
Lipid Nanocarriers for Anti-HIV Therapeutics: A Focus on Physicochemical Properties and Biotechnological Advances





Abstract
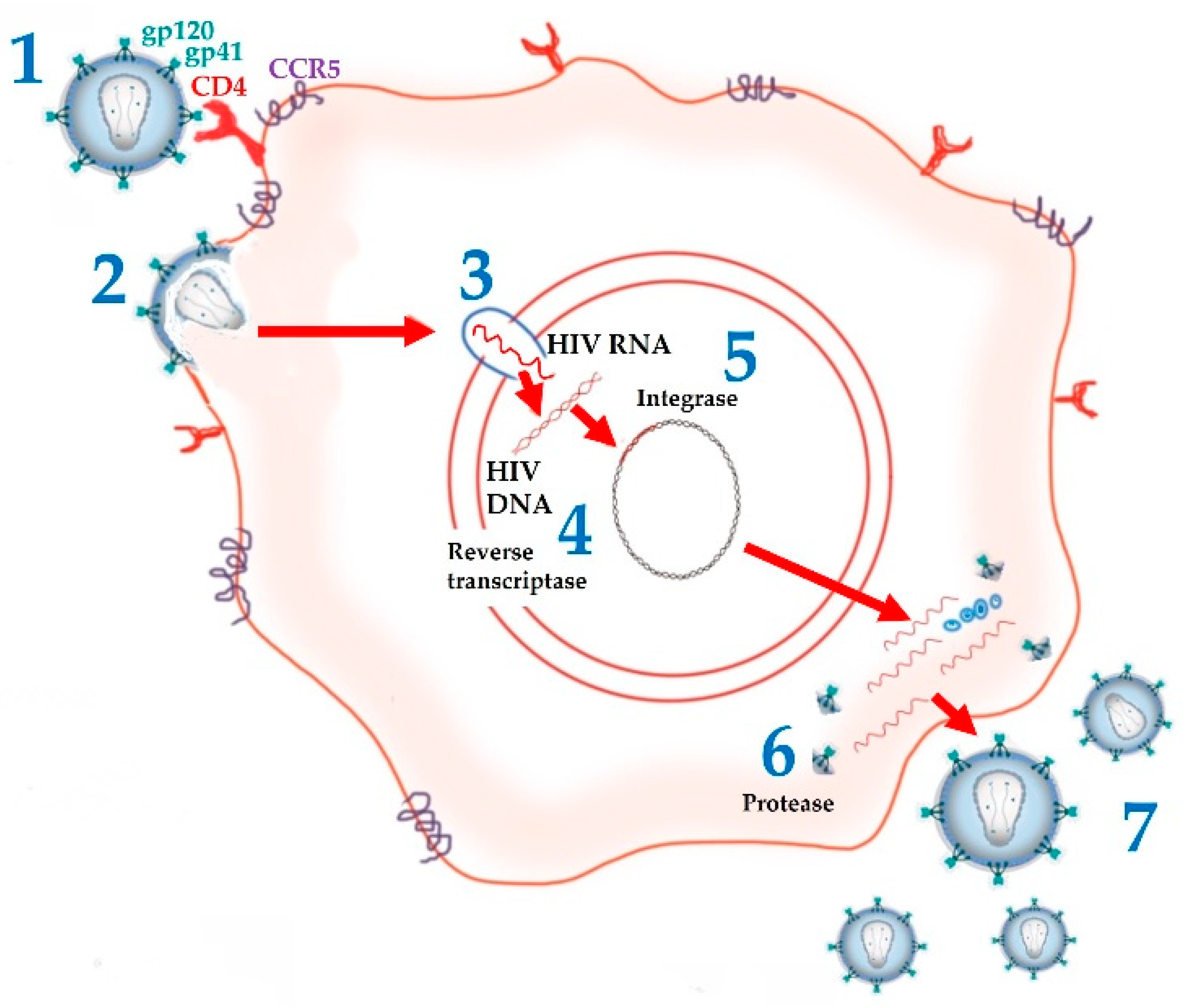
:1. Introduction
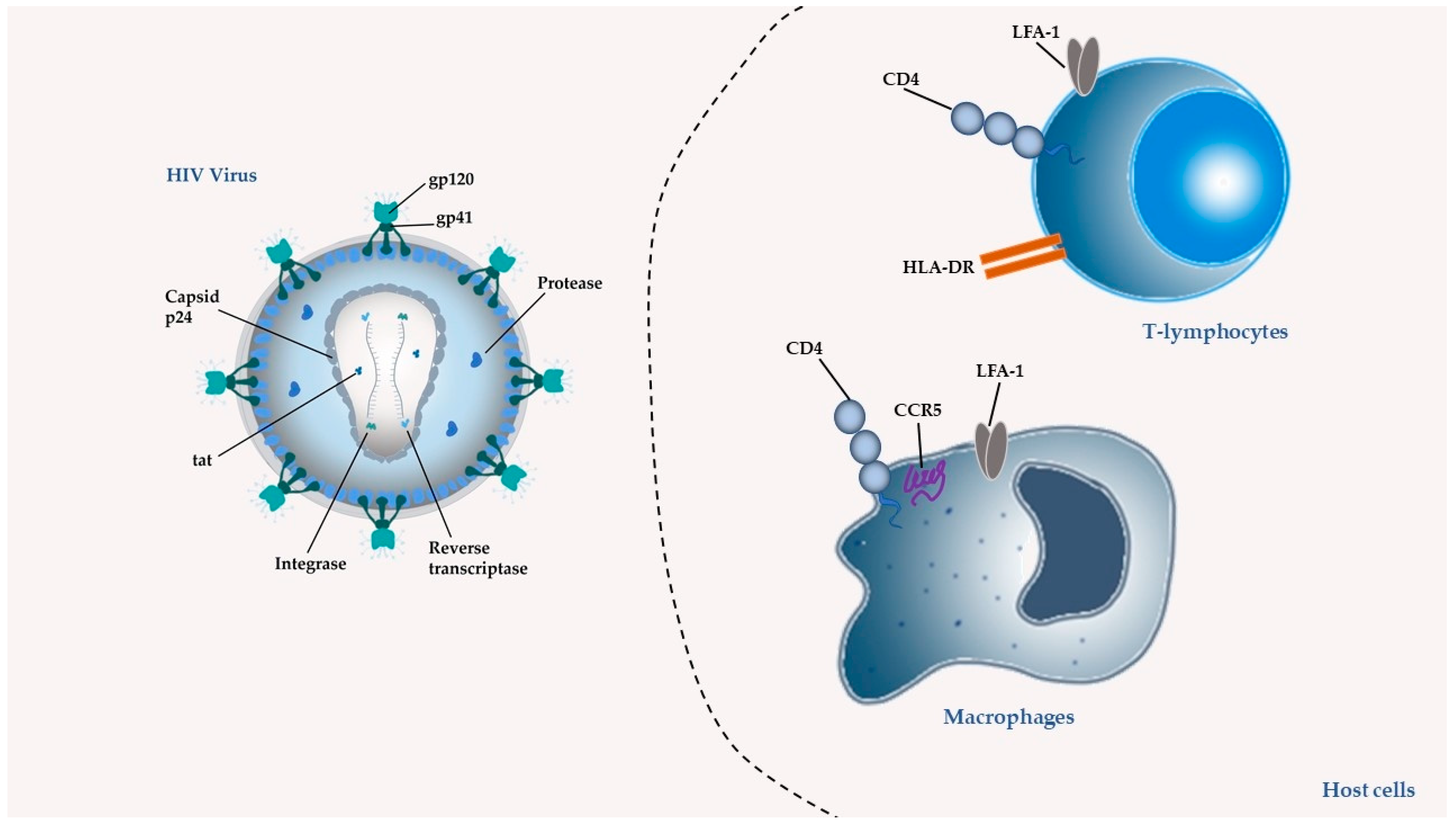
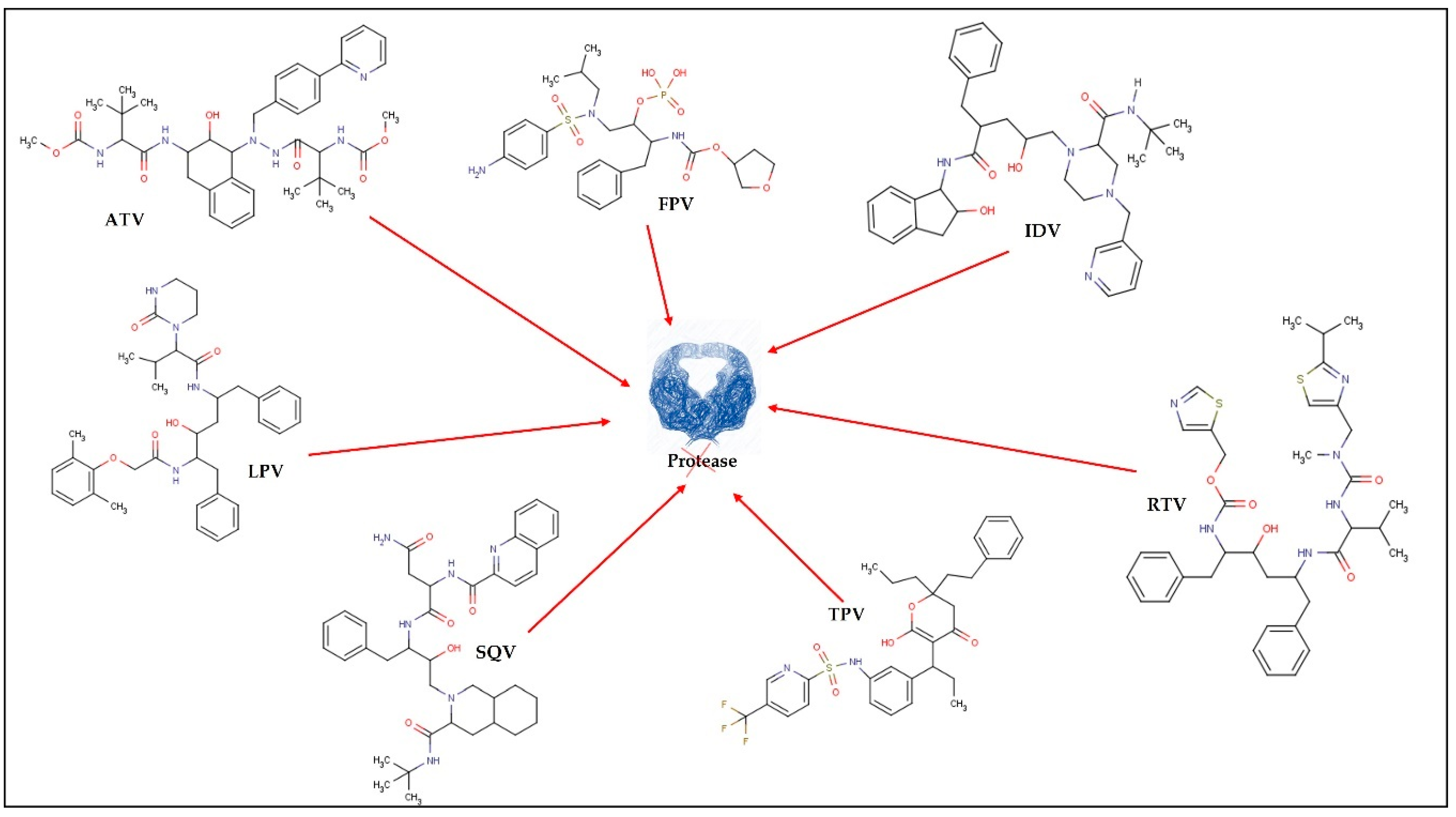
2. ARV Agents: Mechanism of Action and Limitations
3. Lipid-Based Nanocarriers for Delivery of ARV Agents
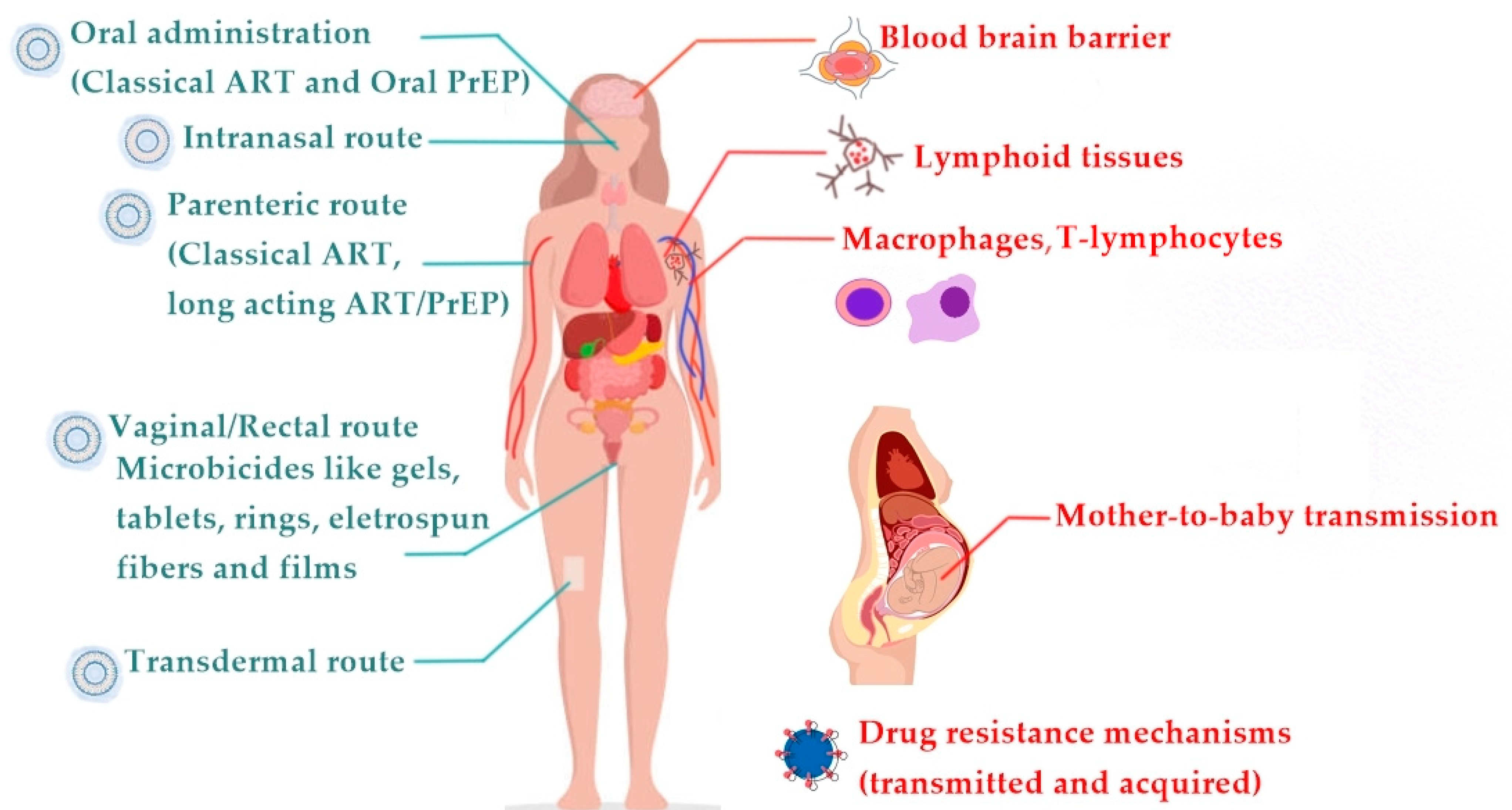
3.1. Tuning the Physicochemical Properties of Lipid-Based Nanocarriers to Overcome Biological Barriers
3.2. Targeting Anatomical and Cellular Reservoirs
4. Biotechnological Advances in ARV Delivery
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
List of Abbreviation
| 3TC | lamivudine |
| AβP | amyloid β-peptide |
| ABC | abacavir |
| AD | Alzheimer’s disease |
| AGO | Argonaute protein |
| AIDS | acquired immunodeficiency syndrome |
| APP | amyloid-β precursor proteinARV–antiretroviral |
| ATV | atazanavir |
| Au | gold |
| AUC | area under the curve |
| AV | aloe vera |
| AZT | zidovudine |
| AZT-M | zidovudine myristate |
| AZTTP | azidothymidine 5′-triphosphate |
| BBB | blood-brain barrier |
| BHT | butylated hydroxy toluene |
| Brij®-35 | polyoxyethylene (23) lauryl ether |
| BSA | bovine serum albumin |
| CAB | cabotegravir |
| Capmul® MCM | mono/diglyceride of caprylic acid |
| Capmul® MCM EP | glycerol monocaprylocaprate |
| Capmul® PG 8 | propylene glycol monocaprylate |
| CapryolTM 90 | propylene glycol monocaprylate |
| Captex® P 500 | triglycerides and esters prepared from fractionated vegetable oil sources and fatty acids from coconuts and palm kernel oils |
| CC50 | concentration at which 50% cells are viable |
| cART | combined antiretroviral therapy |
| CaSki | epidermoid cervical cancer cell line |
| CCR5 | C-C chemokine receptor type 5 |
| CD4 | cluster of differentiation 4 |
| Chol | cholesterol |
| Cmax | maximum concentration |
| CNS | central nervous system |
| Compritol® 888 ATO | glycerol dibehenate |
| COVID-19 | coronavirus disease 2019 |
| Cremophor® EL | castor oil fatty acids, ethoxylated glycerol ester |
| Cremophor® RH 40 | polyoxyl 40 hydrogenated castor oil |
| CTAB | cetyltrimethylammonium bromide |
| d4T | stavudine |
| DCP | dicetyl phosphate |
| ddC | zalcitabine |
| ddI | didanosine |
| ddI EC | enteric coated didanosine |
| Dex–Prot | dextran–protamine; |
| D.L. | drug loading |
| DLV | delavirdine |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DMPE | 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine |
| DMPG | 1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| DNA | deoxyribonucleic acid |
| DODAB | dioctadecyl dimethylammonium bromide |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| DOR | doravirine |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| DPPE-PEG2000 | 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethylene glycol)-2000] |
| DPPG | 1,2-dipalmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| DPTAP | 1,2-dipalmitoyl-3-trimethylammonium-propane (chloride salt) |
| DRV | darunavir |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| DSPE | 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine |
| DSPG | 1,2-distearoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| DTG | dolutegravir |
| Dynasan® 114 | trimyristin |
| Dynasan® 118 | glyceryl tristearate |
| EDPPC | cationic 1,2-dipalmitoylethyl-phosphatidylcholine |
| E.E. | entrapment efficiency |
| EFV | efavirenz |
| EPC | egg phosphatidylcholine |
| ETR | etravirine |
| EVG | elvitegravir |
| FIs | fusion inhibitors |
| FPV | fosamprenavir |
| FTC | emtricitabine |
| FTR | fostemsavir tromethamine |
| GC | glyceryl caprylate |
| Gelucire® 44/14 | lauroyl polyoxyl-32 glycerides |
| Gelucire® 50/13 | stearoyl polyoxyl-32 glycerides |
| GI | gastrointestinal |
| GMO | glyceryl monooleate/monoolein |
| GMS | glyceryl monostearate |
| gp41 | glycoprotein gp41 |
| gp120 | glycoprotein gp120 |
| GRAS | generally recognized as safe |
| HAART | highly effective antiretroviral therapy |
| HBMECs | human brain microvascular endothelial cells |
| HEC-1-A | human endometrial cancer-1 |
| HG | hydrogel |
| HIV | human immunodeficiency virus |
| HSA | human serum albumin |
| HSPC | hydrogenated soy phosphatidylcholine |
| IBA | ibalizumab-uiyk |
| IDV | indinavir |
| II | integrase inhibitors |
| Labrasol® | caprylocaproyl polyoxyl-8 glycerides |
| LauroglycolTM | 90 propylene glycol monolaurate |
| LDL | low-density lipoproteins |
| LFA-1 | lymphocyte function-associated antigen 1 |
| Lipoid® S 75 | fat-free soybean phospholipids with 70% PC |
| logP | partition coefficient |
| LP | liposome |
| LPV | lopinavir |
| mAb | 83-14 monoclonal antibody |
| Maisine® 35-1 | glyceryl monolinoleate |
| MAL | maleimide |
| Man | mannose |
| MCC | microcrystalline cellulose |
| ME | microemulsion |
| Miglyol® 812 | medium-chain triglycerides |
| MLN | multiple lipid nanoparticles |
| MNP | magnetic nanoparticles |
| MonosteolTM | palmitate/stearate of propylene glycol |
| mPEG | methoxyl poly(ethylene glycol) |
| MPEG 2000 | mono methoxy PEG 2000 |
| MPS | mononuclear phagocyte system |
| mRNA | messenger RNA |
| MVC | maraviroc |
| MYS-25 | polyethylene glycol 25 stearate |
| N.D. | no data |
| NE | nanoemulsion |
| NFT | neurofibrillary tangles |
| NK | natural killer |
| NLC | nanostructured lipid carriers |
| NNRTI | non-nucleoside reverse transcriptase inhibitors |
| NP | nanoparticles |
| NRTI | nucleoside reverse transcriptase inhibitors |
| NVP | nevirapine |
| OA | oleic acid |
| Oleic Plurol® | polyglyceryl 6 dioleate |
| OPG | O-palmitoylgalactose |
| OPM | O-palmitoylmannose |
| o/w | oil-in-water |
| PA | phenylalanine |
| PAA | poly(acrylic acid) |
| PBS | phosphate-buffered saline |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PE-PEG2000 | dipalmitoylphosphatidylethanolamine-N-[poly(ethylene glycol)2000] |
| PEG | polyethylene glycol |
| PEG-8-L | octaoxyehtylene laurate ester |
| Pept-DRV-SLN | peptide grafted-darunavir loaded SLN |
| PGDS | polyglyceryl-6-distearate |
| P-gp | P-glycoprotein |
| PI | protease inhibitors |
| PLL | poly(L-lysine hydrochloride |
| Plurol® Oleique CC 497 | polyglyceryl-3 dioleate |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| PPIX | protoporphyrin IX |
| Precirol® ATO 15 | glyceryl palmitostearate |
| PrEP | pre-exposure prophylaxis |
| ProddINP | glycerolipidic prodrug of ddI |
| PS | phosphatidylserine |
| PVA | poly vinyl alcohol |
| RAL | raltegravir |
| RES | reticuloendothelial system |
| RISC complex | RNA-induced silencing complex |
| RNAi | RNA interference |
| RPV | rilpivirine |
| RT | reverse transcriptase |
| RTV | ritonavir |
| SA | stearylamine |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SDS | sodium dodecyl sulfate |
| SEDDS | self-emulsifying drug delivery systems |
| SFV | sifuvirtide |
| SGR | somatic gene recombination |
| siRNA | small interference ribonucleic acid |
| SL | soy lecithin |
| SLN | solid lipid nanoparticles |
| SM | sphingomyelin |
| SMEDDS | self-micro emulsifying delivery systems |
| Smix | surfactant and cosurfactant mix |
| SNEDDS | self-nanoemulsifying drug delivery systems |
| Softisan® 100 | hydrogenated coco-glycerides |
| Solutol® HS15 | polyoxyl 15 hydroxystearate |
| SPC | soy phosphatidylcholine |
| SQV | saquinavir |
| SQVM | saquinavir mesylate |
| S-SNEOF | solid self-nanoemulsifying oily formulations |
| TAM | thymidine analog mutations |
| T-20 | enfuvirtide |
| TDF | tenofovir disoproxil fumarate |
| TFV | tenofovir |
| TPV | tipranavir |
| Transcutol® HP | diethyleneglycol monoethyl ether |
| Transcutol® P | diethylene glycol monoethyl ether |
| UNAIDS | Joint United Nations Program on HIV infection/AIDS |
| U.S. FDA | United States Food and Drug Administration |
| VE | α-tocopherol |
| w/o | water-in-oil |
| w/w | weight/weight |
| wt | weight |
References
- Boyapalle, S.; Mohapatra, S.; Mohapatra, S. Nanotechnology Applications to HIV Vaccines and Microbicides. J. Glob. Infect. Dis. 2012, 4, 62–68. [Google Scholar] [CrossRef] [PubMed]
- das Neves, J.; Amiji, M.M.; Bahia, M.F.; Sarmento, B. Nanotechnology-based systems for the treatment and prevention of HIV/AIDS. Adv. Drug Deliv. Rev. 2010, 62, 458–477. [Google Scholar] [CrossRef]
- UNAIDS. Global HIV&AIDS Statistics—2019 Fact Sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 9 July 2021).
- Nelson, A.G.; Zhang, X.; Ganapathi, U.; Szekely, Z.; Flexner, C.W.; Owen, A.; Sinko, P.J. Drug delivery strategies and systems for HIV/AIDS pre-exposure prophylaxis and treatment. J. Control. Release 2015, 219, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, U.; Jain, N.K. Non-polymeric nano-carriers in HIV/AIDS drug delivery and targeting. Adv. Drug Deliv. Rev. 2010, 62, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Simon, V.; Ho, D.D.; Abdool Karim, Q. HIV/AIDS epidemiology, pathogenesis, prevention, and treatment. Lancet 2006, 368, 489–504. [Google Scholar] [CrossRef] [Green Version]
- Mallipeddi, R.; Rohan, L.C. Progress in antiretroviral drug delivery using nanotechnology. Int. J. Nanomed. 2010, 5, 533–547. [Google Scholar]
- Mamo, T.; Moseman, E.A.; Kolishetti, N.; Salvador-Morales, C.; Shi, J.; Kuritzkes, D.R.; Langer, R.; von Andrian, U.; Farokhzad, O.C. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine 2010, 5, 269–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatham, L.M.; Rannard, S.P.; Owen, A. Nanoformulation strategies for the enhanced oral bioavailability of antiretroviral therapeutics. Ther. Deliv. 2015, 6, 469–490. [Google Scholar] [CrossRef]
- das Neves, J. Novel Approaches for the Delivery of Anti-HIV Drugs-What Is New? Pharmaceutics 2019, 11, 554. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.M.; Silva, J.; Real Oliveira, M.E.C.D.; Lúcio, M. Lipid-based colloidal carriers for topical application of antiviral drugs. In Design of Nanostructures for Versatile Therapeutic Applications; Grumezescu, A.M., Ed.; William Andrew Publishing: Oxford, UK, 2018. [Google Scholar] [CrossRef]
- Cavalcanti, S.M.T.; Nunes, C.; Costa Lima, S.A.; Soares-Sobrinho, J.L.; Reis, S. Optimization of nanostructured lipid carriers for Zidovudine delivery using a microwave-assisted production method. Eur. J. Pharm. Sci. 2018, 122, 22–30. [Google Scholar] [CrossRef]
- Garg, M.; Asthana, A.; Agashe, H.B.; Agrawal, G.P.; Jain, N.K. Stavudine-loaded mannosylated liposomes: In-vitro anti-HIV-I activity, tissue distribution and pharmacokinetics. J. Pharm. Pharmacol. 2006, 58, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Dutta, T.; Jain, N.K. Reduced hepatic toxicity, enhanced cellular uptake and altered pharmacokinetics of stavudine loaded galactosylated liposomes. Eur. J. Pharm. Biopharm. 2007, 67, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Jain, N.K. Reduced hematopoietic toxicity, enhanced cellular uptake and altered pharmacokinetics of azidothymidine loaded galactosylated liposomes. J. Drug Target. 2006, 14, 1–11. [Google Scholar] [CrossRef]
- Gaur, P.K.; Mishra, S.; Bajpai, M.; Mishra, A. Enhanced oral bioavailability of efavirenz by solid lipid nanoparticles: In vitro drug release and pharmacokinetics studies. Biomed. Res. Int 2014, 2014, 363404. [Google Scholar] [CrossRef] [Green Version]
- Harvie, P.; Désormeaux, A.; Bergeron, M.C.; Tremblay, M.; Beauchamp, D.; Poulin, L.; Bergeron, M.G. Comparative pharmacokinetics, distributions in tissue, and interactions with blood proteins of conventional and sterically stabilized liposomes containing 2′,3′-dideoxyinosine. Antimicrob. Agents Chemother. 1996, 40, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Heiati, H.; Tawashi, R.; Phillips, N.C. Solid lipid nanoparticles as drug carriers II. Plasma stability and biodistribution of solid lipid nanoparticles containing the lipophilic prodrug 3′-azido-3′-deoxythymidine palmitate in mice. Int. J. Pharm. 1998, 174, 71–80. [Google Scholar] [CrossRef]
- Jain, S.; Tiwary, A.K.; Jain, N.K. Sustained and targeted delivery of an anti-HIV agent using elastic liposomal formulation: Mechanism of action. Curr. Drug Deliv. 2006, 3, 157–166. [Google Scholar] [CrossRef]
- Nayak, D.; Boxi, A.; Ashe, S.; Thathapudi, N.C.; Nayak, B. Stavudine loaded gelatin liposomes for HIV therapy: Preparation, characterization and in vitro cytotoxic evaluation. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 73, 406–416. [Google Scholar] [CrossRef]
- Perazzolo, S.; Shireman, L.M.; Koehn, J.; McConnachie, L.A.; Kraft, J.C.; Shen, D.D.; Ho, R.J.Y. Three HIV Drugs, Atazanavir, Ritonavir, and Tenofovir, Coformulated in Drug-Combination Nanoparticles Exhibit Long-Acting and Lymphocyte-Targeting Properties in Nonhuman Primates. J. Pharm. Sci. 2018, 107, 3153–3162. [Google Scholar] [CrossRef]
- das Neves, J.; Nunes, R.; Rodrigues, F.; Sarmento, B. Nanomedicine in the development of anti-HIV microbicides. Adv. Drug Deliv. Rev. 2016, 103, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Augustine, R. Challenges in oral drug delivery of antiretrovirals and the innovative strategies to overcome them. Adv. Drug Deliv. Rev. 2016, 103, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Amiji, M.M.; Vyas, T.K.; Shah, L.K. Role of nanotechnology in HIV/AIDS treatment: Potential to overcome the viral reservoir challenge. Discov. Med. 2006, 6, 157–162. [Google Scholar] [PubMed]
- Fernandes, E.; Soares, T.B.; Goncalves, H.; Lucio, M. Spectroscopic Studies as a Toolbox for Biophysical and Chemical Characterization of Lipid-Based Nanotherapeutics. Front. Chem. 2018, 6, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attama, A.A.; Mumuni, A.; Builders, P.F. Lipid Nanoparticulate Drug Delivery Systems: A Revolution in Dosage Form Design and Development. In Recent Advances in Novel Drug Carrier Systems; Sezer, A.D., Ed.; Intechopen: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Venkatesan, N.; Betageri, G.V. Liposomes as nanocarriers for anti-HIV therapy. Drug Deliv. Transl. Res. 2013, 3, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Diksha, M.; Anil, B.J. Lipid Based Nanocarriers for Delivery of Anti-HIV Drugs: A Mini Review. Nanosci. Nanotechnol. Asia 2018, 8, 172–185. [Google Scholar] [CrossRef]
- Huda, A.; Prabha, S. Lipid Based Anti-Retroviral Nanocarriers: A Review of Current Literature and Ongoing Studies. Drug Deliv. Lett. 2017, 7, 71–82. [Google Scholar] [CrossRef]
- Melhuish, A.; Lewthwaite, P. Natural history of HIV and AIDS. Medicine 2018, 46, 356–361. [Google Scholar] [CrossRef]
- FDA. FDA-Approved HIV Medicines. Available online: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines (accessed on 9 July 2021).
- Lisziewicz, J.; Tőke, E.R. Nanomedicine applications towards the cure of HIV. Nanomed. Nanotechnol. Biol. Med. 2013, 9, 28–38. [Google Scholar] [CrossRef]
- Garg, A.B.; Nuttall, J.; Romano, J. The future of HIV microbicides: Challenges and opportunities. Antivir. Chem. Chemother. 2009, 19, 143–150. [Google Scholar] [CrossRef]
- Nuttall, J. Microbicides in the prevention of HIV infection: Current status and future directions. Drugs 2010, 70, 1231–1243. [Google Scholar] [CrossRef]
- Kumar, L.; Verma, S.; Prasad, D.N.; Bhardwaj, A.; Vaidya, B.; Jain, A.K. Nanotechnology: A magic bullet for HIV AIDS treatment. Artif. Cells Nanomed. Biotechnol. 2015, 43, 71–86. [Google Scholar] [CrossRef]
- Zdanowicz, M.M. The pharmacology of HIV drug resistance. Am. J. Pharm. Educ. 2006, 70, 100. [Google Scholar] [CrossRef]
- Clutter, D.S.; Jordan, M.R.; Bertagnolio, S.; Shafer, R.W. HIV-1 drug resistance and resistance testing. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2016, 46, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral Resistance. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [Green Version]
- Blaise, P.; Clevenbergh, P.; Vaira, D.; Moutschen, M.; Dellamonica, P. HIV resistance to antiretroviral drugs: Mechanisms, genotypic and phenotypic resistance testing in clinical practice. Acta Clin. Belg. 2002, 57, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Margeridon-Thermet, S.; Shafer, R.W. Comparison of the Mechanisms of Drug Resistance among HIV, Hepatitis B, and Hepatitis C. Viruses 2010, 2, 2696–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammaranond, P.; Sanguansittianan, S. Mechanism of HIV antiretroviral drugs progress toward drug resistance. Fundam. Clin. Pharmacol. 2012, 26, 146–161. [Google Scholar] [CrossRef]
- Roy, U.; Rodríguez, J.; Barber, P.; das Neves, J.; Sarmento, B.; Nair, M. The potential of HIV-1 nanotherapeutics: From in vitro studies to clinical trials. Nanomedicine 2015, 10, 3597–3609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siccardi, M.; Martin, P.; McDonald, T.O.; Liptrott, N.J.; Giardiello, M.; Rannard, S.; Owen, A. Nanomedicines for HIV therapy. Ther. Deliv. 2013, 4, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. Off. J. Am. Coll. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Wang, T.; Song, J.; Pu, D.; He, D.; Li, J.; Yang, J.; Li, K.; Zhong, C.; Zhang, J. Antiviral Drug Delivery System for Enhanced Bioactivity, Better Metabolism and Pharmacokinetic Characteristics. Int. J. Nanomed. 2021, 16, 4959–4984. [Google Scholar] [CrossRef] [PubMed]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood–Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.J.; Neves, J.d.; Sarmento, B. Nanoparticle-based drug delivery to improve the efficacy of antiretroviral therapy in the central nervous system. Int. J. Nanomed. 2014, 9, 1757–1769. [Google Scholar] [CrossRef] [Green Version]
- Danta, C.C.; Piplani, P. Investigation of Molecular Properties of Antiretroviral Agents to Enhance CNS Penetration Abilities for the Treatment of Cognitive Impairment in HIV-Associated Neurocognitive Disorder. ACS Chem. Neurosci. 2020, 11, 2034–2038. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Kraft, J.C.; Li, B.; Yu, J.; Freeling, J.; Koehn, J.; Ho, R.J. Nanodrug formulations to enhance HIV drug exposure in lymphoid tissues and cells: Clinical significance and potential impact on treatment and eradication of HIV/AIDS. Nanomedicine 2016, 11, 545–564. [Google Scholar] [CrossRef] [Green Version]
- Lúcio, M.; Lopes, C.M.; Fernandes, E.; Gonçalves, H.; Real Oliveira, M.E.C.D. Chapter 4. Organic Nanocarriers for Brain Drug Delivery. In Nanoparticles for Brain Drug Delivery; Vitorino, C., Jorge, A., Pais, A.A.C.C., Eds.; Jenny Stanford Publishing Pte. Ltd.: Singapore, 2021. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Faria, M.J.; Machado, R.; Ribeiro, A.; Goncalves, H.; Real Oliveira, M.; Viseu, T.; das Neves, J.; Lucio, M. Rational Development of Liposomal Hydrogels: A Strategy for Topical Vaginal Antiretroviral Drug Delivery in the Context of HIV Prevention. Pharmaceutics 2019, 11, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Tiwary, A.K.; Jain, N.K. PEGylated elastic liposomal formulation for lymphatic targeting of zidovudine. Curr. Drug Deliv. 2008, 5, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Pokharkar, V.; Patil-Gadhe, A.; Palla, P. Efavirenz loaded nanostructured lipid carrier engineered for brain targeting through intranasal route: In-vivo pharmacokinetic and toxicity study. Biomed. Pharm. 2017, 94, 150–164. [Google Scholar] [CrossRef]
- Vyas, A.; Jain, A.; Hurkat, P.; Jain, A.; Jain, S.K. Targeting of AIDS related encephalopathy using phenylalanine anchored lipidic nanocarrier. Colloids Surf. B Biointerfaces 2015, 131, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Soares, T.B.; Loureiro, L.; Carvalho, A.; Oliveira, M.; Dias, A.; Sarmento, B.; Lucio, M. Lipid nanocarriers loaded with natural compounds: Potential new therapies for age related neurodegenerative diseases? Prog. Neurobiol. 2018, 168, 21–41. [Google Scholar] [CrossRef]
- Phillips, N.C.; Skamene, E.; Tsoukas, C. Liposomal encapsulation of 3′-azido-3′-deoxythymidine (AZT) results in decreased bone marrow toxicity and enhanced activity against murine AIDS-induced immunosuppression. J. Acquir. Immune Defic. Syndr. 1991, 4, 959–966. [Google Scholar]
- Rao, K.S.; Ghorpade, A.; Labhasetwar, V. Targeting anti-HIV drugs to the CNS. Expert Opin. Drug Deliv. 2009, 6, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Sana, K.; Poorva, J.; Sourabh, J.; Richa, J.; Saurabh, B.; Aakanchha, J. Topical Delivery of Erythromycin Through Cubosomes for Acne. Pharm. Nanotechnol. 2018, 6, 38–47. [Google Scholar] [CrossRef]
- Barriga, H.M.G.; Holme, M.N.; Stevens, M.M. Cubosomes: The Next Generation of Smart Lipid Nanoparticles? Angew. Chem. Int. Ed. 2019, 58, 2958–2978. [Google Scholar] [CrossRef] [Green Version]
- Ahirrao, M.; Shrotriya, S. In vitro and in vivo evaluation of cubosomal in situ nasal gel containing resveratrol for brain targeting. Drug Dev. Ind. Pharm. 2017, 43, 1686–1693. [Google Scholar] [CrossRef]
- Boge, L.; Bysell, H.; Ringstad, L.; Wennman, D.; Umerska, A.; Cassisa, V.; Eriksson, J.; Joly-Guillou, M.-L.; Edwards, K.; Andersson, M. Lipid-Based Liquid Crystals As Carriers for Antimicrobial Peptides: Phase Behavior and Antimicrobial Effect. Langmuir 2016, 32, 4217–4228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Angelova, A.; Angelov, B.; Drechsler, M.; Garamus, V.M.; Willumeit-Römer, R.; Zou, A. Sterically stabilized spongosomes for multidrug delivery of anticancer nanomedicines. J. Mater. Chem. B 2015, 3, 7734–7744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, Y.C.; Ko, H.F. Targeting delivery of saquinavir to the brain using 83-14 monoclonal antibody-grafted solid lipid nanoparticles. Biomaterials 2013, 34, 4818–4830. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, N.; Zastre, J.; Wong, H.L.; Wu, X.Y.; Bendayan, R. Solid lipid nanoparticles enhance the delivery of the HIV protease inhibitor, atazanavir, by a human brain endothelial cell line. Pharm. Res. 2008, 25, 2262–2271. [Google Scholar] [CrossRef]
- Garg, B.; Katare, O.P.; Beg, S.; Lohan, S.; Singh, B. Systematic development of solid self-nanoemulsifying oily formulations (S-SNEOFs) for enhancing the oral bioavailability and intestinal lymphatic uptake of lopinavir. Colloids Surf. B Biointerfaces 2016, 141, 611–622. [Google Scholar] [CrossRef]
- Delshadi, R.; Bahrami, A.; McClements, D.J.; Moore, M.D.; Williams, L. Development of nanoparticle-delivery systems for antiviral agents: A review. J. Control. Release 2021, 331, 30–44. [Google Scholar] [CrossRef]
- Phillips, N.C.; Tsoukas, C. Liposomal encapsulation of azidothymidine results in decreased hematopoietic toxicity and enhanced activity against murine acquired immunodeficiency syndrome. Blood 1992, 79, 1137–1143. [Google Scholar] [CrossRef]
- Désormeaux, A.; Harvie, P.; Perron, S.; Makabi-Panzu, B.; Beauchamp, D.; Tremblay, M.; Poulin, L.; Bergeron, M.G. Antiviral efficacy, intracellular uptake and pharmacokinetics of free and liposome-encapsulated 2′,3′-dideoxyinosine. AIDS 1994, 8, 1545–1553. [Google Scholar] [CrossRef]
- Ramana, L.N.; Sharma, S.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Stealth anti-CD4 conjugated immunoliposomes with dual antiretroviral drugs—Modern Trojan horses to combat HIV. Eur. J. Pharm. Biopharm. 2015, 89, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Aji Alex, M.R.; Chacko, A.J.; Jose, S.; Souto, E.B. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur. J. Pharm. Sci. 2011, 42, 11–18. [Google Scholar] [CrossRef]
- Ansari, H.; Singh, P. Formulation and in-vivo Evaluation of Novel Topical Gel of Lopinavir for Targeting HIV. Curr. HIV Res. 2018, 16, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Bhalekar, M.; Upadhaya, P.; Madgulkar, A. Formulation and characterization of solid lipid nanoparticles for an anti-retroviral drug darunavir. Appl. Nanosci. 2017, 7, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.C.; Chung, C.Y. Solid lipid nanoparticles comprising internal Compritol 888 ATO, tripalmitin and cacao butter for encapsulating and releasing stavudine, delavirdine and saquinavir. Colloids Surf. B Biointerfaces 2011, 88, 682–690. [Google Scholar] [CrossRef]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef]
- Vyas, S.P.; Subhedar, R.; Jain, S. Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. J. Pharm. Pharmacol. 2006, 58, 321–326. [Google Scholar] [CrossRef]
- Bazzoli, C.; Jullien, V.; Tiec, C.L.; Rey, E.; Mentré, F.; Taburet, A.-M. Intracellular Pharmacokinetics of Antiretroviral Drugs in HIV-Infected Patients, and their Correlation with Drug Action. Clin. Pharmacokinet. 2010, 49, 17–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Boffito, M.; Khoo, S.; Smit, E.; Back, D. Stopping antiretroviral therapy. AIDS 2007, 21, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Skanji, R.; Andrieux, K.; Lalanne, M.; Caron, J.; Bourgaux, C.; Degrouard, J.; Brisset, F.; Gueutin, C.; Chacun, H.; Dereuddre-Bosquet, N.; et al. A new nanomedicine based on didanosine glycerolipidic prodrug enhances the long term accumulation of drug in a HIV sanctuary. Int. J. Pharm. 2011, 414, 285–297. [Google Scholar] [CrossRef]
- Sudhakar, B.; Krishna, M.C.; Murthy, K.V.R. Factorial design studies of antiretroviral drug-loaded stealth liposomal injectable: PEGylation, lyophilization and pharmacokinetic studies. Appl. Nanosci. 2016, 6, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.X.; Bi, D.Z.; Wang, J.; Wang, Y.Z.; Hu, H.G.; Deng, Y.H. Pharmacokinetics and tissue distribution of zidovudine in rats following intravenous administration of zidovudine myristate loaded liposomes. Pharmazie 2005, 60, 840–843. [Google Scholar] [PubMed]
- Desai, J.; Thakkar, H. Darunavir-Loaded Lipid Nanoparticles for Targeting to HIV Reservoirs. AAPS Pharmscitech 2018, 19, 648–660. [Google Scholar] [CrossRef]
- Garg, B.; Beg, S.; Kumar, R.; Katare, O.P.; Singh, B. Nanostructured lipidic carriers of lopinavir for effective management of HIV-associated neurocognitive disorder. J. Drug Deliv. Sci. Technol. 2019, 53. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Chotai, N.; Misra, A.; Omri, A. Systematic Approach for the Formulation and Optimization of Solid Lipid Nanoparticles of Efavirenz by High Pressure Homogenization Using Design of Experiments for Brain Targeting and Enhanced Bioavailability. Biomed Res. Int. 2017, 2017, 5984014. [Google Scholar] [CrossRef]
- Jindal, A.B.; Bachhav, S.S.; Devarajan, P.V. In situ hybrid nano drug delivery system (IHN-DDS) of antiretroviral drug for simultaneous targeting to multiple viral reservoirs: An in vivo proof of concept. Int. J. Pharm. 2017, 521, 196–203. [Google Scholar] [CrossRef]
- Khan, S.A.; Rehman, S.; Nabi, B.; Iqubal, A.; Nehal, N.; Fahmy, U.A.; Kotta, S.; Baboota, S.; Md, S.; Ali, J. Boosting the Brain Delivery of Atazanavir through Nanostructured Lipid Carrier-Based Approach for Mitigating NeuroAIDS. Pharmaceutics 2020, 12, 1059. [Google Scholar] [CrossRef] [PubMed]
- Rojekar, S.; Fotooh Abadi, L.; Pai, R.; Mahajan, K.; Kulkarni, S.; Vavia, P.R. Multi-organ targeting of HIV-1 viral reservoirs with etravirine loaded nanostructured lipid carrier: An in-vivo proof of concept. Eur. J. Pharm. Sci. 2021, 164, 105916. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, H.S.; Mahajan, M.S.; Nerkar, P.P.; Agrawal, A. Nanoemulsion-based intranasal drug delivery system of saquinavir mesylate for brain targeting. Drug Deliv. 2014, 21, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, K.; Afzal, S.M.; Surender, G.; Kishan, V. Tween 80 containing lipid nanoemulsions for delivery of indinavir to brain. Acta Pharm. Sin. B 2013, 3, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Vyas, T.K.; Shahiwala, A.; Amiji, M.M. Improved oral bioavailability and brain transport of Saquinavir upon administration in novel nanoemulsion formulations. Int. J. Pharm. 2008, 347, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Scheerer, S.; Geyer, M.A.; Howell, S.B. Direct cerebrospinal fluid delivery of an antiretroviral agent using multivesicular liposomes. J. Infect. Dis. 1990, 162, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Makabi-Panzu, B.; Lessard, C.; Beauchamp, D.; Desormeaux, A.; Poulin, L.; Tremblay, M.; Bergeron, M.G. Uptake and binding of liposomal 2′,3′-dideoxycytidine by RAW 264.7 cells: A three-step process. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1995, 8, 227–235. [Google Scholar] [CrossRef]
- Katragadda, A.; Bridgman, R.; Betageri, G. Effect of liposome composition and cholesterol on the cellular uptake of stavudine by human monocyte/macrophages. Cell. Mol. Biol. Lett. 2000, 5, 483–494. [Google Scholar]
- Gagné, J.F.; Désormeaux, A.; Perron, S.; Tremblay, M.J.; Bergeron, M.G. Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. Biochim. Biophys. Acta 2002, 1558, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Kinman, L.; Brodie, S.J.; Tsai, C.C.; Bui, T.; Larsen, K.; Schmidt, A.; Anderson, D.; Morton, W.R.; Hu, S.L.; Ho, R.J. Lipid-drug association enhanced HIV-1 protease inhibitor indinavir localization in lymphoid tissues and viral load reduction: A proof of concept study in HIV-2287-infected macaques. J. Acquir. Immune Defic. Syndr. 2003, 34, 387–397. [Google Scholar] [CrossRef]
- Garg, M.; Garg, B.R.; Jain, S.; Mishra, P.; Sharma, R.K.; Mishra, A.K.; Dutta, T.; Jain, N.K. Radiolabeling, pharmacoscintigraphic evaluation and antiretroviral efficacy of stavudine loaded 99mTc labeled galactosylated liposomes. Eur. J. Pharm. Sci. 2008, 33, 271–281. [Google Scholar] [CrossRef]
- Lalanne, M.; Paci, A.; Andrieux, K.; Dereuddre-Bosquet, N.; Clayette, P.; Deroussent, A.; Re, M.; Vassal, G.; Couvreur, P.; Desmaele, D. Synthesis and biological evaluation of two glycerolipidic prodrugs of didanosine for direct lymphatic delivery against HIV. Bioorg. Med. Chem. Lett. 2007, 17, 2237–2240. [Google Scholar] [CrossRef]
- Kapitza, S.B.; Michel, B.R.; van Hoogevest, P.; Leigh, M.L.; Imanidis, G. Absorption of poorly water soluble drugs subject to apical efflux using phospholipids as solubilizers in the Caco-2 cell model. Eur. J. Pharm. Biopharm. 2007, 66, 146–158. [Google Scholar] [CrossRef]
- Kaur, C.D.; Nahar, M.; Jain, N.K. Lymphatic targeting of zidovudine using surface-engineered liposomes. J. Drug Target. 2008, 16, 798–805. [Google Scholar] [CrossRef]
- Clayton, R.; Ohagen, A.; Nicol, F.; Del Vecchio, A.M.; Jonckers, T.H.; Goethals, O.; Van Loock, M.; Michiels, L.; Grigsby, J.; Xu, Z.; et al. Sustained and specific in vitro inhibition of HIV-1 replication by a protease inhibitor encapsulated in gp120-targeted liposomes. Antivir. Res. 2009, 84, 142–149. [Google Scholar] [CrossRef]
- Ramana, L.N.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Development of a liposomal nanodelivery system for nevirapine. J. Biomed. Sci. 2010, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Franquelim, H.G.; De-Sousa, F.F.; Veiga, A.S.; Santos, N.C.; Castanho, M.A.R.B. Cationic liposomes are possible drug-delivery systems for HIV fusion inhibitor sifuvirtide. Soft Matter 2011, 7, 11089–11092. [Google Scholar] [CrossRef]
- Zidan, A.S.; Rahman, Z.; Khan, M.A. Product and process understanding of a novel pediatric anti-HIV tenofovir niosomes with a high-pressure homogenizer. Eur. J. Pharm. Sci. 2011, 44, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ramana, L.N.; Sharma, S.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Investigation on the stability of saquinavir loaded liposomes: Implication on stealth, release characteristics and cytotoxicity. Int. J. Pharm. 2012, 431, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Khan, M.A.; Burgess, D.J. A two-stage reverse dialysis in vitro dissolution testing method for passive targeted liposomes. Int. J. Pharm. 2012, 426, 211–218. [Google Scholar] [CrossRef]
- Zidan, A.S.; Spinks, C.; Fortunak, J.; Habib, M.; Khan, M.A. Near-infrared investigations of novel anti-HIV tenofovir liposomes. AAPS J. 2010, 12, 202–214. [Google Scholar] [CrossRef] [Green Version]
- Zidan, A.S.; Spinks, C.B.; Habib, M.J.; Khan, M.A. Formulation and transport properties of tenofovir loaded liposomes through Caco-2 cell model. J. Liposome Res. 2013, 23, 318–326. [Google Scholar] [CrossRef]
- Spinks, C.B.; Zidan, A.S.; Khan, M.A.; Habib, M.J.; Faustino, P.J. Pharmaceutical characterization of novel tenofovir liposomal formulations for enhanced oral drug delivery: In vitro pharmaceutics and Caco-2 permeability investigations. Clin. Pharm. 2017, 9, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Patel, G.M.; Shelat, P.K.; Lalwani, A.N. QbD based development of proliposome of lopinavir for improved oral bioavailability. Eur. J. Pharm. Sci. 2017, 108, 50–61. [Google Scholar] [CrossRef]
- Ahammed, V.; Narayan, R.; Paul, J.; Nayak, Y.; Roy, B.; Shavi, G.V.; Nayak, U.Y. Development and in vivo evaluation of functionalized ritonavir proliposomes for lymphatic targeting. Life Sci. 2017, 183, 11–20. [Google Scholar] [CrossRef]
- Figueira, T.N.; Domingues, M.M.; Illien, F.; Cadima-Couto, I.; Todorovski, T.; Andreu, D.; Sagan, S.; Castanho, M.A.R.B.; Walrant, A.; Veiga, A.S. Enfuvirtide-Protoporphyrin IX Dual-Loaded Liposomes: In Vitro Evidence of Synergy against HIV-1 Entry into Cells. ACS Infect. Dis. 2020, 6, 224–236. [Google Scholar] [CrossRef]
- Jain, S.; Tiwary, A.K.; Sapra, B.; Jain, N.K. Formulation and evaluation of ethosomes for transdermal delivery of lamivudine. AAPS Pharmscitech 2007, 8, E111. [Google Scholar] [CrossRef] [Green Version]
- Chettupalli, A.K.; Ananthula, M.; Amarachinta, P.R.; Bakshi, V.; Yata, V.K. Design, Formulation, In-Vitro and Ex-Vivo Evaluation of Atazanavir Loaded Cubosomal Gel. Biointerface Res. Appl. Chem. 2021, 11, 12037–12054. [Google Scholar] [CrossRef]
- Hosny, K.M. Nanosized Cubosomal Thermogelling Dispersion Loaded with Saquinavir Mesylate to Improve Its Bioavailability: Preparation, Optimization, in vitro and in vivo Evaluation. Int. J. Nanomed. 2020, 15, 5113–5129. [Google Scholar] [CrossRef]
- Tomitaka, A.; Arami, H.; Huang, Z.; Raymond, A.; Rodriguez, E.; Cai, Y.; Febo, M.; Takemura, Y.; Nair, M. Hybrid magneto-plasmonic liposomes for multimodal image-guided and brain-targeted HIV treatment. Nanoscale 2017, 10, 184–194. [Google Scholar] [CrossRef]
- Saiyed, Z.M.; Gandhi, N.H.; Nair, M.P.N. Magnetic nanoformulation of azidothymidine 5′-triphosphate for targeted delivery across the blood-brain barrier. Int. J. Nanomed. 2010, 5, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Nunes, R.; Bogas, S.; Faria, M.J.; Gonçalves, H.; Lúcio, M.; Viseu, T.; Sarmento, B.; das Neves, J. Electrospun fibers for vaginal administration of tenofovir disoproxil fumarate and emtricitabine in the context of topical pre-exposure prophylaxis. J. Control. Release 2021, 334, 453–462. [Google Scholar] [CrossRef]
- Alukda, D.; Sturgis, T.; Youan, B.C. Formulation of tenofovir-loaded functionalized solid lipid nanoparticles intended for HIV prevention. J. Pharm. Sci. 2011, 100, 3345–3356. [Google Scholar] [CrossRef] [Green Version]
- Shegokar, R.; Singh, K.K. Stavudine entrapped lipid nanoparticles for targeting lymphatic HIV reservoirs. Die Pharm. 2011, 66, 264–271. [Google Scholar]
- Kuo, Y.C.; Chung, J.F. Physicochemical properties of nevirapine-loaded solid lipid nanoparticles and nanostructured lipid carriers. Colloids Surf. B Biointerfaces 2011, 83, 299–306. [Google Scholar] [CrossRef]
- Joshi, K.S.; Sharma, C.P.; Kalarikkal, N.; Sandeep, K.; Thomas, S.; Pothen, L.A. Evaluation of in-vitro cytotoxicity and cellular uptake efficiency of zidovudine-loaded solid lipid nanoparticles modified with Aloe Vera in glioma cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 66, 40–50. [Google Scholar] [CrossRef]
- Javan, F.; Vatanara, A. Encapsulation of ritonavir in solid lipid nanoparticles: In-vitro anti-HIV-1 activity using lentiviral particles. J. Pharm. Pharmacol. 2017, 69, 1002–1009. [Google Scholar] [CrossRef]
- Cavalcanti, S.M.T.; Nunes, C.; Lima, S.A.C.; Soares-Sobrinho, J.L.; Reis, S. Multiple Lipid Nanoparticles (MLN), a New Generation of Lipid Nanoparticles for Drug Delivery Systems: Lamivudine-MLN Experimental Design. Pharm. Res. 2017, 34, 1204–1216. [Google Scholar] [CrossRef]
- Mahajan, K.Y.; Rojekar, S.V.; Desai, D.V.; Kulkarni, S.S.; Vavia, P.R. Efavirenz loaded nanostructured lipid carriers for efficient and prolonged viral inhibition in HIV-infected macrophages. Pharm. Sci. 2020. [Google Scholar] [CrossRef]
- Shaikh, N.A.; Lala, R.R. Formulation development of dolutegravir sodium loaded nano lipid carriers for improved solubility and permeability. Int. J. Pharm. Sci. Res. 2021, 12, 3654–3665. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.; des Rieux, A.; Préat, V.; Rodríguez-Gascón, A. Dextran-protamine coated nanostructured lipid carriers as mucus-penetrating nanoparticles for lipophilic drugs. Int. J. Pharm. 2014, 468, 105–111. [Google Scholar] [CrossRef]
- Dixit, G.R.; Mathur, V.B. Formulation and characterization of solid microemulsion of darunavir for enhanced solubility and dissolution. Int. J. Pharm. Sci. Res. 2015, 6, 3990–3999. [Google Scholar] [CrossRef]
- Carvalho, A.L.; Silva, J.A.; Lira, A.A.; Conceição, T.M.; Nunes Rde, S.; de Albuquerque Junior, R.L.; Sarmento, V.H.; Leal, L.B.; de Santana, D.P. Evaluation of Microemulsion and Lamellar Liquid Crystalline Systems for Transdermal Zidovudine Delivery. J. Pharm. Sci. 2016, 105, 2188–2193. [Google Scholar] [CrossRef]
- Kotta, S.; Khan, A.W.; Ansari, S.H.; Sharma, R.K.; Ali, J. Anti HIV nanoemulsion formulation: Optimization and in vitro-in vivo evaluation. Int. J. Pharm. 2014, 462, 129–134. [Google Scholar] [CrossRef]
- McConville, C.; Friend, D. Development and characterisation of a self-microemulsifying drug delivery systems (SMEDDSs) for the vaginal administration of the antiretroviral UC-781. Eur. J. Pharm. Biopharm. 2013, 83, 322–329. [Google Scholar] [CrossRef]
- Senapati, P.C.; Sahoo, S.K.; Sahu, A.N. Mixed surfactant based (SNEDDS) self-nanoemulsifying drug delivery system presenting efavirenz for enhancement of oral bioavailability. Biomed. Pharm. 2016, 80, 42–51. [Google Scholar] [CrossRef]
- De Melo-Diogo, D.; Pais-Silva, C.; Dias, D.R.; Moreira, A.F.; Correia, I.J. Strategies to Improve Cancer Photothermal Therapy Mediated by Nanomaterials. Adv. Healthc. Mater. 2017, 6, 1700073. [Google Scholar] [CrossRef]
- Chenthamara, D.; Subramaniam, S.; Ramakrishnan, S.G.; Krishnaswamy, S.; Essa, M.M.; Lin, F.-H.; Qoronfleh, M.W. Therapeutic efficacy of nanoparticles and routes of administration. Biomater. Res. 2019, 23, 20. [Google Scholar] [CrossRef]
- Raoufi, E.; Bahramimeimandi, B.; Salehi-Shadkami, M.; Chaosri, P.; Mozafari, M.R. Methodical Design of Viral Vaccines Based on Avant-Garde Nanocarriers: A Multi-Domain Narrative Review. Biomedicines 2021, 9, 520. [Google Scholar] [CrossRef]
- Danaei, M.; Kalantari, M.; Raji, M.; Samareh Fekri, H.; Saber, R.; Asnani, G.P.; Mortazavi, S.M.; Mozafari, M.R.; Rasti, B.; Taheriazam, A. Probing nanoliposomes using single particle analytical techniques: Effect of excipients, solvents, phase transition and zeta potential. Heliyon 2018, 4. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef]
- Kerns, E.H.; Di, L. Chapter 5—Lipophilicity. In Drug-Like Properties: Concepts, Structure Design and Methods; Kerns, E.H., Di, L., Eds.; Academic Press: San Diego, CA, USA, 2008; pp. 43–47. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, J.; Lu, Y.; He, W.; Li, X.; Wu, W. Biotinylated liposomes as potential carriers for the oral delivery of insulin. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 167–176. [Google Scholar] [CrossRef]
- Benziger, D.P.; Edelson, J. Absorption from the vagina. Drug Metab. Rev. 1983, 14, 137–168. [Google Scholar] [CrossRef]
- das Neves, J.; Palmeira-de-Oliveira, R.; Palmeira-de-Oliveira, A.; Rodrigues, F.; Sarmento, B. Vaginal Mucosa and Drug Delivery. In Mucoadhesive Materials and Drug Delivery Systems; Khutoryanskiy, V.V., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 99–132. [Google Scholar] [CrossRef]
- das Neves, J.; Amiji, M.; Sarmento, B. Mucoadhesive nanosystems for vaginal microbicide development: Friend or foe? Wires Nanomed. Nanobiotechnol. 2011, 3, 389–399. [Google Scholar] [CrossRef]
- Parboosing, R.; Maguire, G.E.M.; Govender, P.; Kruger, H.G. Nanotechnology and the treatment of HIV infection. Viruses 2012, 4, 488–520. [Google Scholar] [CrossRef] [Green Version]
- Pollock, S.; Dwek, R.A.; Burton, D.R.; Zitzmann, N. N-Butyldeoxynojirimycin is a broadly effective anti-HIV therapy significantly enhanced by targeted liposome delivery. AIDS 2008, 22, 1961–1969. [Google Scholar] [CrossRef]
- Flasher, D.; Konopka, K.; Chamow, S.M.; Dazin, P.; Ashkenazi, A.; Pretzer, E.; Düzgünes, N. Liposome targeting to human immunodeficiency virus type 1-infected cells via recombinant soluble CD4 and CD4 immunoadhesin (CD4-IgG). Biochim. Biophys. Acta 1994, 1194, 185–196. [Google Scholar] [CrossRef]
- Bestman-Smith, J.; Gourde, P.; Désormeaux, A.; Tremblay, M.J.; Bergeron, M.G. Sterically stabilized liposomes bearing anti-HLA-DR antibodies for targeting the primary cellular reservoirs of HIV-1. Biochim. Biophys. Acta Biomembr. 2000, 1468, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Kinman, L.; Bui, T.; Larsen, K.; Tsai, C.C.; Anderson, D.; Morton, W.R.; Hu, S.L.; Ho, R.J. Optimization of lipid-indinavir complexes for localization in lymphoid tissues of HIV-infected macaques. J. Acquir. Immune Defic. Syndr. 2006, 42, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Oliveira, A.C.N.; Fernandes, J.; Gonçalves, A.; Gomes, A.C.; Real Oliveira, M.E.C.D. Lipid-based Nanocarriers for siRNA Delivery: Challenges, Strategies and the Lessons Learned from the DODAX: MO Liposomal System. Curr. Drug Targets 2019, 20, 29–50. [Google Scholar] [CrossRef]
- Berkhout, B.; ter Brake, O. Towards a durable RNAi gene therapy for HIV-AIDS. Expert Opin. Biol. Ther. 2009, 9, 161–170. [Google Scholar] [CrossRef]
- Haasnoot, J.; Westerhout, E.M.; Berkhout, B. RNA interference against viruses: Strike and counterstrike. Nat. Biotechnol. 2007, 25, 1435–1443. [Google Scholar] [CrossRef]
- Kim, S.-S.; Peer, D.; Kumar, P.; Subramanya, S.; Wu, H.; Asthana, D.; Habiro, K.; Yang, Y.-G.; Manjunath, N.; Shimaoka, M.; et al. RNAi-mediated CCR5 silencing by LFA-1-targeted nanoparticles prevents HIV infection in BLT mice. Mol. Ther. 2010, 18, 370–376. [Google Scholar] [CrossRef]
- Karlsen, T.A.; Brinchmann, J.E. Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol. Ther. 2013, 21, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Krebs, M.D.; Alsberg, E. Localized, targeted, and sustained siRNA delivery. Chem. Weinh. Bergstr. Ger. 2011, 17, 3054–3062. [Google Scholar] [CrossRef]
- Kaeser, G.E.; Chun, J. Mosaic Somatic Gene Recombination as a Potentially Unifying Hypothesis for Alzheimer’s Disease. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Chai, G.; Gleeson, J.G. A newly discovered mechanism driving neuronal mutations in Alzheimer’s disease. Nature 2018, 563, 631–632. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ARV Therapeutic Class | Mechanism of Action | ARV Single Agents and Some ARV Associations | |
|---|---|---|---|
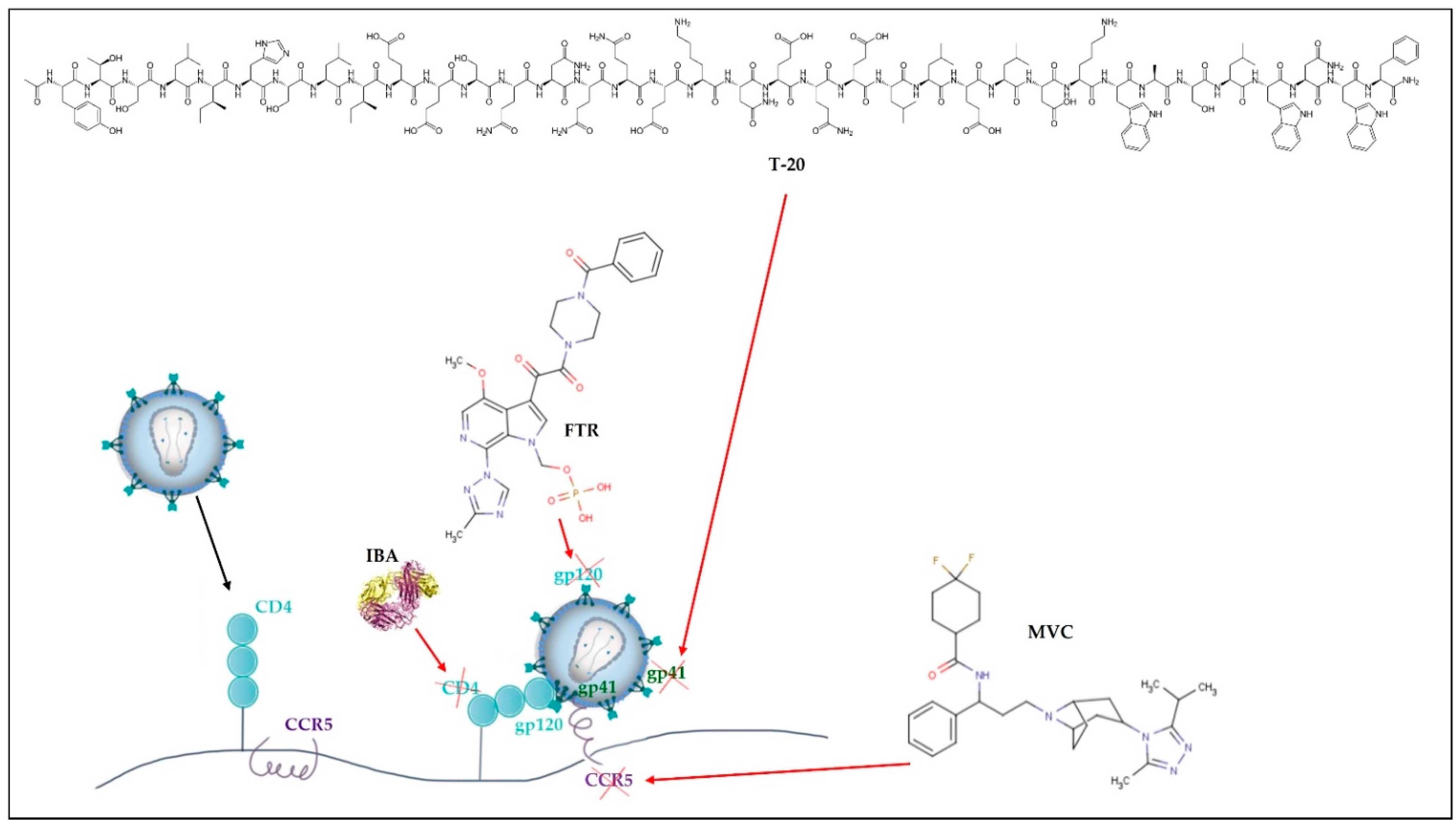
| Cell entry inhibitors | CCR5 antagonists | Block CCR5 coreceptors present on the surface of specific immune cells, preventing HIV from entering the cells. | MVC |
| Attachment inhibitors | Bind to the gp120 protein on the viral outer surface, blocking HIV entry into CD4 cells. | FTR | |
| Post-attachment inhibitors | Block CD4 receptors present on the surface of specific immune cells, preventing HIV from entering the cells. | IBA | |
| Fusion inhibitors (FI) | Interferes with HIV binding, fusion, and cell entrance by preventing the gp41 glycoprotein from being exposed to the virus-host cell membrane. | T-20 | |
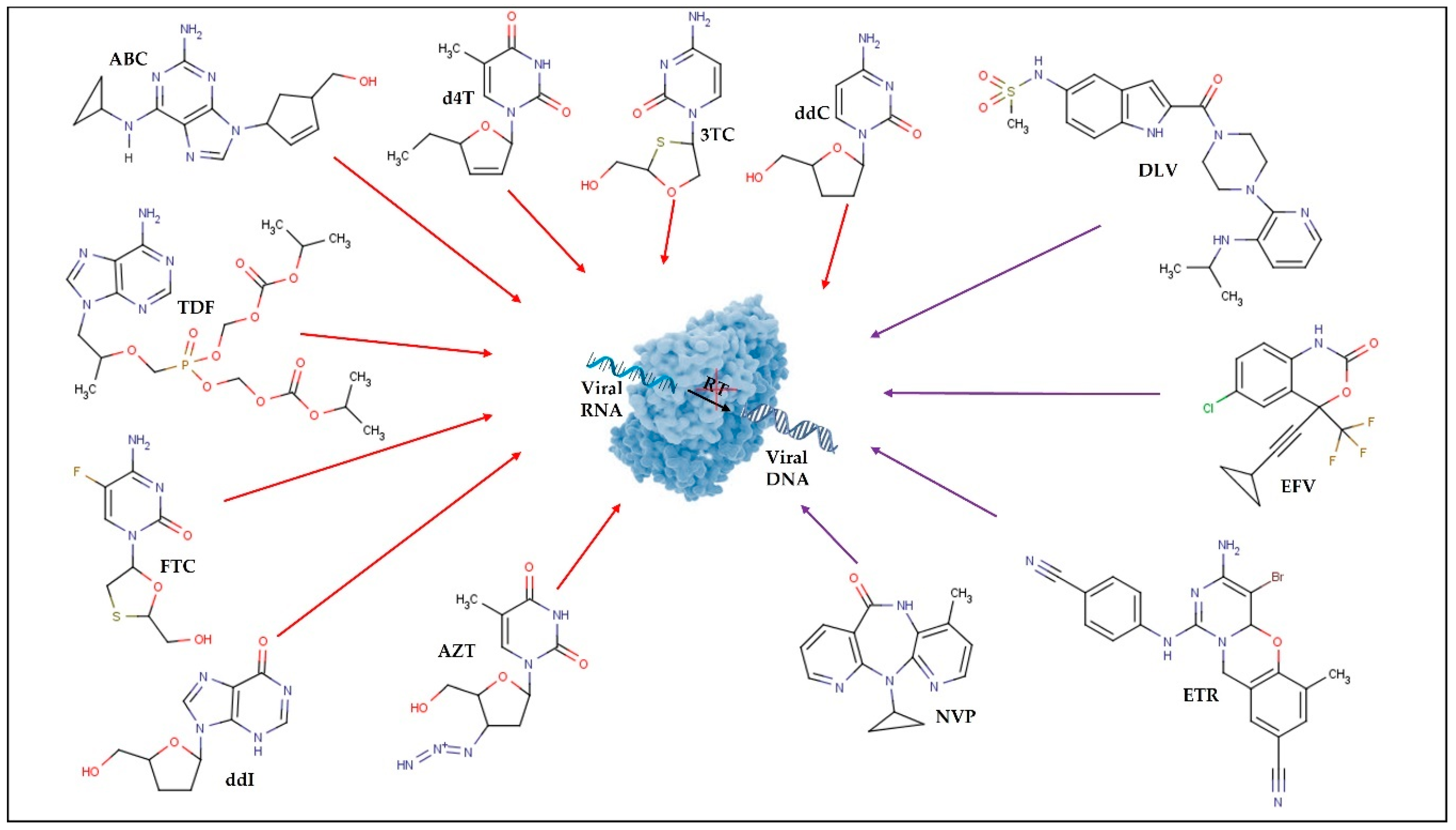
| Nucleoside reverse transcriptase inhibitors (NRTI) | Block the viral RT, inhibiting HIV replication. | 3TC; ABC; AZT; d4T; ddI; ddI EC; ddC (F.M.); FTC; TDF; 3TC+AZT; ABC+3TC; ABC+AZT+3TC; TDF+FTC | |
| Non-nucleoside reverse transcriptase inhibitors (NNRTI) | Bind to viral RT and subsequently modify it, limiting HIV replication. | DOR; EFV; RPV; ETR; NVP; DLV | |
| Integrase inhibitors (II) | Inhibition of viral integrase. Prevents the incorporation of HIV proviral DNA strands into the host cell genome. | RAL; DTG; EVG; CAB | |
| Protease inhibitors (PI) | Inhibition of viral protease. Prevents the cleavage of some viral proteins and the maturation of virions, resulting in non-viral particles. | TPV; IDV; RTV; DRV; FPV; ATV; LPV+RTV; SQVM+RTV | |
| Type of Lipid Nanocarriers | Description and Main Characteristics | Advantages/Disadvantages for ARV Delivery | References |
|---|---|---|---|
LIPOSOMES ETHOSOMES  |
|
| [19,53,57,58] |
CUBOSOMES |
|
| [59,60,61,62,63] |
LIPID NANOPARTICLES  |
|
| [7,64,65] |
LIPID EMULSIONS Nanoemulsion O/W  SNEDDS W/O/W |
|
| [29,66] |
| Year | Composition | ARV | Physicochemical Characterization | Outcomes | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| E.E. (%) | D.L. (%) | ζ-Potential (mV) | Size (nm) | |||||
| Liposomes | ||||||||
| 1990 | DOPC:DPPG:Chol:Triolein (5:1:8:1) | ddC e logP = −1.35 | 48.3 ± 2.6 | N.D. | N.D. | 39,600 ± 11,900 | ↑ residence time in CNS ↑ controlled release | [91] * |
| 1991/92 | DPPC:DMPG (10:1) or DSPC:DMPG (10:1) | AZT a logP = 0.05 | N.D. | LP w/DPPC: 9.35 ± 0.45 LP w/DSPC: 8.98 ± 0.56 | N.D. | N.D. | ↑ ARV activity ↓ hematopoietic toxicity | [57,68] * |
| 1994 | DSPC:DSPG (10:3) | ddI a logP = −1.24 | 85 ± 15 | 21 ± 1 | N.D. | 180 ± 20 | ↑ bioavailability ↓ systemic exposure ↓ effective fighting the virus compared to free ddI | [69] *,** |
| 1995 | DPPC:DCP:Chol (4:1:5) | ddC logP = −1.35 | N.D. | 35 | N.D. | 300 | ↑ ddC retention in macrophages | [92] ** |
| 1996 | DSPC:DSPG (10:3) or DSPC:DSPG:DSPE-PEG (10:3:1.45) | ddI a logP = −1.24 | N.D. | 26 ± 4 | N.D. | 150 ± 10 | Better pharmacokinetic profile ↑ viral reservoirs targeting ↑ bioavailability | [17]*,** |
| 2000 | EPC, DMPG, SM, DMPC, DPPC, DSPC, DMPC:Chol (4:1; 2:1 and 1:1), DSPC:Chol (4:1; 2:1 and 1:1), DPPC:Chol (2:1), DPPC:Chol:PS (6:3:1), DPPC:Chol: DCP (6:3:1) | d4T logP = −0.72 | 35 to 50 | N.D. | +, − and neutral charges | 600 to 1400 | ↑ maximal uptake by DPPC liposomes in macrophages and monocytes ↑ uptake with negative charged liposomes | [93] ** |
| 2002 | DPPC:DPPG:DSPE-PEG-MAL (10:3:0.83) | IDV g logP = 2.9 | 11 ± 4 | N.D. | N.D. | 100 to 120 | ↑ [IDV] to lymphoid tissues | [94] * |
| 2003 | EPC:Chol (3:1) | IDV g logP = 2.9 | 97.5 ± 2.5 at pH 7.4 ≈ 20 at pH 5.5 | 19.5 ± 0.5 at pH 7.4 ≈4 at pH 5.5 | N.D. | 69 ± 7 | ↑ CD4+ T cells ↓ viral load in lymph nodes and plasma | [95] * |
| 2005 | SPC:AZT-M:VE (6:2:0.1) | AZT-M a | Liophilization Before: 99.4 ± 0.8 After: 98.3 ± 1.2 | N.D. | - charges | Liophilization Before:88.5 ± 4.5 After:89.6 ± 6.3 | ↑ [ARV] in organs of RES and brain | [81] * |
| 2006 | EPC:Chol:SA (7:2:1) Uncoated LP or coated w/OPM: OPM-LP | d4T a logP = −0.72 | Uncoated LP: 49.6 ± 1.2 OPM-LP: 47.2 ± 3.3 | N.D. | Uncoated LP: + charge OPM-LP: charges ↓ close to neutrality | Uncoated LP: 120 ± 1.5 OPM-LP: 140 ± 2.3 | ↑ targeting ↑ residence time in HIV viral reservoirs ↑ d4T half-life ↑ pharmacological activity ↑ distribution ↓ elimination | [13] *,** |
| 2007 | EPC:Chol:DMPE (7:2:1) Uncoated LP or coated w/OPG: OPG-LP | d4T a logP = −0.72 | Uncoated LP: 46.2 ± 0.69 OPM-LP: 47.1 ± 1.2 | N.D. | Uncoated LP: +8.21 ± 0.15 OPG-LP: +3.2 ± 0.21 | Uncoated LP: 122.3 ± 0.3 OPM-LP: 129.5 ± 0.3 | ↑ d4T half-life ↑ residence time ↑ hepatic cellular d4T uptake | [14] *,**,*** |
| 2008 | EPC:Chol:DMPE (7:2:1) Uncoated LP or coated w/OPG: OPG-LP | d4T a logP = −0.72 | Uncoated LP: 49.6 ± 1.2 OPG-LP: 48.7 ± 0.2 | N.D. | N.D. | Uncoated LP: 120 ± 4 OPG-LP: 126 ± 4 | Inhibition of HIV p24 protein with uncoated LP and OPG-LP ↑ accumulation of OPG-LP in the liver, spleen, and MPS ↓ uptake of OPG-LP in bone | [96] *,** |
| 2006 | EPC:Chol:PE (7:2:1) Uncoated LP or coated w/OPG: OPG-LP) | AZT a logP = 0.05 | Uncoated LP: 54.3 ± 3.3 OPG-LP: 53.9 ± 2.1 | N.D. | Uncoated LP: + charge OPM-LP: charges ↓ close to neutrality | Uncoated LP: 120.0 ± 2.1 OPG-LP: 136.9 ± 1.9 | ↑ AZT half-life ↑ residence time ↑ bioavailability | [15] *,** |
| 2006 | SPC:Span80® (85:15) SPC:PEG-8-L (85:15) | AZT c logP = 0.05 | LP w/Span80®: 63.5 ± 2.9 LP w/PEG-8-L: 57.1 ± 3.1 | N.D. | LP w/Span80: −2.8 ± 0.4 LP w/PEG-8-L: −16.7 ± 0.7 | LP w/Span80®: 132 ± 15 LP w/PEG-8-L: 116 ± 10 | Better pharmacokinetic profile ↑ accumulation of AZT in target RES organs ↑ AZT half-life ↑ residence time, targeting, and controlled release | [19] *,** |
| 2007 | DPPC Note: intended for oral administration | ddI logP = −1.24 | N.D. | N.D. | N.D. | 1160 ± 129 | ↑ bioavailability | [97] ** |
| 2007 | PC:POPG (3:1) | IDV logP = 2.9 SQV logP = 3.8 | N.D. | N.D. | N.D. | 130 to 150 | ↑ liposomal solubilization of both drugs ↑ drug concentration in the media (10- and 750-fold for IDV and SQV, respectively) | [98] ** |
| 2008 | Plain-LP: SPC:PE:Span 80 (42.5:42.5:15) PEG-LP: SPC:PE:Span 80:MPEG 2000 (42.5:42.5:15:33.3) | AZT c logP = 0.05 | Plain-LP: 63.5 ± 2.9 PEG-LP: 72.3 ± 4.5 | N.D. | Plain-LP: −2.8 ± 0.4 PEG-LP: −18.2 ± 0.8 | Plain-LP: 132 ± 14 PEG-LP: 158 ± 15 | ↑ cellular uptake in lymphoid cells ↑ biodistribution ↑ residence time and sustained drug release | [53] *,** |
| 2008 | LP: SPC:Chol (7:3) + charge LP: SPC:Chol:SA (7:3:1) - charge LP: SPC:Chol:DCP (7:3:1) w/Mannose: SPC:Chol:Man (7:3:2.5) | AZT g logP = 0.05 | LP: 18.5 ± 1.2 + charge LP: 24.2 ± 0.9 − charge LP: 22.4 ± 1.4 Man-LP: 20.0 ± 2.5 | N.D. | LP: +10.3 ± 1.8 +charge LP: +54.4 ± 2.3 −charge LP: −34.8 ± 4.45 Man-LP: +14.7 ± 3.9 | LP: 122 ± 6 + charge LP: 126 ± 3 − charge LP: 128 ± 4 Man-LP: 127 ± 1.2 | ↓ release in Man-LP as compared to LP ↑ uptake ↑ localization of Man-LP in the lymph nodes and spleen | [99] *,** |
| 2009 | HSPC:Chol:mPEG–DSPE (55:40:5) | PI1 | N.D. | N.D. | N.D. | N.D. | ↑ and longer antiviral activity Facilitated specific uptake by non-phagocytic HIV-infected cells | [100] ** |
| 2010 | EPC:Chol (9:1) | NVP logP = 2.5 | 78.1 | 7.81 | N.D. | <200 | ↑ E.E. Quick in vitro release from liposomes | [101] ** |
| 2011 | DPPC | ProddINP b logP = 0.05 | 99 | 8.83 | −0.8 ± 0.5 | 187 to 208 | ↑ ddI blood half-life (3-fold) ↑ accumulation as prodrug at 24 h in various organs compared to plain drug | [79] *,** |
| 2011 | DPPC:EDPPC (1:1) | SFV logP = −19.5 | N.D. | N.D. | N.D. | N.D. | Strong affinity of SFV for DPPC:EDPPC ↑ Affinity with ↑ cationic EDPPC Fusion w/viral/raft-mimicking vesicles | [102] ** |
| 2011 | Chol:SA (194:1; 39:1; 22:1; 16:1;4:1) w/Span 20®/Span 40®/Span 60® | TFV logP = −1.6 | 3.46 to 65.26 | N.D. | +4.79 to +17.13 | 36.13 to 114.9 | The composition had a significant impact on TFV release Size and ζ were inversely proportional to the homogenization parameters, in contrast to the E.E. and conductivity TFV distributed within both the aqueous and lipid phases | [103] ** |
| 2012 | EPC:DSPE-PEG | SQV logP = 3.8 | 32.2 ± 2.9 | N.D. | −35.50 ± 1.66 | 176.6 ± 6.8 | ↓ cytotoxicity with PEGylated liposomes | [104] ** |
| 2012 | DMPC:Chol:DPTAP (55:27:18) DPPC:Chol:DPTAP (55:27:18) DSPC:Chol:DPTAP (55:27:18) DSPC:Chol:SA (60:30:10) | TFV logP = −1.6 | N.D. | N.D. | DMPC:Chol:DPTAP: +71.11 ± 5.72 DPPC:Chol:DPTAP: +62.50 ± 2.64 DSPC:Chol:DPTAP: +59.76 ± 2.49 DSPC:Chol:SA: +31.54 ± 1.90 | DMPC:Chol:DPTAP: 166.8 ± 18.1 DPPC:Chol:DPTAP: 158.1 ± 32.0 DSPC:Chol:DPTAP: 159.0 ± 35.5 DSPC:Chol:SA: 158.5 ± 34.7 | In the two-stage reverse dialysis method proposed, no drug leakage occurred during the 1st stage in LP containing high phase transition temperature lipids and high Chol content In the 2nd stage, significant differences in TFV release rate occurred in LP with different compositions | [105] ** |
| 2010/13 | Chol:Phospholipon 100H:SA (1:1:0; 5:5:1; 3:3:1; 2.3:2.3:1; 2:2:1; 2:1:1) | TFV. logP = −1.6 | 1.28 ± 0.24 (1:1:0) to 70.8 ± 2.55 (2:1:1) | 0.39 ± 0.087 (1:1:0) to 17.71 ± 1.87 (2:1:1) | −3.43 (1:1:0) to +93.5 (5:5:1) | 46.6 (1:1:0) to 2,200 (2:1:1) | ↑ permeation of TFV (Caco-2 cell model) | [106,107] ** |
| 2016 | LP DSPE:Stearic Acid:Chol (1:1:1) Stealth LP DSPE:Stearic Acid:Chol w/PEG 10000 | RTV a logP = 3.9 | LP: 98 ± 0.5 Stealth LP: 94.12 ± 0.29 | LP: 11.92 ± 0.06 Stealth LP: 11.45 ± 0.03 | LP: −33 ± 0.4 Stealth LP: −43.6 ± 1.8 | LP: 49 ± 0.3 Stealth LP: 116.6 ± 0.1 | Stealth LP prolongs RTV release to 34 h ↑ half-life of RTV for stealth LP LP and pure RTV showed dose dependent pharmacokinetics | [80] *,** |
| 2017 | Phospholipon 100H:Chol:SA (3:3:1 and 2:2:1) Note: intended for oral administration | TFV logP = −1.6 | (3:3:1): 39.8 ± 8.1 (2:2:1): 68.1 ± 2.6 | N.D. | + charge | N.D. | ↑ cellular permeability (10 times higher) ↑ E.E. | [108] ** |
| 2017 | HSPC:Chol (7:3) | LPV b logP = 5.94 | 90.47 ± 0.32 | N.D. | −24.8 ± 0.21 | 659.7 ± 23.1 | ↑ LPV release at 60 min (95% for LPV loaded proliposomes vs. 55% for free LPV) ↑ intestinal permeation (≈1.99 fold) compared to pure LPV) ↑ oral bioavailability (2.24- and 1.16-fold) than pure LPV and commercial LPV/RTV, respectively. | [109] *,**,*** |
| 2015 | EPC:Chol:DSPE-PEG (9:1:1) | NVP logP = 2.5 and SQV logP = 3.8 | NVP: 44 ± 2 SQV: 44 ± 1 | N.D. | −29 ± 2 | 160 ± 2 | ↑ inhibition of viral proliferation at lower doses compared to free drugs NVP is mainly released in the early phases and SQV in the later phases of infection | [70] ** |
| 2017 | SPC:Chol (2:1) Plain or coated w/biotin | RTV b logP = 3.9 | Plain LP: 62.3 ± 1.7 Biotin-LP: 61.6 ± 1.8 | N.D. | Plain LP: −18.9 ± 2.0 Biotin-LP: −26.1 ± 2.5 | Plain LP: 126.6 ± 6.2 Biotin-LP: 149.8 ± 6.8 | ↑ release from biotin coated liposomes compared to conventional ones ↑ [RTV] in lymphatic tissues | [110] *,** |
| 2018 | DSPC:DSPE-mPEG2000 (9:1) | ATV g logP = 4.5 RTV logP = 3.9 TFV logP = −1.6 | ATV: 99 ± 8.2 RTV: 92 ± 7.1 TFV: 10 ± 0.8 | N.D. | N.D. | 6 to 62 | ↑ residence time in plasma and peripheral blood mononuclear cells | [21] * |
| 2019 | DPPC Note: intended for vaginal administration | TDF logP = 2.65 FTC logP = −0.43 | 84 | 1 | Zwitterio-nic | 134 ± 13 | ↑ TDF permeation and ↑ sustained release Non-cytotoxic in CaSki (epidermoid cervical cancer cell line) and HEC-1-A (Human Endo-metrial Cancer-1) | [52] ** |
| 2020 | POPC POPC:DPPE-PEG2000 (9:1) | T20 logP = −14.7 PPI xT20 + PPIX | N.D. | N.D. | Zwitterio- nic charge was predominantly affected by PPIX | Unloaded POPC: 110 nm Unloaded POPC:DPPE-PEG2000 (9:1): 120 nm Size was affected by PPIX | ↑ entry inhibitors (T20 and PPIX) synergy compared to combination in free aqueous form | [111] ** |
| Ethosomes | ||||||||
| 2007 | SPC w/ethanol | 3TC c logP = −1.4 | 57.2 ± 4.1 | N.D. | −8.2 ± 1.5 | 102 ± 13 | ↑ cellular uptake ↑ transdermal flux (25 times higher) ↑ elasticity contributes to enhanced skin permeation | [112] *,*** |
| Cubosomes | ||||||||
| 2021 | GMO:CTAB:poloxamer 407(245:9:1, 219:9:1) | ATV g logP = 4.5 | 61 ± 4.6 (219:9:1) to 93 ± 1.2 (245:9:1) | N.D. | −29.41 (219:9:1) to −24.53 (245:9:1) | 253 ± 5.6 (219:9:1) to 150 ± 8.7 (245:9:1) | ↑ATV absorption and bioavailability (4.6 folds) compared to oral administration ↑ transdermal drug permeation due to bio-adhesive characteristic and permeation enhancement effect | [113] *,**,*** |
| 2020 | GMO:CTAB: poloxamer 407 (18:15:1) | SQV b,d logP = 3.8 | 72 ± 2 higher concentrations of GMO favored drug entrapment | N.D. | N.D. | 120 ± 2 ↑ particle size with ↑ GMO and ↓ Poloxamer 407 | ↑ SQV bioavailability (12-fold and 2.5-fold) when compared with oral and intranasal administration of free SQV | [114] *,*** |
| Hybrid liposomal nanocarriers | ||||||||
| 2017 | SPC and gelatin nanoparticles (SG-LP) | d4T logP = −0.72 | Gelatin NP (SG): 56.0 ± 1.7 SG-LP: 55.1 ± 2.1 | N.D. | SG-LP: −44.6 ± 1.36 | SG-LP: 232.9 ± 1.5 | ↑ controlled release ↑ uptake and hemocompatibility ↑ d4T half-life ↓ blood viremia | [20] ** |
| 2017 | LP DPPC or DPPC:Chol (1:1, 4:1, 2:1) Magneto-plasmonic LP MNP@Au coated w/PEG | TDF logP = 2.65 | ↑ E.E. w/higher drug ratio (≈30% for LP:TDF (1:34)) ↑ E.E. w/smaller Chol content (≈60% for DPPC) | N.D. | N.D. | ↓ with increasing Chol | ↑ TDF release for LP without Chol ↑ transmigration across an in vitro BBB model by magnetic targeting ↓ viral replication of HIV infected microglial cells | [115] ** |
| 2010 | LP SPC:Chol (1.2:1) Magnetic LP LP + magnetic AZTTP NP | AZTTP | 54.5 ± 6 | N.D. | N.D. | ∼150 nm | ↑ permeability (3-fold) for magnetic AZTTP LP than free AZTTP Efficient taken up by monocytes ↑ transendothelial migration in presence of an external magnetic field compared to normal/non-magnetic monocytes | [116] ** |
| 2021 | LP DMPC:DOPE:Chol (7:2:1) inside PVA nanofibers | TDF h logP = 2.65 FTC logP = −0.43 | 100 | 4 (FTC) and 2.8 (TDF) | LP −0.67 ± 0.01 | 211 ± 24 | Rapid onset of local drug levels upon single vaginal administration of fibers to mice comparing to the continuous daily use for 5 days of oral TDF/FTC Drug concentrations in vaginal fluids were fairly sustained up to 1–4 h, which could be translatable into a quite wide protection time window in humans | [117] * |
| Year | Composition | ARV | Physicochemical Characterization | Outcomes | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| E.E. (%) | D.L. (%) | ζ-Potential (mV) | Size (nm) | |||||
| SLN | ||||||||
| 1998 | Lipid phase: Trilaurin:DPPC:DMPG (0.69:0.28: 0.03 wt:wt) SLN coated w/PE-PEG2000 [10% (mol ratio)] | AZT a logP = 0.05 | N.D. | N.D. | Plain SLN: −20 ± 5 PE-PEG SLN: −6 ± 4 | Plain SLN: 183 ± 48 PE-PEG SLN: 182 ± 44 | ↓ release rate in SLN-PE-PEG ↑ bioavailability ↑ accumulation of SLN in the liver | [18] *,** |
| 2006 | Lipid phase: Trilaurin or Tristearin:Chol:PC:SA (1:0.5:1:0.1) | AZT a logP = 0.05 | Trilaurin SLN: 57.8 ± 6.2 Tristearin SLN: 59.7 ± 6.1 | N.D. | +charges | Trilaurin SLN: 130 ± 18 Tristearin SLN: 142 ± 22 | ↑ uptake in hepatocytes ↑ controlled release (12–15% in 24 h) | [76] *,** |
| 2008 | Lipid phase: stearic acid Aqueous phase: Pluronic® F68 (3%) Note: intended for enhanced brain delivery | ATV logP = 4.5 | 98.9 ± 0.8 98.2 ± 1.3 89.3 ± 2.7 | 1 2 5 | 18.43 ± 0.70 | 167 ± 8.3 | Burst ATV release of≈17% by 1 h and gradual release up to 40% by 24 h ↑ uptake and accumulation of ATV when delivered by SLN (human brain endothelial cell monolayer) compared to the free ATV | [65] ** |
| 2011 | Lipid phase: Compritol® 888 ATO/tripalmitin/cacao butter (wt of 8%) Aqueous phase: PC (7%), cholesteryl hemisuccinate (5%), taurocholate (2.5%) and 1-butanol (9.2%) | d4T logP = −0.72 DLV logP = 2.8 SQV logP = 3.8 | SQV > DLV > d4T | N.D. | N.D. | 142–308 ↑ % Compritol® 888 ATO: ↑ d4T-SLN mean size and ↓ DLV-SLN and SQV-SLN mean size | ↑ E.E. for d4T Sustained drug release: d4T > DLV > SQV | [74] ** |
| 2011 | Lipid phase: Compritol® 888 ATO Aqueous phase: Pluronic® F127 (2.5%) | LPV b,f logP = 5.94 | >99 | N.D. | −26.5 ± 0.45 | 230.4 ± 5.6 | Slow-release in both media pH 6.8 and pH 1.2 ↑ bioavailability and targeting ↑ % LPV secreted into the lymph | [71] *,** |
| 2011 | Lipid phase: Softisan® 100 Aqueous phase: BSA and PAA (negative moiety) Additional layer of PLL (positively charged) and heparin (negatively charged) Note: intended for topical/vaginal administration | TFV logP = −1.6 | 8.3 ± 0.7 | 0.083 | −51.07 ± 4.44 | 153.66 ± 11.33 | Non-cytotoxic (human vaginal epithelial cell line) and easily functionalized ↑ solubility | [118] ** |
| 2011 | Lipid phase: Dynasan® 114 Solutol® HS15 and Plurol® Oleique CC 497 (hydrophobic surfactants 3%) Aqueous phase: Poloxamer 118 and Tween® 80 (aqueous surfactant solution 0.5%) | d4T f logP = −0.72 | 96 ± 4.42 | N.D. | −34.48 | 75 ± 1.22 | ↑ residence in splenic tissues ↑ uptake by macrophages compared to free drug | [119] *,*** |
| 2011 | Lipid phase: Compritol® 888 ATO/steric acid (4%) Aqueous phase: DODAB (1.8%), Tween® 80 (1%), lecithin (0.2%) and 1-butanol (0.5%) SLN coated with HSA | NVP logP = 2.5 | NVP-SLN: ≈77 (maximum achieved) | N.D. | NVP-SLN: + ↑ [HSA] ↓ ζ in SLN to values close to neutrality | NVP-SLN: 153.1 HSA/NVP-SLN: 189.2 | ↑ E.E. with SLN ↓ HBMECs viability with NVP-SLN ↑ HBMECs viability with HSA-NVP-SLN | [120] ** |
| 2013 | Lipid phase: Dynasan® 114/palmitic acid (4%) DSPE-PEG2000 (1.25%) Note: wt fractions of palmitic acid in Dynasan-palmitic acid mixture were 0, 0.33, 0.67, 1 Aqueous phase: cholesteryl hemisuc-cinate (0.4%), poloxamer 407, Tween® 80 and SDS (1%) Note: wt fractions of poloxamer 407 in poloxamer 407-Tween® 80 were 0, 0.5, and 1, 0.1% (w/v) Note: intended for brain delivery mAb-grafted SQV-loaded SLN | SQV logP = 3.8 | ≈55 to 80 | N.D. | >−30 | 120 to 450 | ↑ BBB permeation ↑ bioavailability and ↑ solubility ↑ controlled release ↑ E.E. High biocompatibility of mAb-grafted SLN to HBMECs ↑ HBMECs uptake ↓ lymphatic uptake | [64] ** |
| 2014 | Lipid phase: GMS Aqueous phase: Tween® 80 (1.25%) | EFV b logP = 4.6 | 86 | N.D. | −15.9 | 124.5±3.2 | ↑ 5.32-fold in Cmax and ↑ 10.98-fold in AUC w/EFV-SLN compared to EFV suspension | [16] * |
| 2015 | Lipid phase: tristearin: HSPC:DSPE (11.2:12.6:0.3, molar ratio) Aqueous phase: Tween® 80 (0.5%) SLN conjugated with PA | EFV a logP = 4.6 | SLN: 72.1 ± 0.4 PA-SLN: 63.5 ± 0.6 | N.D. | SLN: −28.8 ± 1.2 PA-SLN: −36.2 ± 1.0 | SLN: 113 ± 0.2 PA-SLN: 163 ± 0.5 | Able to permeate BBB ↑ bioavailability ↑ controlled release | [55] *,** |
| 2015 | Lipid phase: Gelucire® 44/14 (30%) and Compritol® 888 ATO (20%) Aqueous phase: Lipoid® S 75 (25%) and poloxamer 188 (5%) | EFV b logP = 4.6 | 85.6 | 39.4 | −35.55 | 168.92 ± 31.2 | Prolonged and biphasic in PBS pH 6.8 ↑ bioavailability ↑ [EFV] in the spleen ↑ biodistribution in lymphatic organs | [75] *,** |
| 2016 | Lipid phase: stearic acid Aqueous phase: PVA SLN modified with Aloe Vera | AZT logP = 0.05 | SLN: 66.5 SLN-AV: 84 | SLN: 18.01 SLN-AV: 29.6 | AZT-SLN: −12.18 to 13.1 AZT-SLN-AV: −14.2 to 15.41 | SLN: 222 to 227 SLN-AV: 402 to 434 | ↑ solubility ↑ cellular uptake and no cytotoxicity (C6 glioma cells) ↑ E.E. | [121] ** |
| 2017 | Lipid phase: GC and GMS (1.5 g) Note: N.D. of GC:GMS Span 80 (1%) Aqueous phase: Tween® 80 (2%) | DRV logP = 1.89 | SLN: 74.23 Freeze-dried SLN: 69.8 | Post-freeze drying: 9.37 | Freeze-dried SLN: −22 ± 2 | SLN: 210 Freeze-dried SLN: 270 | Sustained release of DRV until 12 h Apparent permeability across rat intestine: 24 × 10−6 (cm/s) at 37 °C and 5.6 × 10−6 (cm/s) at 4 °C Endocytic uptake | [73] **,*** |
| 2017 | Lipid phase: tripalmitin Aqueous phase: poloxamer 188 (1%) | EFV d logP = 4.6 | 64.9 | N.D. | −21.2 | 108.3 | Burst release followed by a prolonged release ↑ [EFV] in brain ↑ brain targeting efficiency (more 150 times) and better absorption of the EFV (70 times more) with intranasal as compared to orally administered marketed formulation (capsule) | [84] *,** |
| 2017 | Lipid phase: GMS:SL (1:1) Aqueous phase: Poloxamer118 (0.5%) or Tween® 80 (0.25%) | RTV logP = 3.9 | 21.4−53.3 | N.D. | −39.35 ± 1.2 to −50.80 ± 4.8 | 178 to 254 | ↑ E.E. and mean size using poloxamer 118 as surfactant ↑ controlled release RTV-SLN can maintain inhibition of virus production | [122] ** |
| 2017 | Preconcentrate: PGDS or stearic acid and poloxamer 118 (0.1–1.0%) Diluted with water | NVP a logP = 2.5 | >90 | N.D. | PGDS-SLN: −26.8 ± 2.1 | Stearic acid-SLN: 940.2 ± 1.54 PGDS-SLN: 70 to 1100 (average particle size ~212 nm) | Biphasic release profile with an initial burst release ↑ [NVP] in the liver, kidneys, and brain Targeting for multiple viral reservoirs | [85] *,** |
| 2018 | Lipid phase: Compritol® ATO 888 (0.5%) Aqueous phase: poloxamer 407 (0.25%), Labrasol® (0.25%) Topical formulation for transdermal delivery SLN based HG | LPV c logP = 5.94 | 69.78 | N.D. | −17.7 ± 0.54 | 48.86 ± 4.6 | ↑ sustained LPV release from SLN based HG (71.197 ± 0.006% after 12 h) compared to plain HG of the drug released (98.406 ± 0.007% after 4 h) SLN based HG resulted in the highest Cmax (20.3127 ± 6056 μg/mL) compared to plain HG (8.0655 ± 1.6369 μg/mL) and oral LPV suspension (4.2550 ± 1 6.380 μg/mL) | [72] *,**,*** |
| 2018 | Lipid phase: hydrogenated castor oil (castor wax) Aqueous phase: sodium oleate (3.5%) Grafted SLN: peptide (100 μg):SLN (1 μM) (Pept-DRV-SLN) | DRV b logP = 1.89 | DRV-SLN: 90.10 ± 1.15 Pept-DRV-SLN 90.16 ± 1.25 | DRV-SLN: 13.06 ± 1.18 Pept-DRV-SLN 13.18 ± 1.23 | DRV-SLN: −50.1 ± 1.17 Pept-DRV-SLN −35.45 ± 1.10 | D-SLN: 189.45 ± 2.10 Pept-D-SLN 195.11 ± 1.53 | ↑ DRV release in SLN compared to a plain drug suspension ↑ permeability in Caco-2 cells (4-fold) than free drug ↑ uptake in HIV host cells (molt-4 cells were taken as a model containing CD4 receptors) as compared to non-CD4 receptor-bearing Caco-2 cells ↑ bioavailability than free DRV: ↑ uptake in various organs (also in HIV reservoirs like spleen and brain) with Pept-DRV-SLN ↑ binding with the HIV host cells | [82] *,** |
| NLC | ||||||||
| 2011 | Lipid phase: OA (1%) and Compritol® 888 ATO/steric acid (4%) Aqueous phase: DODAB (1.8%), Tween® 80 (1%), lecithin (0.2%) and 1-butanol (0.5%) NLC coated with HSA | NVP logP = 2.5 | NVP-NLC: ≈68 (maximum achieved) | N.D. | NVP-NLC: + ≈+17.5 ↑ [HSA] ↓ ζ of NLC to values close to neutrality | NVP-NLC: 159.6 HSA/NVP-NLC: N.D. | NLC promote a fast release | [120] ** |
| 2017 | NLC Lipid phase: Compritol® ATO 888:OA (1:3) Tween® 80 (44 mg) MLN Lipid phase: Compritol® ATO 888:OA:Span® 80 (1:3:1.8) Tween® 80 | 3TC. logP = −1.4 | 3TC-NLC: 34 ± 1 3TC-MLN: 20 ± 2 | 3TC-NLC: 0.3 ± 0.01 3TC-MLN: 1.08 ± 0.06 | Unloaded-NLC: −42.9 ± 0.7 3TC-NLC: −45 ± 2 Unloaded MLN: −24.5 ± 0.4 3TC-MLN: −21 ± 2 | Unloaded-NLC: 229 ± 2 3TC-NLC: 218 ± 4 Unloaded MLN: 426 ± 9 3TC-MLN: 450 ± 10 | Sustained and controlled 3TC release under gastric and plasma-simulated conditions (≈45 h in MLN) Low cytotoxicity (T lymphocytes) for both formulations ↑ loading capacity and storage stability with MLN | [123] ** |
| 2018 | Lipid phase: Precirol®ATO 5:Miglyol® 812 (3:1) Tween® 80 (158 mg) AZT-NLC prepared by hot ultrasonication method and M-AZT-NLC prepared by the one-step microwave-assisted method | AZT logP = 0.05 | AZT-NLC: 44 ± 3 M-AZT-NLC: 22 ± 2 | AZT-NLC: 0.31± 0.04 M-AZT-NLC: 1.41 ± 0.02 | - AZT-NLC: −29 ± 2 M-AZT-NLC: −20 ± 1 | AZT-NLC: 266 ± 4 M-AZT-NLC: 113 ± 3 | Controlled release of AZT Suitable for oral administration | [12] ** |
| 2019 | Lipid phase: Compritol® 888 ATO:OA (3.7:1) Aqueous phase: Tween® 80 (0.04) | LPV b logP = 5.94 | 83.6 | N.D. | + 21.2 | 196.6 | ↑ bioavailability ↑ [LPV] in the brain ↑ uptake and ↓ cytotoxicity (Caco-2 cells and macrophages) | [83] *,** |
| 2020 | Lipid phase: Precirol® ATO 5:LauroglycolTM 90 (70:30) Cremophor® RH 40 (3%) | ATV b logP = 4.5 | 71.09 ± 5.84 | 8.12 ± 2.7 | −11.7 ± 0.47 | 227.6 ± 5.4 | Fast release (60%) in the initial 2 h, followed by sustained release ↑ permeation of ATV (2.36-fold) across the rat intestine as compared to the free drug 2.75-fold greater Cmax in the brain and a 4-fold improvement in brain bioavailability as compared to the free drug | [86] *,**,*** |
| 2020 | Lipid phase: MonosteolTM (71.5%): Capmul® PG 8 (28.5%) Aqueous phase: Tween® 80 (0.43%) and poloxamer 188 (1.3%) Cationic NLC Lipid phase: same composition Aqueous phase: same composition + CTAB (1% w/w of lipid phase) | EFV logP = 4.6 | EFV-NLC: 91.18 ± 2.9 Cationic EFV-NLC: 90.21 ± 2.3 | EFV-NLC: 10.94 ± 0.35 Cationic EFV-NLC: 11.04 ± 0.17 | Plain NLC: −15.16 ± 0.69 EFV-NLC: −15.8 ± 1.21 Cationic EFV-NLC: +23.86 ± 0.49 | Plain NLC: 114.53 ± 5.63 EFV-NLC: 116.5 ± 9.59 Cationic EFV-NLC: 105.6 ± 4.93 | ↑ solubility Excellent cytocompatibility (CC50 13.23±0.54 µg/mL) Uptake of cationic NLC by THP-1 macrophages ↑ retention/sustained release and ↑ inhibition of HIV-1 (2.32-fold) in infected macrophages with cationic NLC compared to the free drug ↑ anti-HIV-efficacy (2.29-fold) with cationic NLC | [124] ** |
| 2021 | Lipid phase: Precirol® ATO 5:Capmul® MCM (40:60) CapryolTM 90 (N.D.) Aqueous phase: Lutrol® F 127 (1%) | ETR a logP = 4.5 | >90 | 5 to 10 | −20 ± 2.3 | 351.7 ± 3.36 | ↑ cellular uptake and ↑ anti-HIV efficacy Overall better pharmacokinetics as compared to the free drug ↑ [ETR] several-fold in the liver, ovary, lymph node, and brain as compared to the free drug | [87] *,** |
| 2021 | Lipid phase: GMS/Gelucire® 50/13/Dynasan® 118:Capmul® MCM EP (80:20) (8%) Span® 80 Aqueous phase: Tween® 80, sodium cholate; PEG 6000 (1%), propylene glycol (1%), BHT (0.4%) Note: Surfactant mixture [(Tween® 80: Span® 80 (70:30)]: 5% | DTG logP = 2.2 | 88.09 | N.D. | −16.6 | 123.1 | Sustained release over 48 h ↑ DTG permeation through rat intestine (≈94.02%) as compared to plain drug suspension (only 55.62%) after 8 h | [125] **,*** |
| 2014 | Lipid phase: Precirol® ATO 15 (10%) and Miglyol® 812 (1%) Aqueous phase: Tween® 80 (1%) and poloxamer 188 (1 or 0.5%) Coating on NLC: Dex–Prot | SQV logP = 3.8 | All formulations: 99 | N.D. | Uncoated NLC: −36 ± 6 to −22 ± 4 Dex-Prot NLC: −0.5 ± 4 to +12 ± 4 | Uncoated NLC: 152 ± 1 to 936 ± 1 Dex-Prot NLC: 244 ± 1 to 1326 ± 1 | ↑ permeability (up to 9-fold) with Dex–Prot NLC in comparison to uncoated NLC (Caco-2/HT29-MTX co-culture monolayer model) | [126] ** |
| 2017 | Lipid phase: Precirol® ATO 5:Captex® P 500 (7:3) Aqueous phase: MYS-25 (2%) | EFV a,d logP = 4.6 | 95.78 ± 0.42 | N.D. | −18.7 ± 1.0 | 161 ± 2.8 | EFV release of 92.45% after 24 h The therapeutic concentration of EFV in the CNS following intranasal administration No toxicity of encapsulated EFV compared to free EFV | [54] *,** |
| Year | Composition | ARV | Physicochemical Characterization | Outcomes | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| E.E. (%) | D.L. (%) | ζ-Potential (mV) | Size (nm) | |||||
| Microemulsions (ME) | ||||||||
| 2015 | o/w ME Lipid phase: Capmul® MCM (75%), Cremophor® RH 40 Aqueous Phase Transcutol® P Cremophor RH 40: Transcutol® P (1:1)(40%) Solid ME Absorbing agent (aerosil 200) ME:aerosil 200 (1:1) | DRV logP = 1.89 | 99.42 | N.D. | N.D. | 40.68 | ↑ solubilityGreater intestinal permeability than the free drug ↑ intestinal permeability with ↑ [oil phase] | [127] *** |
| 2016 | Lipid phase: isopropyl myristate (10%), Labrasol® (30%); Oleic Plurol® (10%) | AZT c logP = 0.05 | N.D. | N.D. | N.D. | N.D. | ↑ AZT permeated (≈2-fold) as compared to control—HGNo apparent skin irritation; little histological changes in mice skin | [128] *,** |
| Nanoemulsions (NE) | ||||||||
| 2008 | o/w Lipid phase: Flax-seed oil or safflower oil (1 mL) Aqueous phase: EPC (3%) and deoxycholic acid (1%) | SQV a,b logP = 3.8 | N.D. | N.D. | - SQV-Flax-seed NE: −43.28 ± 3.79 SQV-Safflower NE: −49.55 ± 5.02 | SQV-Flax-seed NE:218.0 ± 13.9SQV-Safflower NE:140.0 ± 12.6 | ↑ SQV (3-fold) in systemic circulation when loaded in Flax-seed NE than in the free form↑ bioavailability↑ brain distribution (Cmax 5-fold and AUC 3-fold) higher in the brain with Flax-seed NE than the free drug | [90] * |
| 2014 | NE Lipid phase: Capryol® 90, Geucire® 44/14 (13.728%) Aqueous phase: Transcutol® HP (3.432%) and water (79.98%) Oil:Smix (1:6) | EFV b logP = 4.6 | N.D. | N.D. | N.D. | 26.427 ± 1.960 | >80% release within 6 h ↑ AUC0→24h (43.53 μg h/mL) compared to EFV suspension (20.65 μg h/mL)EFV absorption resulted in 2.6-fold increase in bioavailability in comparison to the free EFV | [129] *,** |
| 2014 | o/w NE Lipid phase: Capmul MCM (6%) Aqueous phase: Tween® 80 (6%), PEG 400 (2%) and water (86%) | SQVM d logP = 3.8 | 96.8 ± 1.2 | N.D. | −10.3 ± 1.67 | 176.3 ± 4.21 | ↑ Diffusion of SQVM across nasal mucosa than the free drug No significant adverse effect in cilia toxicity study ↑ [SQVM] brain after intranasal administration of NE than intravenous delivery of free drugEffective CNS targeting | [88] *,** |
| 2013 | o/w NE Lipid phase: soya bean oil (10%), Chol (0 or 0.3%), EPC-80 (1.2%), α-tocopherol (0.25%) OA (0.3%) Aqueous phase: glycerol (2.25%), Tween® 80 (0 to 1%), and double-distilled water (10%) | IDV a | No Chol, no Tween® 80: 99.1 ± 0.2 Chol and no Tween® 80: 98.9 ± 0.03 Tween®80 (1%): 98.97 ± 0.2 | N.D. | No Chol and no Tween 80 NE: −35.8 ± 6.04 Chol and no Tween 80 NE: −31.3 ± 1.80 Tween 80 (1%) NE: −40.1 ± 4.05 | No Chol and no Tween 80 NE:329.5 ± 3.08 Chol and no Tween 80 NE:237.0 ± 5.08 Tween 80 (1%) NE:196.0 ± 3.54 | NE containing Chol and higher [Tween 80] (1%) had lower globule size, relative better release, and higher ζ↑ brain uptake of IDV in Tween 80 (1%) NE compared to Chol and no Tween 80 NE↑ brain level of IDV administered by Tween 80 (1%) NE compared to the free drug (2.44-fold)↑ IDV brain-specific accumulation | [89] *,** |
| Self-emulsifying drug delivery systems (SEDDS) | ||||||||
| 2013 | SMEDDS Lipid phase: Capmul® MCM (C8) (10%), Cremophor® RH 40 (81%) Aqueous phase: PEG 300 (9%) Fill SMEDDS into a hard gelatine capsule | Micronized UC-781 | N.D. | N.D. | +20.5 ± 0.52 to +32.0 ± 0.02 | Smallest droplet sizes (14.9 ± 0.9,12.8 ± 0.4 and 16.1 ± 0.7) with the lowest oilcontent (1:9, 2:8 and 3:7 oil to surfactant/Cosurfactant ratios)The concentration of oil above 40% w/w: droplet size increased to as high as 100 nm | ↓ solubility as the ↑ oil component (higher solubility of UC-781 in the surfactant and cosurfactant compared to oil)↑ in droplet size as ↑ oil UC781 had no significant effect on droplet size, polydispersity index, or zeta potentialFaster UC-781 release (100% by 60 min) from SMEDDS↑ absorption across the model membrane than UC781 powder | [130] ** |
| 2016 | SNEDDS Lipid phase: Eucalyptus oil (12%), Smix (Cremophor® EL and Brij®35, 1:1) (12–18%) Aqueous phase: Transcutol® P (0–24%) | EFZ b logP = 4.6 | N.D. | N.D. | N.D. | 21.97 ± 1.3 to 113.9 ± 4.8 | ↓ mean globule size as ↑ surfactant (Smix) ↑ mean globule size as ↑ co-surfactant (Trancutol®) Faster EFZ release (>80% by 30 min) from SNEDDS compared to the free drug (18.3% by 30 min)↑ oral bioavailability (2.63-fold) than the free drug | [131] *,** |
| 2016 | SNEOF Lipid phase: Maisine® 35-1 (0.7) Aqueous phase: Tween®80:Transcutol® HP (1:0.6) S-SNEOF Aeroperl® (absorbing agent) Compressed tablet (MCC) | LPV b logP = 5.94 | 99.45 ± 0.59 | N.D. | N.D. | SNEOF: 53.16SNEOF tablets: 80 | ↑ LPV release (60% by 10 min) Lymphatic uptake of LPV from SNEOF↑ rate and extent of oral bioavailability than the free drug | [66] *,** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria, M.J.; Lopes, C.M.; das Neves, J.; Lúcio, M. Lipid Nanocarriers for Anti-HIV Therapeutics: A Focus on Physicochemical Properties and Biotechnological Advances. Pharmaceutics 2021, 13, 1294. https://doi.org/10.3390/pharmaceutics13081294
Faria MJ, Lopes CM, das Neves J, Lúcio M. Lipid Nanocarriers for Anti-HIV Therapeutics: A Focus on Physicochemical Properties and Biotechnological Advances. Pharmaceutics. 2021; 13(8):1294. https://doi.org/10.3390/pharmaceutics13081294
Chicago/Turabian StyleFaria, Maria J., Carla M. Lopes, José das Neves, and Marlene Lúcio. 2021. "Lipid Nanocarriers for Anti-HIV Therapeutics: A Focus on Physicochemical Properties and Biotechnological Advances" Pharmaceutics 13, no. 8: 1294. https://doi.org/10.3390/pharmaceutics13081294
APA StyleFaria, M. J., Lopes, C. M., das Neves, J., & Lúcio, M. (2021). Lipid Nanocarriers for Anti-HIV Therapeutics: A Focus on Physicochemical Properties and Biotechnological Advances. Pharmaceutics, 13(8), 1294. https://doi.org/10.3390/pharmaceutics13081294