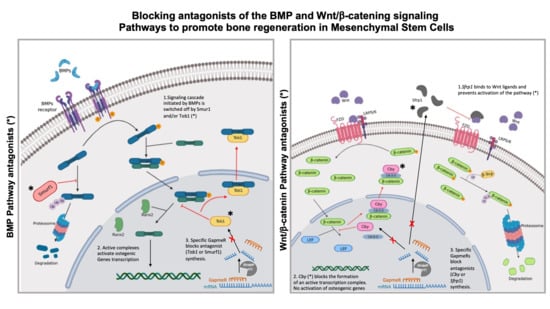
Due to their key function in bone formation, the BMP and Wnt/β-catenin signaling pathways are interesting targets for the development of therapies aimed to treat bone loss associated diseases. We have previously shown that the sustained activation of the BMP pathway through the silencing of
Smurf1 (SMAD specific E3 Ubiquitin Protein Ligase 1), a major inhibitor of this pathway, has a marked positive effect on bone formation [
5,
6]. Importantly, this was achieved by using a particular class of Locked-Nucleic-Acids Antisense-Oligonucleotides (LNA-ASOs) known as GapmeRs, a clinically safe method that would enable the translation of this protocol to the clinical practice. Moreover, using this approach, we were able to overcome the reduced osteogenic potential showed by MSCs from osteoporotic patients [
7]. These results were achieved in the presence of BMP-2 concentrations one million times lower than those used in the clinic [
6], opening the possibility of significantly reducing the adverse effects linked to high doses of these factors [
8,
9,
10,
11,
12,
13,
14], one of the problems hindering the clinical use of regenerative techniques in this field. Whereas the osteogenic induction using the previous method was highly significant, the overall increase in bone formation in vivo through the silencing of
Smurf1 in MSCs did not exceed 30%. In this current work, we set out to increase this efficiency by targeting other extra and intracellular osteogenic inhibitors.
We hypothesize that it would be possible to achieve a greater pro-osteogenic effect by silencing genes coding for other negative regulators of this and other osteogenic pathways, either independently or in combination with
Smurf1.
Tob1 (Transducer of Erb-2 1) is one of the genes that could be useful for this purpose. This gene encodes an inhibitor of the interaction of RUNX2 and SMADs complexes, switching off the osteogenic signal initiated by the binding of BMP proteins to their receptors. Importantly,
Tob1 defective mice show abnormally high levels of bone formation and block osteoporosis induced by estrogen deficiency [
15]. Other signaling networks are coordinated with BMPs to regulate the osteogenic differentiation of MSCs. The one receiving more attention is the Wnt/β-catenin pathway. In fact, several molecules involved in the regulation of Wnt signaling, such as sclerostin, are currently being targeted in bone-building therapies for patients with osteoporosis [
16]. The key switch of this pathway is β-catenin, which, upon binding of Wnt to Wnt receptors, translocates into the nucleus and forms a dimer with LEF (Lymphoid Enhancer Binding Factor). This dimer acts as a transcription factor to promote
Runx2 transcription and the differentiation of MSCs to osteoblasts [
17]. Data from several rare mutations affecting bone mass and from knockout mice [
18] have revealed two more genes whose silencing would enhance bone formation. One encodes for the secreted frizzled related protein 1 (
Sfrp1), that antagonizes canonical Wnt signaling by binding to Wnt ligands, preventing their binding to the receptors and inhibiting their downstream signaling activity [
19]. Mice defective in
Sprf1 have a higher bone mass and
Sfrp1-/- mouse progenitor cells are directly shifted into the osteoblastic lineage. Interestingly,
Sfrp1 has been already proposed as a drug target for the treatment of fracture healing [
20]. On the other hand, bone of osteoporotic patients shows an enhanced differentiation of MSCs to adipocytes [
21]. The product of the gene Chibby Family Member 1 (
Cby1), a nuclear protein that acts as an intracellular antagonist of the Wnt canonical pathway, prevents the formation of the β-catenin/Lef/Tcf [
22] complex, directing multipotent progenitor cells toward the adipocytic differentiation. Since the osteogenic and adipogenic fates of MSCs are mutually exclusive,
Cby1 silencing could block adipogenic differentiation directing MSCs toward osteoblast formation.
In this current work, we tested whether the silencing of genes encoding for inhibitors of the BMP signaling pathway others than Smurf1 (Tob1) and inhibitors of the Wnt/β-catenin pathway (Cby1 and Sfrp1) in MSCs could, either individually or in combination, potentiate the osteogenic priming of MSCs and promote bone mass growth in a more efficient way than the sole silencing of Smurf1. However, since the Wnt/β-catenin pathway is crucial not only for determining cell fate, but also in the regulation of several cell cycle events, we first needed to verify that the silencing of Wnt/β-catenin inhibitors did not alter key processes such as cell proliferation, cell death or cell migration. Additionally, since there is increasing evidence that the canonical and non-canonical Wnt pathways are intersecting networks, we also needed to verify that silencing of the inhibitors of the Wnt/β-catenin pathway selected for this work (Cby1 and Sfrp1) did not influence the activity of the non-canonical Wnt pathways.
We describe here how
Sfrp1 silencing in MSCs can considerably accelerate bone formation both in vitro and in vivo in an ectopic mouse model, something that could be advantageous in clinical practice, for example, to reduce the length of fracture healing. Importantly, the pro-osteogenic effect obtained by the silencing of
Sfrp1 significantly exceeds that achieved by
Smurf1 silencing, particularly in an in vivo setting [
6]. Although alteration of the expression levels of
Sfrp1 in some cell lineages has been linked to different pathological conditions including tumor formation, we show here that transiently silencing
Sfrp1 in MSCs does not seem to lead to any abnormal biological behavior that could compromise the biosafety of these cells. Additionally, no concomitant activation of other Wnt pathways has been detected. Besides its greater osteogenic efficiency, the use of
Sfrp1 silencing for enhancing bone formation has an additional advantage over the silencing of
Smurf1, since
Sfrp1 silencing using LNA-ASOs in MSCs is able to achieve a similar degree of osteogenic induction to that attained by direct BMP-2 stimulation. As BMP-2 is not needed to ensure the activation of the Wnt/β-catenin pathway, this approach would completely avoid the potential side effects currently associated with the use of this recombinant protein in the clinic [
23] and considerably reduce the cost of treatments. Our results also highlight the efficacy of the inhibitors of the Wnt/β-catenin pathway in bone regeneration.
2. Materials and Methods
2.1. Cell Isolation
Human MSCs were isolated from femoral heads of osteoporotic patients undergoing hip replacement surgery due to an osteoporotic fracture, following previously described protocols [
7]. A reduced number of colonies (on average < 10) appeared between five and seven days after seeding. Once the initial culture was 80–90% confluent, cells were expanded to achieve a final cell number between 3–5 × 10
5. Due to the risk of those cells undergoing replicative senescence with subsequent passages, only cells in passage 1 were routinely used for all experiments.
A total of five samples obtained from female patients aged 65 to 85 were included in the study. The study protocol (Identification Code 2016.159) was approved on 17 October 2016 by the Institutional Review Board (Comité Ético de Investigación Clínica de Cantabria).
2.2. Cell Culture and Differentiation
The immortalized murine MSC line C3H10T1/2 was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Waltham, MA, USA) supplemented with 10% FBS and 1% penicillin-streptomycin. For osteogenic differentiation, C3H10T1/2 cells were plated at 12,500 cells/cm2 and hMSCs at 20,000 cells/cm2, allowing them to attach overnight. To induce differentiation, culture medium was replaced with osteogenic media 24 h later. C3H10T1/2 were differentiated with DMEM supplemented with 50 µM ascorbic acid, 20 Mm β-glycerolphosphate, and 1 µM dexamethasone. Human primary MSC osteogenesis was induced with DMEM supplemented with 50 µM ascorbic acid, 10 Mm β-glycerolphosphate, and 0.1 µM dexamethasone.
For C3H10T1/2 adipogenic differentiation, culture medium was replaced with adipogenic media. DMEM (10% FBS and 1% penicillin-streptomycin) supplemented with 1 µM dexamethasone, 2 µM rosiglitazone, 5 µg/mL insulin, and 0.5 mM isobutyl methylxanthine (IBMX).
2.3. GapmeRs Design
Antisense LNA GapmeRs were purchased from Exiqon (Qiagen, Venlo, The Netherlands). As a control, an Antisense LNA GapmeR Negative Control A (Ref. 339516) was used.
2.4. Flow Cytometry
A FACSCanto II flow cytometer and FACSDiva Software version 6.1.2 (BD Bioscience, San Diego, CA, USA) were used for the cytometry assays. GapmeR delivery efficiency after lipofection was tested as described in [
6] by using FAM-Control labeled GapmeRs.
For the apoptosis assay, FITC Annexin V Apoptosis Detection Kit I (556547, BD Bioscience, San Diego, CA, USA) was used following the protocol described by the manufacturer.
2.5. Cell Transfection
Lipofection was performed using Dhamafect (Dharmacon, Horizon Discovery, Cambridge, UK). following instructions from the manufacturer. The C3H10T1/2 cell line was seeded at 12,500 cells/cm2, and human primary cells at 20,000 cells/cm2. Two hours prior to transfection, culture media was replaced with Opti-MEM I media (Invitrogen, Waltham, MA, USA). In order to increase transfection efficiency, no serum supplementation was added to these media for the transfection procedure. The recommended amount of Dharmafect was mixed with Opti-MEM I. On the other hand, to prepare the GapmeR mix, each of the GapmeR or GapmeRs combinations was also mixed with Opti-MEM I. After 5 min of incubation at room temperature (RT), GapmeR mix was added dropwise on top of the Dharmafect mix, and incubated for 20 min at RT. The mix was then added to the cells dropwise. Twenty-four hours after transfection, one volume of DMEM with 10% FBS and 1% penicillin-streptomycin was added to the wells and incubated at 37 °C for another 24 h. Forty-eight hours after transfection, cells were washed once with PBS and fresh complete culture media was added.
For in vivo experiments, right after transfection, 50,000 cells were seeded onto discoidal alginate scaffolds prepared as previously described [
6]. Cell-loaded scaffolds were incubated a minimum of 20 min to allow cell attachment to the scaffold prior to implantation in the recipient mice. Longer incubation times (up to 24 h) could also be applied if necessary. In this occasion, cells were incubated with the scaffold for 14 h prior implantation.
2.6. Proliferation Assay
For the proliferation assay, cells were transfected in 6-well plates and harvested 48 h after transfection. After estimation of cell number, different cell concentrations (100, 200, 300, 400 cells per well) were seeded onto a 96-well plate in triplicate and allowed to attach overnight. Then, culture medium was changed, and cells were allowed to proliferate for the required number of days. Plates were harvested at days 1, 3, 5, 7, and 9. To determine cell viability, the medium was changed to DMEM (10% FBS and 1% penicillin-streptomycin) containing 0.5 mg/mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Cells were incubated 4 h at 37 °C. After this incubation, the medium was removed, 100 μL of 2-propanol was added to the wells, and the plate was incubated at 37 °C for 10 min. When the incubation time was finished, absorbance at 570 nm was measured with a plate lector Eon (BioTek, Winooski, VT, USA).
For the Ki67 immunofluorescence assay, cells were seeded on 10 mm diameter crystals, pretreated with gelatin 0.1%, then transfected as described above. A Ki-67 monoclonal antibody (SP6: MA5-14520, Invitrogen, Waltham, MA, USA) was used as a primary antibody. A mouse anti-rabbit secondary antibody labeled with FITC was used for subsequent detection (sc-2359, Santa Cruz Biotechnology Inc., Dallas, TX, USA). Crystals were observed under a fluorescence microscope, where pictures were taken and analyzed with ImageJ 1.53 c software (
http://imagej.nih.gov/ij) to score positive cells.
2.7. Migration Assay
To study cell migration capability, a wound healing assay was performed. For this, cells were seeded at a high confluence in 6-well plates (25,000 cells/cm2), and transfected following the protocol already described. Forty-eight hours after transfection, MesenPro medium was added to the cells and 400 µM wide wounds were performed in each plate. Pictures were taken at 3 h intervals for 72 h. For each of the samples, six different areas along the wound were recorded and analyzed. Wound area was measured every 12 h. Images were analyzed using the ImageJ 1.53 software to evaluate the wound size by quantification of the empty areas occupied by the lesion.
2.8. Staining Quantification
Alizarin Red quantification was performed following a previously described method [
24]. Oil Red quantification was also performed following previously described protocols [
25]. Absorbance lecture of the plates was performed using an Eon Microplate Spectrophotometer (BioTek, Winooski, VT, USA).
2.9. Gene Expression
mRNA was isolated from cell cultures, after washing cells twice with PBS and collecting them with TRIzol reagent (Invitrogen, Waltham, MA, USA) by scraping the plate surface. RNA extraction was done following the protocol of the TRIzol manufacturer. Between 1 and 1.5 μg of RNA were isolated from a confluent 24-well plate. RNA retro-transcription was performed with the PrimeScript RT Reagent Kit (RR037A, Takara Bio Inc, Shiga, Japan) following the manufacturers’ instructions. A total of 500 ng of RNA was used in a 10 μL reverse transcription reaction. The resulting cDNA was diluted four times with ddH2O. One μL of this solution was used for each PCR reaction.
To measure the gene expression levels, semi-quantitative PCRs were performed using Taqman assays (Applied Biosystems, Waltham, MA, USA). To test gene silencing in the murine cell line C3H10T1/2, the following Taqman assays were used: Mm00503802_m1 (Cby1), Mm00489161_m1 (Sfrp1), Mm00547102_m1 (Smurf1), and Mm01496371_m1 (Tob1). To analyze gene expression of the Runx2, Alpl, Bglap, Lpl, and Pparg genes, the following assays were used: Mm00501578_m1, Mm01187117_m1, Mm03413726_mH, Mm00440940_m1, and Mm00434764_m1, respectively. Mouse housekeeping genes GAPDH (Assay Mm99999915_G1) and RPL13A (Assay Mm0162986_gH) were used for normalization.
For the gene expression analyses in the primary hMSCs the following Taqman assays were used: Hs00360360_m1 (Cby1), s00610060_m1 (Sfrp1), Hs00410929_m1 (Smurf1), Hs03986111_s1 (Tob1), Hs00231692_m1 (Runx2), Hs00758162_m1 (Alpl), and Hs01587814_g1 (Bglap). Human GAPDH (Hs99999905_m1) and RPL13A (Hs04194366_g1) genes were used for normalization.
Amplification conditions for all experiments were as follows: One initial denaturation cycle of 10′ at 90 °C, followed by 40 cycles with 15 seconds denaturation at 90 °C and 1 min extension at 60 °C.
2.10. Western Blot Analysis
To prepare the protein extracts, 1.2 million cells per condition were transfected. Forty-eight hours after transfection, cells were washed, harvested, and centrifuged. The supernatant was discarded, and cells were resuspended in 100 µL per million of cells of Pierce RIPA buffer (89900, Pierce Biotecnology, Waltham, MA, USA) supplemented with protease inhibitors to obtain a cell extract. On average, 100 μg of proteins were obtained for each of the conditions. Routinely, 15 to 20 μg of protein extract per well were loaded onto a precast NuPAGE 4–12% Bis-Tris Gel (Invitrogen, Waltham, MA, USA).
For the cytosolic and nuclear protein fractions collection, NE-PER Nuclear and Cytoplasmatic Extraction Reagents were used (Pierce Biotecnology, Waltham, MA, USA) following the protocol provided by the manufacturer. Protein concentration was determined with the BCA Protein Assay (Pierce Biotecnology, Waltham, MA, USA). The same amount of protein extract per condition was loaded onto the polyacrylamide gel. Once the run was completed, proteins were transferred to a nitrocellulose membrane using an iBlot 2 Dry Blotting System (Invitrogen, Waltham, MA, USA) and iBlot 2 NC Regular Stacks (Invitrogen, Waltham, MA, USA). Membranes were blocked 1 h at RT with 5% BSA, and incubated overnight at 4 °C with specific primary antibodies. Primary antibodies used were GAPDH (MAB347, MERK, Poole, Dorset, UK), p-JNK (sc-6245, Santa Cruz Biotechnology Inc., Dallas, TX, USA), p-PKC δ/θ (#9376, Cell Signaling Technology Inc., Danvers, MA, USA ), and β-catenin (ab6302, Abcam, Cambridge, UK).
Membranes were then washed with TBS-T and incubated 1 h with specific secondary antibodies: IRDye 680LT Goat anti-rabbit (926-68021, LI-COR, Lincoln, NE, USA) and IRDye 800CW Donkey anti-Mouse (926-32212, LI-COR, Lincoln, NE, USA). Membranes were revealed with the LI-COR Odyssey Imaging System, and membrane signals revealed were analyzed and quantified with the Image Studio 5.2 program (LI-COR, Lincoln, NE, USA).
2.11. In Vivo Experiments
All surgical procedures were performed under isoflurane anesthesia in sterile conditions as previously described [
26]. Discoidal alginate scaffolds (4 mm in diameter and 1 mm thickness) were implanted subcutaneously in C57/BL6 12 weeks old female mice on both sides of the dorsal line, leaving at least 1 cm separation between them. Scaffolds were seeded with un-transduced mouse MSCs (mMSCs) or mMSCs transduced with the Control (Ctrl),
Smurf1,
Sfrp1,
Cby1, and
Tob1 GapmeRs and their combinations following the transfection conditions and experimental procedures previously described for in situ transfection. Scaffolds with no cells were also used as the control. Scaffolds were incubated with the cells in the standard growing media used for those cells for a minimum of 20 min and a maximum of 24 h before implantation. A fourteen hours incubation was used in this occasion. Scaffolds used have a dose of 3 μg BMP2 as previously described [
6]. All scaffolds carrying cells transfected with the different GapmeRs were surgically implanted in the same animal to avoid individual variations. As an example, three animals were implanted with all the control scaffolds plus one scaffold seeded with cells transfected with the
Smurf1 GapmeR, one scaffold seeded with cells transfected with the
Sfrp1 GapmeR and one scaffold with cells transfected with a combination of the two GapmeRs tested (
Smurf1/
Sfrp1). A total of nine animals were used for the procedure. Three different experimental groups including three animals per group were established in the experiment, each one carrying a different GapmeR combination. Each of the animals received six different scaffolds. Insertion of the scaffold was performed through a cut in the skin of approximately 0.5 cm. To close the incision, the skin was stapled. Twelve weeks after the procedure, animals were euthanized by CO
2, implants were removed from the mice, and fixed in 10% formaldehyde, decalcified, and embedded in paraffin. Five micron sections were prepared for the different analyses.
All experiments performed in animals were reviewed and approved on August 2016 by the Institutional Bioethics and Animal Care Committee of the University of Cantabria (Project identification code 2015721, PI08/16) and the competent authority (Consejería de Agricultura y Ganadería de Cantabria, Santander, Spain).
2.12. Histological Analysis
The implants extracted from the mice were fixed in 10% formaldehyde for 6 h and decalcified in 20% EDTA in PBS (pH 7.4) for a week at 4 °C with two weekly changes of the descaling solution. Once decalcified, they were embedded in paraffin and 5 micron sections were obtained that were deparaffined and stained following standard protocols with topographic hematoxylin/eosin staining [
25], and Masson-Goldner Trichrome [
26] or Sirius Red histochemical stains [
27] for collagen detection. For the indirect immunoenzymatic technique, the deparaffined and rehydrated sections in Tris buffered saline (TBS) (pH 7.4, 0.01 M Trizma base, 0.04 M Tris hydrochloride, 0.15 M NaCl) were subjected to antigen retrieval in citrate buffer (pH 6) for 5 min at 90 °C and subsequently blocked in 2% FBS in 0.2% TBS-Triton (blocking buffer). For the study of osteogenic differentiation and neovascularization, the sections were incubated with polyclonal anti-osteocalcin (OCN) antiserum (1/100) (Millipore) and polyclonal anti-CD34 antiserum (1/100) (Abyntek Biopharma, Derio, Spain ), respectively, in blocking buffer overnight at 4 °C. The sections were then washed with TBS and incubated with a donkey anti-rabbit IgG antibody conjugated with biotin (1/500) (Millipore, Poole, Dorset, UK) for 60 min and then with streptavidin-peroxidase (1/500) (Millipore, Poole, Dorset, UK) for 60 min. Peroxidase activity was demonstrated in 0.005% 3,3‘-diaminobenzidine (Sigma-Aldrich, Poole, Dorset, UK) and 0.01% hydrogen peroxide in Tris-HCl buffer (0.05 M, pH 7.6). The specificity of the immunostaining with both antisera was confirmed by replacing them with normal serum. Vascular density and OCN staining was evaluated using computer-based image analysis software (Leica Q-win V3 Pro-Image Analysis System). To measure vascular density, the CD34 positive staining blood vessels were counted in five different areas in implant sections for each sample and expressed in absolute value as the number of vessels per unit area (microscopic field). OCN staining was measured by applying a fixed threshold to select for positive staining within the implant region. Positive pixel areas were divided by the total surface size (mm
2) of the implant. Values were normalized to those measured from Control GapmeR group and are reported as relative staining intensities.
2.13. Statistical Analysis
Error bars on graphs represent the standard error of the mean values. Depending on sample size, statistical significance was calculated using the Students’ t test (for n > 5) or the Mann–Whitney U test (for n = 3). To assay the bone formation in the mouse ectopic model, statistical analysis was performed with SPSS.25 software by means of a one-way analysis of variance (ANOVA) with a Tukey multiple comparison post-test. Significance was set at p < 0.05.
4. Discussion
The numbers and potency of MSCs are key to maintaining normal bone physiology, therefore, modifications aimed to increase MSCs osteogenic potential could result in being highly useful to develop efficient therapies to promote bone regeneration. Modulating the two main osteogenic pathways, the BMP and the Wnt/β-catenin signaling pathways, in order to increase or extend their activity seems a reasonable approach to achieve this goal. We have previously shown that transiently silencing
Smurf1, one of the main inhibitors of the BMP pathway, using LNA-ASOs, leads to a significant increase in bone formation, even in the presence of low BMP doses [
6]. This modification of MSCs allowed us to increase bone formation by at least 30%. In this current work, we set out to further improve this system in order to achieve superior bone regeneration by targeting other inhibitors of the two main osteogenic pathways.
The relevance of the canonical Wnt signaling pathway in bone formation is underscored by the direct relation of mutations that activate or inactivate this route with sclerosteosis and osteoporosis, respectively. Different approaches have been designed to stimulate this osteogenic pathway, either through the upregulation of key players of the signaling cascade or through the inhibition of endogenous antagonists. A good example of the latest is the recently, FDA approved humanized anti-sclerostin monoclonal antibody Romosozumab (Amgen, Thousand Oaks, CA, USA), used for the treatment of osteoporosis. The suitability of other members of this route as potential targets for bone regeneration techniques are currently under investigation. Extracellular modulators of the Wnt/β-catenin pathway also seem to have a key role in bone regeneration. One of these modulators,
Sfrp1, acts as an inhibitor of the Wnt canonical pathway by binding to Wnt ligands and preventing the activation of this pathway. Although the manipulation of the Wnt signaling cascade could bring several potential benefits, as shown by the use of Romosozumab, alterations of this route have also been linked to various pathologies including cancer, and thus, these manipulations should be performed with extreme caution [
34]. While
Sfrp1 loss of expression in other cell types has been linked to abnormal cell proliferation, we did not detect significant changes in either cell proliferation or apoptosis in MSCs upon
Sfrp1 silencing. Another possible problem that could be encountered after
Sfrp1 silencing is the increase in cell migration and invasion related to an induction in a partial epithelial-mesenchymal transition [
35]. This has been reported in a non-malignant cell line (mammary epithelial cell line) and was reflected by an increased motility in a scratch wound assay noticeable after just 8 h incubation. Using a similar assay, we did not find either a significant increase in the motility of MSCs transfected with an LNA-ASO specific for
Sfrp1 after 72 h of incubation. Therefore, our results would indicate that the transient silencing of this gene in MSCs does not seem to produce an increase in the tumorigenic potential of the transfected MSCs. This is particularly important since these cells are ultimately intended for therapeutic use. It is noteworthy that although, as previously shown by us,
Smurf1 silencing using LNA-ASOs led to a significant increase in the osteogenic potential of MSCs, the wound healing experiments showed that these MSCs where
Smurf1 or
Cby1 have been silenced show a slightly reduced migration capacity, something that could have a negative effect on fracture healing, particularly in aged or osteoporotic patients in which MSCs already feature a reduced migration ability [
36,
37]. Besides, this reduced migration could also limit the application of these cells in some treatments where migration of exogenous cells to the site of damage is required.
Since the canonical Wnt signaling pathway seems to crosstalk with other Wnt pathways, to be able to use Sfrp1 silencing in MSCs, either alone or in combination with other inhibitors of osteogenic pathways, as a strategy to safely potentiate bone regeneration, the effect of its silencing in other signaling pathways should also be assessed. As expected, upon Sfrp1 silencing, we did detect a significant increase in the nuclear β-catenin, indicating an activation of the Wnt/β-catenin signaling pathway, however, no apparent changes were detected in the selected mediators of the other two Wnt pathways that activate diverse biological functions including proliferation, apoptosis, and invasion. The same would apply to the case of Cby1, the other Wnt/β-catenin inhibitor silenced in this study. These results would agree with the outcome of our Ki67, MTT, and Annexin V analyses, which showed no alteration of those processes upon silencing of Sfrp1, or any of the other targeted genes, in MSCs. Overall, we could not find any indication that transiently silencing Sfrp1 in MSCs using LNA-ASOs would be linked to any biosafety concern.
Of all the inhibitors and inhibitor combinations tested, our in vitro results clearly show that
Sfrp1 silencing has the more marked effect in osteogenesis, overcoming those of our previous selected target,
Smurf1, and the other inhibitors tested in this work. RUNX2 is a master regulator of osteoblastic differentiation and plays a critical role in the early stages of this process, however, its expression needs to be downregulated to allow for the expression of later genes and terminal differentiation of osteoblasts [
38]. We observed that MSCs where
Sfrp1 expression had been silenced in combination with
Smurf1 and
Cby1 showed a significant downregulation of
Runx2 expression at 11 days of differentiation. On the other hand,
Sfrp1 silencing either alone or in combination with
Smurf1 also led to a clear increase in the
Alpl expression, although this was statistically significant only for the
Smurf1/
Sfrp1 GapmeR combination. Only the combination of
Sfrp1 with
Smurf1 led to a significant up-regulation of the later osteogenic marker
Bglap. It is important to note that the earlier downregulation of
Runx2 and upregulation of late osteogenic genes seen with the
Smurf1/
Sfrp1 GapmeR combination would be indicative of an accelerated osteogenesis. The upregulation of
Alpl at the gene expression level was also in agreement with the quantified ALPL activity. In this case,
Sfrp1 and
Smurf1 were the only GapmeRs able to significantly increase this activity when tested individually. Altogether, these data suggest an increased osteogenic potential of cells treated with the
Sfrp1 GapmeR alone or in combination with the
Smurf1 GapmeR. An enhanced mineralization upon
Sfrp1 silencing, as measured by Alizarin Red staining, further supports this notion. Despite the efficient bone formation elicited by the silencing of
Smurf1 in vivo [
6], neither significant changes in
Alpl expression nor increased mineralization were detected in vitro when this gene was silenced. It is important to highlight, though, that, except for
Smurf1, the silencing of the other anti-osteogenic genes also led to an increase in mineralization compared to the control. Interestingly, when cells were grown in adipogenic media, we observed a decrease in viability of cells transfected with
Smurf1/
Sfrp1 or with
Smurf1/
Tob1 GapmeRs, although the initial decrease in proliferation seems to be recovered later on. Since this initial phase would coincide with the clonal expansion of adipogenic progenitors prior to proliferation, this might reflect a reduced adipogenic capacity. This idea would be further supported by a reduced Oil Red staining of the cells transfected with
Sfrp1 and
Smurf1 growing under adipogenic conditions, as expected by the upregulation of the canonical Wnt/β-catenin pathway [
39].
The in vitro results were further validated in our in vivo ectopic model where
Sfrp1 silencing also performed significantly better than the rest of the targeted genes. This ectopic system allowed us to specifically evaluate the contribution of the implanted cells to bone formation. The analysis of the cell-loaded scaffolds showed that all types of MSCs transplanted were able to form bone tissue to a greater or a lesser extent after 12 weeks, however, those cells where
Sfrp1 had been silenced, either alone or together with
Smurf1 were able to produce a significantly higher amount of bone matrix in vivo, as reflected by the larger areas of collagen fibers shown by Masson–Goldner trichromic and Sirius Red stainings and by the analysis of the samples under fluorescent light. Immunohistochemistry analysis to detect the presence of osteocalcin (OCN), encoded by the
Bglap gene in the different scaffolds, showed a significantly higher signal than the control in all samples, except for
Cby1 when compared to the control sample (
Supplementary Figure S4b). Since OCN is produced by osteoblasts and is used as a late marker in the process of bone formation, these results suggest the presence of a more mature bone matrix in those samples. Importantly, unlike the rest of the samples tested, the scaffolds seeded with MSCs where
Sfrp1 had been silenced reached a degree of angiogenesis similar to that of the MSCs transfected with the Ctrl GapmeR. Since normal angiogenesis is key for correct bone healing [
40], this further supports the choice of
Sfrp1 as the best target to achieve efficient bone regeneration through the LNA-ASO mediated silencing of osteogenesis inhibitors in MSCs. It is noteworthy that although the simultaneous silencing of
Smurf1 and
Sfrp1 seems to have, in vitro, an additive effect in the expression of osteogenic markers as well as in the alkaline phosphatase activity, no significant improvement in bone formation was detected upon simultaneous silencing of
Smurf1 and
Sfrp1 in our in vivo experiments, suggesting that the sole silencing of
Sfrp1 would suffice to efficiently prime MSCs in vivo toward the osteogenic differentiation path.
It has been suggested that the anabolic effect of the parathyroid hormone (PTH), an FDA approved osteo-anabolic drug commonly used for osteoporosis treatment, requires
Sfrp1 downregulation to stimulate Wnt signaling and osteoblastogenesis [
41]. Thus,
Sfrp1 silencing would have a similar effect to that of the PTH but would lack the detrimental side effects of this drug that highly limit the length of the treatment [
42], something extremely important due to the chronic character of that pathology. Furthermore, our results using hMSC-OP show that
Sfrp1 silencing in those cells can achieve a similar or even greater osteogenic induction than that obtained through BMP-2 stimulation, therefore, the use of
Sfrp1 inhibition would have the additional advantage of avoiding the need of concomitantly administrating BMP-2 in bone regeneration therapies based on MSC transplantation and thus, of avoiding the unwanted side effects linked to the use of this protein in the clinic.
In the last few years,
Sfrp1 has been the focus of several studies related to bone regeneration. In fact, reports in mice where this gene has been inactivated have shown that fracture healing is potentiated in the absence of this gene [
20]. This, and other works confirming the importance of
Sfrp1 in osteogenesis, have led to the development of different approaches aimed to achieve a safe
Sfrp1 inhibition that could be used for bone regeneration therapies. One study showed that diphenyl sulfone sulfonamide, a small-molecule inhibitor of
Sfrp1 that disrupts the interaction between Wnt and
Sfrp1, is able to stimulate bone formation ex vivo [
43]. More recently, the discovery that different miRNAs such as miR542-3p and miR144 have a positive effect on osteogenesis through
Sfrp1 downregulation [
44,
45] has opened the possibility of using these molecules as therapeutic agents to treat bone loss associated diseases. However, miRNA activity is pleiotropic, with a single miRNA repressing numerous targets, which in many cases are not even completely identified. This lack of specificity would also apply to drugs such as the aforementioned sulfone sulfonamide. Thus, in therapies based on the use of drugs or miRNAs mimics, it might be challenging to delineate all the possible effects in cells other than MSCs. This would be an obvious concern from the biosafety point of view.
Due to its ability to selectively modulate gene expression and the fact that they are a clinically safe method, LNA-ASOs have countless clinical applications. This is highlighted by the increasing number of oligonucleotide-based drugs gaining approval [
46]. Despite the promising results of these molecules, the application of LNA-ASOs in the clinic has been hindered until recently by the difficulties in achieving efficient delivery to target organs. This would be particularly important in the case of
Sfrp1, since it has the ability to, depending on the cellular context, act as an agonist or as an antagonist of the Wnt signaling pathway [
47]. In this work, we have shown that the transient silencing of
Sfrp1 using these molecules is enough to efficiently prime MSCs toward the osteogenic route and favor an efficient bone matrix formation, even in osteoporotic MSCs where this osteogenic capacity is already limited. Treatment of a chronic pathology such as osteoporosis needs a very stringent biosafety profile. Although we have found no alteration of cell proliferation, apoptosis, or migration upon
Sfrp1 silencing in vitro, given the known involvement of Wnt signaling in certain types of cancer, translation of this method to the clinic will require further molecular analysis such as analysis of important tumor suppressor genes (p53), karyotyping, etc. and extensive in vivo pre-clinical studies to ensure long-term safety of the treatment. Additionally, to increase the biosafety of treatments involving Wnt modulators, bone should be targeted as specifically as possible to prevent systemic stimulation of the pathway. Combination of GapmeRs specifically designed for
Sfrp1 silencing with one of the targeted delivery methods that are currently being developed would open the possibility of specifically silencing this gene in bone marrow MSCs and thus safely undertake the treatment of systemic bone loss such as that associated with osteoporosis.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





