
Review: Continuous Manufacturing of Small Molecule Solid Oral Dosage Forms


Abstract
:1. Introduction
2. Background
- For investment in pharmaceutical manufacturing innovation, including CM in R&D and production.
- For the manufacturing of products using CM processes.
- Expedited approvals (similar to break-through designations) for products using CM for new products, generics or those moving from batch processes.
- Some form of certification for companies where the increased rigour and control in CM process allows simpler and faster regulatory approvals.
3. Regulatory Aspects of CM
3.1. International Conference on Harmonisation (ICH)
3.2. Major Regulatory Agencies
3.2.1. FDA
3.2.2. MHRA
3.2.3. EMA
3.2.4. PMDA
3.3. Other Regulatory Considerations
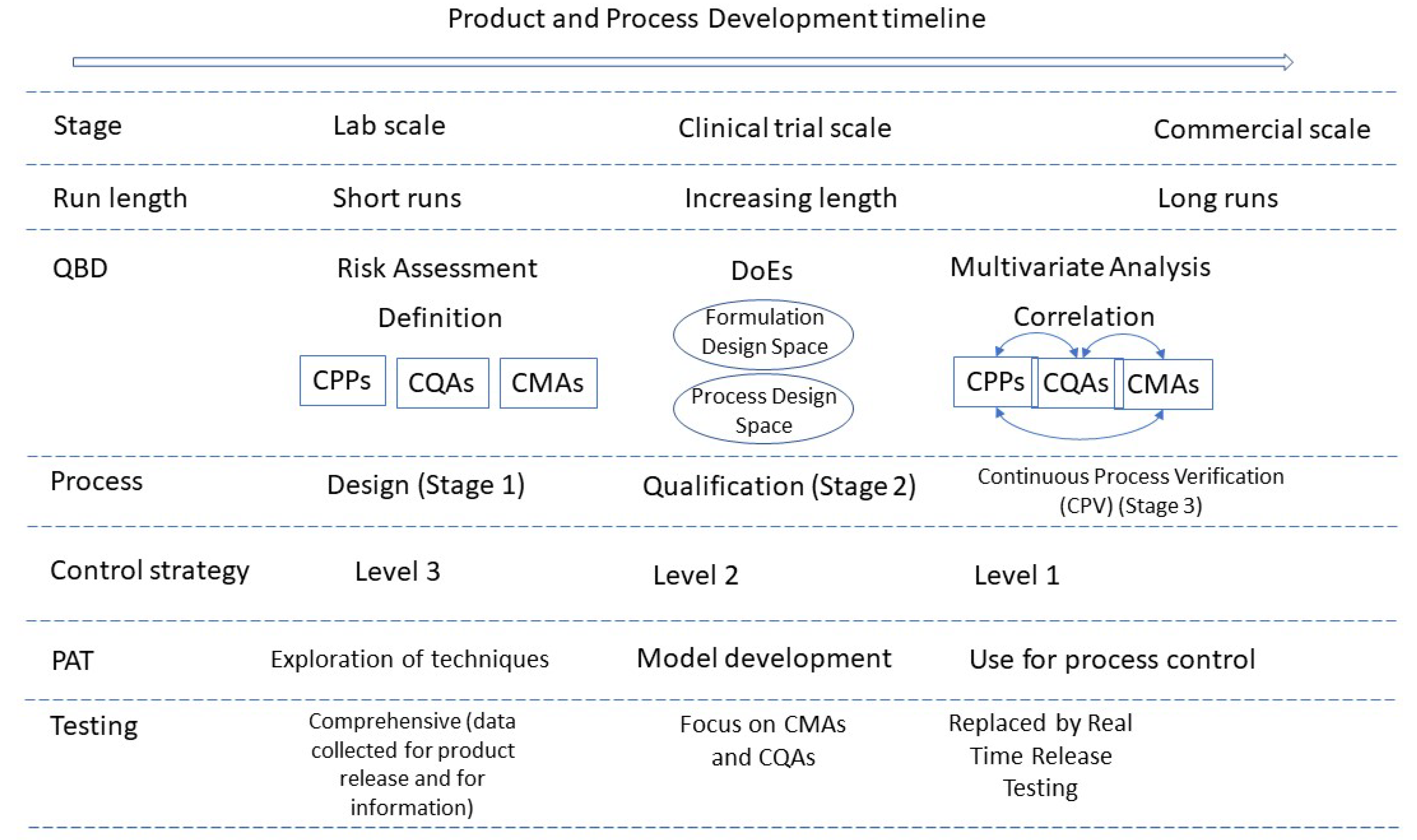
4. Quality Considerations of CM Processes
4.1. Control Strategy
4.2. Batch and Lot Definitions
- The amount of material produced within a specified period of time (the most common definition);
- A fixed quantity of raw material processed.
- Or by operational considerations such as:
- The input API batch size;
- The length of an operator’s shift.
- Increasing the run time (within validated limits). Consideration needs to be given to issues such as the build-up of material during longer runs (in transfer lines, filters, granulator barrel walls, punch surfaces, etc.), equipment overheating, etc.
- Parallel operation of multiple items of kit. Need to assure the uniformity of output from each process.
- Increasing the throughput of the process. This has an impact on the process dynamics and may affect aspects such as sampling frequency and size for PAT equipment, Residence Time Distribution (RTD) and material diversion (see Section 4.3) and will likely require that the control strategy is rebuilt.
- Change of size of equipment. Also, likely to require a completely different control strategy and complete re-validation
4.3. Traceability and Residence Time Distribution (RTD)
4.4. Process Disturbances and Diversion of Material
- Deviations that fall outside pre-set ranges of material attributes and process parameters as determined by development studies (e.g., response surfaces, design spaces);
- Prediction of quality attributes from process modelling with real-time inputs of process parameters and measurements;
- Direct measurement of quality attributes in real-time (use of PAT).
4.5. Real Time Release Testing (RTRT)
4.5.1. Blend Mixing and Tablet Content Uniformity
4.5.2. Moisture Content
4.5.3. Solid State Chemistry
4.5.4. Drug Release (Dissolution)
4.5.5. Related Impurities
4.6. Process Validation and Verification
4.7. Models Used in CM
4.8. Data Requirements
4.9. Summary
5. CM Equipment
5.1. Unit Operations
5.1.1. Feeders
5.1.2. Powder Blenders/Mixers
5.1.3. Granulators
5.1.4. Dryers
5.1.5. Coaters
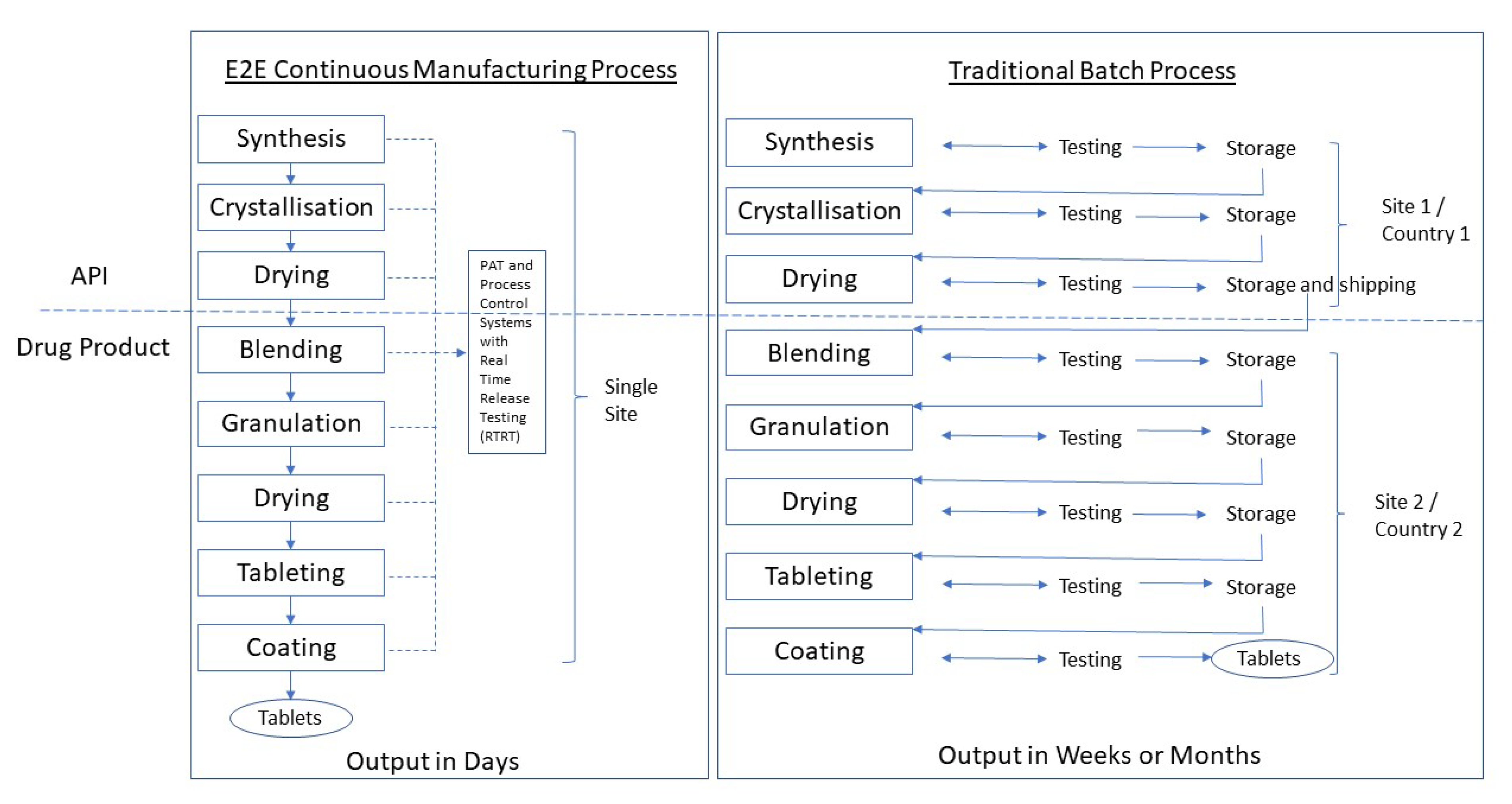
5.2. Integrated Continuous and End-to-End (E2E) Manufacturing
5.3. Alternative Continuous Manufacturing Approaches
6. Process Analytical Technology (PAT)
- Direct measurement of the CQA;
- Prediction of the CQA based on a first-principles model that is fed measurements of related variables and is running in parallel with CM unit operations;
- Prediction of the CQA based on an empirical or semi-empirical model (e.g., response surface map, chemometrics model) that is fed measurements of other variables;
- Operation of the CPPs to lie within a design space (ie a specified set shown in offline studies to provide assurance) [97].
- At-line: measurement where the sample is removed, isolated from and analysed near the process stream.
- On-line: measurement where the sample is diverted from the manufacturing process and may be returned to the process stream.
- In-line: measurement where the sample is not removed from the process stream and can be invasive or non-invasive.
7. Material Property Requirements for Continuous Manufacturing
8. Industry/Academia/Government Collaborations to Progress Continuous Manufacturing
8.1. International Symposia for Continuous Manufacture of Pharmaceuticals (ISCMP)
8.2. Consortia Working to Progress CM
8.2.1. Novartis—MIT Center for Continuous Manufacturing
8.2.2. CMAC
8.2.3. C-SOPS
8.2.4. RCPE
8.2.5. IIAPM
8.2.6. MMIP/MMIC
- To create a ‘digital twin’ of the CM process and via virtual formulation experiments to select equipment and initial processing conditions for CM and thereby give confidence that CM DC process will work. This will be useful evidence that a company might use to justify an investment in CM.
- To enhance understanding of raw materials to build a CM control strategy to allow RTRT and develop robust formulation processes on a modular flexible CM DC platform.
- To de-risk and accelerate CM DC technology by modularising equipment and standardising control systems moving away from supplier-specific approaches [120].
8.2.7. SSPC
8.2.8. NIIMBL
8.2.9. UK-CPI National Formulation Centre, National Industrial Biotechnology Centre and National Biologics Manufacturing Centre
8.2.10. AMED
8.3. Other Supportive Organisations and Guidance
8.3.1. United States Pharmacopeia
8.3.2. ASTM
8.3.3. ISPE
8.3.4. NIPTE
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleinebudde, P.; Khinast, J.; Rantanen, J. (Eds.) Continuous Manufacturing of Pharmaceuticals; Wiley: Hoboken, NJ, USA, 2017; ISBN 978-1-119-00132-4. [Google Scholar]
- Food and Drug Administration. Quality Considerations for Continuous Manufacturing Guidance for Industry; FDA: Silver Spring, MD, USA, 2019; pp. 1–27. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-considerations-continuous-manufacturing (accessed on 2 July 2021).
- Lee, S.L.; O’Connor, T.F.; Yang, X.; Cruz, C.N.; Chatterjee, S.; Madurawe, R.D.; Moore, C.M.; Yu, L.X.; Woodcock, J. Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production. J. Pharm. Innov. 2015, 10, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Vanhoorne, V.; Vervaet, C. Recent progress in continuous manufacturing of oral solid dosage forms. Int. J. Pharm. 2020, 579, 119194. [Google Scholar] [CrossRef]
- Badman, C.; Cooney, C.L.; Florence, A.; Konstantinov, K.; Krumme, M.; Mascia, S.; Nasr, M.; Trout, B.L. Why We Need Continuous Pharmaceutical Manufacturing and How to Make It Happen. J. Pharm. Sci. 2019, 108, 3521–3523. [Google Scholar] [CrossRef] [PubMed]
- Jhamb, K. Continuous Manufacturing in Pharmaceuticals: Implications for the Generics Market. Drug Dev. Deliv. 2019, 19, 1–5. [Google Scholar]
- Rogers, L.; Jensen, K.F. Continuous manufacturing—The Green Chemistry promise?: Critical Review. Green Chem. 2019, 21, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Shanley, A. Continuous Manufacturing: Addressing the Tough Questions. Pharm. Tech. 2019, 43, 1–6. [Google Scholar]
- Woodcock, J. Modernizing Pharmaceutical Manufacturing—Continuous Manufacturing as a Key Enabler. In Proceedings of the First International Symposium on the Continuous Manufacturing of Pharmaceuticals (ISCMP), Cambridge, MA, USA, 20–21 May 2014; Available online: https://iscmp.mit.edu/sites/default/files/documents/ISCMP%202014%20-%20Keynote_Slides.pdf (accessed on 2 July 2021).
- Burcham, C.L.; Florence, A.J.; Johnson, M.D. Continuous Manufacturing in Pharmaceutical Process Development and Manufacturing. Ann. Rev. Chem. Biomol. Eng. 2018, 9, 253–281. [Google Scholar] [CrossRef]
- Toro, J.L. Continuous Manufacturing And Its Regulatory Challenge. Contract Pharma 2019, 1–6. Available online: https://www.lachmanconsultants.com/2019/09/continuous-manufacturing-and-its-regulatory-challenge/ (accessed on 2 July 2021).
- Marriott, N. EMA Approves Janssen’s Prezista Continuous Manufacturing Line. European Pharmharmaceutical Review, 27 June 2017, p. 1. Available online: https://www.europeanpharmaceuticalreview.com/news/62587/ema-continuous-manufacturing/ (accessed on 2 July 2021).
- Medendorp, J.; Shapally, S.; Vrieze, D.; Tolton, K. Process Control of Drug Product Continuous Manufacturing Operations—A Study in Operational Simplification and Continuous Improvement. J. Pharm. Innov. 2020, 1–12. [Google Scholar] [CrossRef]
- Srai, J.S.; Badman, C.; Krumme, M.; Futran, M.; Johnston, C. Future supply chains enabled by continuous processing--opportunities and challenges. 20–21 May 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Conway, S.; Hurley, S.; MacConnell, C.; Meyer, R.; Moore, C.; Starbuck, C.; Ramasamy, M.; Ricart, B.; Witulski, F.; Zhang-Plasket, F.; et al. Proof of Operations for Continuous Manufacturing and Real Time Release Testing of Film Coated Tablets. In Proceedings of the Second International Symposium on the Continuous Manufacturing of Pharmaceuticals (ISCMP), Cambridge, MA, USA, 26–27 September 2016; Available online: https://iscmp.mit.edu/sites/default/files/documents/ISCMP%202016%20-%20Merck%20Meyer%202nd%20ISCMP%20Symposium.pdf (accessed on 2 July 2021).
- CMAC; PwC. Business Case Insights for Continuous Manufacturing. 2019. Available online: https://www.cmac.ac.uk/Business_case.htm (accessed on 28 July 2021).
- Srai, J.S.; Settanni, E.; Aulakh, P.K. Evaluating the Business Case for Continuous Manufacturing of Pharmaceuticals: A Supply Network Perspective. In Continuous Pharmaceutical Processing; AAPS Advances in the Pharmaceutical Sciences Series; Nagy, Z., El Hagrasy, A., Litster, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; Volume 42, pp. 477–512. ISBN 978-3-030-41523-5. [Google Scholar]
- Presentations given at the Virtual International Symposium on the Continuous Manufacturing of Pharmaceuticals (ISCMP), 18 February 2021. Available online: https://hopin.com/events/international-symposium-on-continuous-manufacturing-of-pharmaceuticals (accessed on 2 July 2021).
- Nasr, M.M.; Krumme, M.; Matsuda, Y.; Trout, B.L.; Badman, C.; Mascia, S.; Cooney, C.L.; Jensen, K.D.; Florence, A.; Johnston, C.; et al. Regulatory Perspectives on Continuous Pharmaceutical Manufacturing: Moving From Theory to Practice September 26–27, 2016, International Symposium on the Continuous Manufacturing of Pharmaceuticals. J. Pharm. Sci. 2017, 106, 3199–3206. [Google Scholar] [CrossRef]
- International Council for Harmonisation (ICH). ICH Guideline Q8 (R2) on Pharmaceutical Development; ICH: Geneva, Switzerland, 2009; 24p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-11.pdf (accessed on 2 July 2021).
- ICH. ICH Guideline Q9 on Quality Risk Management; ICH: Geneva, Switzerland, 2015; 2p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-3.pdf (accessed on 2 July 2021).
- ICH. ICH Guideline Q10 on Pharmaceutical Quality System; ICH: Geneva, Switzerland, 2008; 20p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human_en.pdf (accessed on 2 July 2021).
- ICH. ICH Quality IWG: Points to Consider for ICH Q8/Q9/Q10 Guidelines; ICH: Geneva, Switzerland, 2012; 17p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use/q9/q10-guidelines_en.pdf (accessed on 2 July 2021).
- ICH. Final Concept Paper ICH Q13: Continuous Manufacturing of Drug Substances and Drug Products; ICH: Geneva, Switzerland, 2018; 2p, Available online: https://database.ich.org/sites/default/files/Q13_EWG_Concept_Paper.pdf (accessed on 2 July 2021).
- Lee, S.L. FDA’s Continuous Manufacturing Journey. Presented at the Virtual International Symposium on the Continuous Manufacture of Pharmaceuticals (ISCMP). 18 February 2021. Available online: https://www.youtube.com/watch?v=BXULaxXjbUI (accessed on 2 July 2021).
- Matsuda, Y. Global Regulatory Landscape. AAPS PharmSciTech 2018, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- FDA. Innovation and Continuous Improvement in Pharmaceutical Manufacturing: Pharmaceutical CGMPs for the 21st Century; FDA: Silver Spring, MD, USA, 2004; 39p, Available online: https://www.semanticscholar.org/paper/Innovation-and-Continuous-Improvement-in-CGMPs-for/3985a6e1bea806660e2fff7c0e9d7495af08674a (accessed on 2 July 2021).
- FDA. Pharmaceutical cGMPs for the 21st Century—A Risk based Approach: Final Report; FDA: Silver Spring, MD, USA, 2004; 32p. Available online: https://www.fda.gov/media/77391/download (accessed on 2 July 2021).
- O’Connor, T.F.; Yu, L.X.; Lee, S.L. Emerging technology: A key enabler for modernizing pharmaceutical manufacturing and advancing product quality. Int. J. Pharm. 2016, 509, 492–498. [Google Scholar] [CrossRef]
- FDA. Advancement of Emerging Technology Applications for Pharmaceutical Innovation and Modernization: Guidance for Industry; FDA: Silver Spring, MD, USA, 2017; 8p. Available online: https://www.fda.gov/files/drugs/published/Advancement-of-Emerging-Technology-Applications-for-Pharmaceutical-Innovation-and-Modernization-Guidance-for-Industry.pdf (accessed on 2 July 2021).
- FDA. Submission of Proposed Recommendations for Industry on Developing Continuous Manufacturing of Solid Dosage Drug Products in Pharmaceutical Manufacturing; Establishment of a Public Docket; FDA: Silver Spring, MD, USA, 2017; 2p. Available online: https://www.regulations.gov/docket?D=FDA-2017-N-2697 (accessed on 2 July 2021).
- FDA. Guidance for Industry: PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance; FDA: Silver Spring, MD, USA, 2004; 19p. Available online: https://www.fda.gov/media/71012/download (accessed on 2 July 2021).
- FDA. Guidance for Industry: Process Validation: General Principles and Practices; FDA: Silver Spring, MD, USA, 2011; 19p. Available online: https://www.fda.gov/files/drugs/published/Process-Validation--General-Principles-and-Practices.pdf (accessed on 2 July 2021).
- MHRA Innovation Office. Available online: https://www.gov.uk/government/groups/mhra-innovation-office (accessed on 2 July 2021).
- Hernán, D. Continuous manufacturing: Challenges and opportunities. EMA perspective. In Proceedings of the Third FDA/PQRI Conference on Advancing Product Quality, Washington, DC, USA, 22 March 2017; Available online: https://pqri.org/wp-content/uploads/2017/02/3-DHernan.pdf (accessed on 2 July 2021).
- EMA. Mandate for Process Analytical Technology Team; EMA: Amsterdam, The Netherlands, 2006; 2p, Available online: https://www.ema.europa.eu/en/documents/other/mandate-process-analytical-technology-team_en.pdf (accessed on 2 July 2021).
- EMA. Mandate of the EMA Innovation Task Force (ITF); EMA: Amsterdam, The Netherlands, 2014; 3p, Available online: https://www.ema.europa.eu/en/documents/other/mandate-european-medcines-agency-innovation-task-force-itf_en.pdf (accessed on 2 July 2021).
- EMA. Guideline on Process Validation for Finished Products—Information and Data to Be Provided in Regulatory Submissions; EMA: Amsterdam, The Netherlands, 2016; 15p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-process-validation-finished-products-information-data-be-provided-regulatory-submissions_en.pdf (accessed on 2 July 2021).
- EMA. Guideline on Real Time Release Testing (Formerly Guideline on Parametric Release); EMA: Amsterdam, The Netherlands, 2012; 10p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-real-time-release-testing-formerly-guideline-parametric-release-revision-1_en.pdf (accessed on 2 July 2021).
- EMA. Guideline on the Use of near Infrared Spectroscopy by the Pharmaceutical Industry and the Data Requirements for New Submissions and Variations; EMA: Amsterdam, The Netherlands, 2014; 28p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-use-near-infrared-spectroscopy-pharmaceutical-industry-data-requirements-new-submissions_en.pdf (accessed on 2 July 2021).
- Matsuda, Y. PMDA Activities for Implementation of Continuous Manufacturing. In Proceedings of the ISPE Continuous Manufacturing Workshop, Arlington, VA, USA, 6–7 June 2018; Available online: https://www.pmda.go.jp/files/000224561.pdf (accessed on 2 July 2021).
- PMDA. PMDA Views on Applying Continuous Manufacturing to Pharmaceutical Products for Industry; PMDA: Tokyo, Japan, 2018; 10p, Available online: https://www.pmda.go.jp/files/000223712.pdf (accessed on 2 July 2021).
- PMDA. Points to Consider Regarding Continuous Manufacturing; PMDA: Tokyo, Japan, 2017; 22p, Available online: https://www.nihs.go.jp/drug/section3/AMED_CM_PtC.pdf (accessed on 2 July 2021).
- Matsuda, Y.; Aoyama, A.; Aoyama, E.; Ikematsu, Y.; Inoue, K.; Ohta, T.; Kawakita, H.; Koide, T.; Sakamoto, T.; Shimono, R.; et al. Research into Quality Assurance of Pharmaceutical Continuous Manufacturing: State of Control in Continuous Pharmaceutical Manufacturing. 2018. Available online: https://www.nihs.go.jp/drug/section3/AMED_CM_CONTROLST.pdf (accessed on 2 July 2021).
- Allison, G.; Cain, Y.T.; Cooney, C.; Garcia, T.; Bizjak, T.G.; Holte, O.; Jagota, N.; Komas, B.; Korakianiti, E.; Kourti, D.; et al. Regulatory and Quality Considerations for Continuous Manufacturing. May 20–21, 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Amidon, G.; Khan, M.A.; Hoag, S.W.; Polli, J.; Raju, G.K.; Woodcock, J. Understanding pharmaceutical quality by design. AAPS J. 2014, 16, 771–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, H.; Kano, M.; Sugiyama, H.; Takeuchi, H.; Terada, K.; Aoyama, A.; Shoda, T.; Demizu, Y.; Shimamura, J.; Yokoyama, R.; et al. Approach to Establishment of Control Strategy for Oral Solid Dosage Forms Using Continuous Manufacturing. Chem. Pharm. Bull. 2021, 69, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Pauli, V.; Elbaz, F.; Kleinebudde, P.; Krumme, M. Methodology for a Variable Rate Control Strategy Development in Continuous Manufacturing Applied to Twin-screw Wet-Granulation and Continuous Fluid-bed Drying. J. Pharm. Innov. 2018, 13, 247–260. [Google Scholar] [CrossRef]
- ICH. ICH Q7: Note for Guidance on Good Manufacturing Practice for Active Pharmaceutical Ingredients; ICH: Geneva, Switzerland, 2000; 48p, Available online: https://www.ema.europa.eu/en/ich-q7-good-manufacturing-practice-active-pharmaceutical-ingredients (accessed on 2 July 2021).
- Billups, M.; Singh, R. Systematic Framework for Implementation of Material Traceability into Continuous Pharmaceutical Tablet Manufacturing Process. J. Pharm. Innov. 2020, 15, 51–65. [Google Scholar] [CrossRef]
- Gao, Y.; Muzzio, F.J.; Ierapetritou, M.G. A review of the Residence Time Distribution (RTD) applications in solid unit operations. Powder Technol. 2012, 228, 416–423. [Google Scholar] [CrossRef]
- Suzuki, Y.; Sugiyama, H.; Kano, M.; Shimono, R.; Shimada, G.; Furukawa, R.; Mano, E.; Motoyama, K.; Koide, T.; Matsui, Y.; et al. Control strategy and methods for continuous direct compression processes. Asian J. Pharm. Sci. 2021, 16, 253–262. [Google Scholar] [CrossRef]
- Engisch, W.; Muzzio, F. Using Residence Time Distributions (RTDs) to Address the Traceability of Raw Materials in Continuous Pharmaceutical Manufacturing. J. Pharm. Innov. 2016, 11, 64–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escotet-Espinoza, M.S.; Moghtadernejad, S.; Oka, S.; Wang, Y.; Roman-Ospino, A.; Schäfer, E.; Cappuyns, P.; Assche, I.V.; Futran, M.; Ierapetritou, M.; et al. Effect of tracer material properties on the residence time distribution (RTD) of continuous powder blending operations. Part I of II: Experimental evaluation. Powder Technol. 2019, 342, 744–763. [Google Scholar] [CrossRef]
- Escotet-Espinoza, M.S.; Moghtadernejad, S.; Oka, S.; Wang, Z.; Wang, Y.; Roman-Ospino, A.; Schäfer, E.; Cappuyns, P.; Assche, I.V.; Futran, M.; et al. Effect of material properties on the residence time distribution (RTD) characterization of powder blending unit operations. Part II of II: Application of models. Powder Technol. 2019, 344, 525–544. [Google Scholar] [CrossRef]
- Almaya, A.; de Belder, L.; Meyer, R.; Nagapudi, K.; Lin, H.H.; Leavesley, I.; Jayanth, J.; Bajwa, G.; DiNunzio, J.; Tantuccio, A.; et al. Control Strategies for Drug Product Continuous Direct Compression-State of Control, Product Collection Strategies, and Startup/Shutdown Operations for the Production of Clinical Trial Materials and Commercial Products. J. Pharm. Sci. 2017, 106, 930–943. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, A.; Singh, R. Residence Time Distribution (RTD)-Based Control System for Continuous Pharmaceutical Manufacturing Process. J. Pharm. Innov. 2019, 14, 316–331. [Google Scholar] [CrossRef]
- Pauli, V.; Kleinebudde, P.; Krumme, M. Predictive Model-Based Process Start-Up in Pharmaceutical Continuous Granulation and Drying. Pharmaceutics 2020, 12, 67. [Google Scholar] [CrossRef] [Green Version]
- Markl, D.; Warman, M.; Dumarey, M.; Bergman, E.L.; Folestad, S.; Shi, Z.; Manley, L.F.; Goodwin, D.J.; Zeitler, J.A. Review of real-time release testing of pharmaceutical tablets: State-of-the art, challenges and future perspective. Int. J. Pharm. 2020, 582, 119353. [Google Scholar] [CrossRef]
- Pawar, P.; Wang, Y.; Keyvan, G.; Callegari, G.; Cuitino, A.; Muzzio, F. Enabling real time release testing by NIR prediction of dissolution of tablets made by continuous direct compression (CDC). Int. J. Pharm. 2016, 512, 96–107. [Google Scholar] [CrossRef]
- Galata, D.L.; Farkas, A.; Könyves, Z.; Mészáros, L.A.; Szabó, E.; Csontos, I.; Pálos, A.; Marosi, G.; Nagy, Z.K.; Nagy, B. Fast, Spectroscopy-Based Prediction of In Vitro Dissolution Profile of Extended Release Tablets Using Artificial Neural Networks. Pharmaceutics 2019, 11, 400. [Google Scholar] [CrossRef] [Green Version]
- Bawuah, P.; Markl, D.; Turner, A.; Evans, M.; Portieri, A.; Farrell, D.; Lucas, R.; Anderson, A.; Goodwin, D.J.; Zeitler, J.A. A Fast and Non-destructive Terahertz Dissolution Assay for Immediate Release Tablets. J. Pharm. Sci. 2020, 111, 2083–2092. [Google Scholar]
- EMA. Orkambi Assessment Report; EMA: Amsterdam, The Netherlands, 2015; 104p, Available online: https://www.ema.europa.eu/en/documents/assessment-report/orkambi-epar-public-assessment-report_en.pdf (accessed on 2 July 2021).
- Pharmamanufacturing.com. Janssen’s Historic FDA Approval. 2016. Available online: https://www.pharmamanufacturing.com/articles/2016/janssens-historic-fda-approval/ (accessed on 2 July 2021).
- Gordon, L. Real-Time Release Testing for Dissolution Has Arrived. 2019. Available online: https://ipsdb.com/happenings/insights/real-time-release-testing-for-dissolution-has-arrived (accessed on 2 July 2021).
- Kourti, T.; Lepore, J.; Liesum, L.; Nasr, M.; Chatterjee, S.; Moore, C.; Korakianiti, E. Scientific and Regulatory Considerations for Implementing Mathematical Models in Quality by Design (QbD) Framework. Pharm. Eng. 2014, 34, 1–21. [Google Scholar]
- ICH. ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management; ICH: Geneva, Switzerland, 2020; 31p, Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-q12-technical-regulatory-considerations-pharmaceutical-product-lifecycle-management_en.pdf (accessed on 2 July 2021).
- Kim, E.J.; Kim, J.H.; Kim, M.S.; Jeong, S.H.; Choi, D.H. Process Analytical Technology Tools for Monitoring Pharmaceutical Unit Operations: A Control Strategy for Continuous Process Verification. Pharmaceutics 2021, 13, 919. [Google Scholar] [CrossRef] [PubMed]
- Teżyk, M.; Milanowski, B.; Ernst, A.; Lulek, J. Recent progress in continuous and semi-continuous processing of solid oral dosage forms: A review. Drug Dev. Ind. Pharm. 2016, 42, 1195–1214. [Google Scholar] [CrossRef]
- Khinast, J. Continuous Manufacturing—Critical Steps and Possible Solutions: Batch Definition, Control Strategy and New PAT. In Proceedings of the 8th Global DDF Summit, Berlin, Germany, 27–29 March 2017; Available online: http://fplreflib.findlay.co.uk/images/pdf/Johannes-Khinast.pdf (accessed on 2 July 2021).
- Van Snick, B.; Kumar, A.; Verstraeten, M.; Pandelaere, K.; Dhondt, J.; Di Pretoro, G.; De Beer, T.; Vervaet, C.; Vanhoorne, V. Impact of material properties and process variables on the residence time distribution in twin screw feeding equipment. Int. J. Pharm. 2019, 556, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.; Harting, J.; Happel, J.; Kleinebudde, P. Implementation of microwave sensors in continuous powder feeding—A novel tool to bridge refill phases. Pharm. Ind. 2017, 79, 576–582. [Google Scholar]
- Pernenkil, L. Continuous Blending of Dry Pharmaceutical Powders. Ph.D. Thesis, Massachusetts Institute of Technology: MIT, Cambridge, MA, USA, 2008. Available online: https://www.researchgate.net/publication/38003180_Continuous_blending_of_dry_pharmaceutical_powders (accessed on 2 July 2021).
- Oka, S.; Sahay, A.; Meng, W.; Muzzio, F. Diminished segregation in continuous powder mixing. Powder Technol. 2017, 309, 79–88. [Google Scholar] [CrossRef]
- Ervasti, T.; Niinikoski, H.; Mäki-Lohiluoma, E.; Leppinen, H.; Ketolainen, J.; Korhonen, O.; Lakio, S. The Comparison of Two Challenging Low Dose APIs in a Continuous Direct Compression Process. Pharmaceutics 2020, 12, 279. [Google Scholar] [CrossRef] [Green Version]
- Kittikunakorn, N.; DiNunzio, J.; Martin, C.; Zhang, F. Processes, Challenges, and the Future of Twin-Screw Granulation for Manufacturing Oral Tablets and Capsules. AAPS News Magazine, March 2018; pp. 12–18. [Google Scholar]
- Nandi, U.; Trivedi, V.; Ross, S.A.; Douroumis, D. Advances in Twin-Screw Granulation Processing. Pharmaceutics 2021, 13, 624. [Google Scholar] [CrossRef]
- Portier, C.; Vervaet, C.; Vanhoorne, V. Continuous Twin Screw Granulation: A Review of Recent Progress and Opportunities in Formulation and Equipment Design. Pharmaceutics 2021, 13, 668. [Google Scholar] [CrossRef]
- Pauli, V.; Roggo, Y.; Kleinebudde, P.; Krumme, M. Real-time monitoring of particle size distribution in a continuous granulation and drying process by near infrared spectroscopy. Eur. J. Pharm. Biopharm. 2019, 141, 90–99. [Google Scholar] [CrossRef]
- Meier, R. Continuous Wet Granulation and Fluid-bed Drying—An Experimental Investigation of a New Revolutionary System. The Medicine Maker. 4 June 2020, pp. 1–10. Available online: https://themedicinemaker.com/white-papers/continuous-wet-granulation-and-fluid-bed-drying-an-experimental-investigation-of-a-new-revolutionary-system (accessed on 2 July 2021).
- Shah, P.; Boesen, A.; Smith, T.J.; Batra, A.; Zhang, Q.; Bi, Y.; Dürig, T. Using Continuous Granulation to Make Robust and High Drug Load APAP Granules. In Proceedings of the AAPS Conference, Washington DC, USA, 2–8 November 2018; M1130-06-048. Available online: https://www.freund-vector.com/technical_papers/using-continuous-granulation-to-make-robust-and-high-drug-load-apap-granules/ (accessed on 2 July 2021).
- Leane, M.; Pitt, K.; Reynolds, G.K.; Dawson, N.; Ziegler, I.; Szepes, A.; Crean, A.M.; Agnol, R.D. Manufacturing classification system in the real world: Factors influencing manufacturing process choices for filed commercial oral solid dosage formulations, case studies from industry and considerations for continuous processing. Pharm. Dev. Technol. 2018, 23, 964–977. [Google Scholar] [CrossRef]
- Siew, A. Exploring the Potential of Continuous Coating. Pharm. Tech. 2015, 39, 44–46. [Google Scholar]
- Gandolfi, N. Proxima® and Croma®, Integrated technologies for Continuous Manufacturing. In Proceedings of the Third International Symposium on the Continuous Manufacture of Pharmaceuticals (ISCMP), London, UK, 3–4 October 2018; Available online: https://issuu.com/luxevents2/docs/nicola_gandolfi_of_ima_s.p.a__proxi (accessed on 2 July 2021).
- Teckoe, J.; Cunningham, C. All Aboard for Continuous Coating. The Medicine Maker. 21 September 2016, pp. 1–4. Available online: https://themedicinemaker.com/business-regulation/all-aboard-for-continuous-coating (accessed on 2 July 2021).
- Alper, J. Continuous Manufacturing for the Modernization of Pharmaceutical Production: Proceedings of a Workshop; National Academies Press: Washington, DC, USA, 2019; ISBN 978-0-309-48781-8. [Google Scholar] [CrossRef]
- Metta, N.; Ghijs, M.; Schäfer, E.; Kumar, A.; Cappuyns, P.; Assche, I.V.; Singh, R.; Ramachandran, R.; Beer, T.D.; Ierapetritou, M.; et al. Dynamic Flowsheet Model Development and Sensitivity Analysis of a Continuous Pharmaceutical Tablet Manufacturing Process Using the Wet Granulation Route. Processes 2019, 7, 234. [Google Scholar] [CrossRef] [Green Version]
- Robinson, K. Continuous manufacturing: The facts and the future. Manufacturing Chemist. 21 January 2019, pp. 1–5. Available online: https://www.manufacturingchemist.com/news/article_page/Continuous_manufacturing_the_facts_and_the_future/150919 (accessed on 2 July 2021).
- RCPE. RCPE Wins Major Order from Bosch Packaging Technology. 7 March 2016. Available online: http://www.rcpe.at/wp-content/uploads/2018/09/Pressinformation-RCPE_BOSCH.pdf (accessed on 2 July 2021).
- Schmidt, A.; de Waard, H.; Moll, K.P.; Kleinebudde, P.; Krumme, M. Simplified end-to-end continuous manufacturing by feeding API suspensions in twin-screw wet granulation. Eur. J. Pharm. Biopharm. 2018, 133, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.A.; Osorio, J.G.; Brancazio, D.; Hammersmith, G.; Klee, D.M.; Rapp, K.; Myerson, A. A compact, portable, re-configurable, and automated system for on-demand pharmaceutical tablet manufacturing. Int. J. Pharm. 2018, 539, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Purdue. DARPA Selects Continuity Pharma, Funds Continuous Manufacturing Technology. Purdue News. 6 October 2020. Available online: https://www.purdue.edu/newsroom/releases/2020/Q4/darpa-selects-continuity-pharma,-funds-continuous-manufacturing-technology.html (accessed on 2 July 2021).
- Mascia, S.; Heider, P.L.; Zhang, H.; Lakerveld, R.; Benyahia, B.; Barton, P.I.; Braatz, R.D.; Cooney, C.L.; Evans, J.M.; Jamison, T.F.; et al. End-to-end continuous manufacturing of pharmaceuticals: Integrated synthesis, purification, and final dosage formation. Angew. Chem. Int. Ed. Engl. 2013, 52, 12359–12363. [Google Scholar] [CrossRef] [PubMed]
- Continuus. Continuus Pharmaceuticals Secures $69.3 Million Government Contract to Manufacture Critical Medicines in the U.S. 21 January 2021. Available online: https://www.prnewswire.com/news-releases/continuus-pharmaceuticals-secures-69-3-million-government-contract-to-manufacture-critical-medicines-in-the-us-301212044.html?tc=eml_cleartime (accessed on 2 July 2021).
- Abaci, A.; Gedeon, C.; Kuna, A.; Guvendiren, M. Additive Manufacturing of Oral Tablets: Technologies, Materials and Printed Tablets. Pharmaceutics 2021, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- GSK. Liquid Dispensing Technology (LDT). 2009. Available online: https://www.gsk.com/media/2758/liquid-dispensing-technology-leaflet.pdf (accessed on 2 July 2021).
- Myerson, A.S.; Krumme, M.; Nasr, M.; Thomas, H.; Braatz, R.D. Control systems engineering in continuous pharmaceutical manufacturing. May 20–21, 2014 Continuous Manufacturing Symposium. J. Pharm. Sci. 2015, 104, 832–839. [Google Scholar] [CrossRef]
- ASTM. Standard Guide for Verification of Process Analytical Technology (PAT) Enabled Control Systems; ASTM: West Conshohocken, PA, USA, 2020; p. E2629-20. [Google Scholar]
- Matos, N.; Ali, S.; Almaya, A.; Biba, E.; Carlin, B.; Cruz, C.; Hausner, D.; Jayjock, E.; Jensen, K.; Khinast, J.; et al. USP Pharmacopeial Perspective for Pharmaceutical Continuous Manufacturing: Stimuli to the Revision Process. Pharmacop. Forum 2018, 44, 1–30. [Google Scholar]
- FDA. FDA Development and Submission of Near Infrared Analytical Procedures; FDA: Silver Spring, MD, USA, 2015; 24p. Available online: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm440247.pdf (accessed on 2 July 2021).
- Wesholowski, J.; Berghaus, A.; Thommes, M. Inline Determination of Residence Time Distribution in Hot-Melt-Extrusion. Pharmaceutics 2018, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Schlindwein, W.; Bezerra, M.; Almeida, J.; Berghaus, A.; Owen, M.; Muirhead, G. In-Line UV-Vis Spectroscopy as a Fast-Working Process Analytical Technology (PAT) during Early Phase Product Development Using Hot Melt Extrusion (HME). Pharmaceutics 2018, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCPE. RCPE Awarded Two FDA Contracts for Next-Generation Pharma Technology. 30 October 2020. Available online: https://www.prnewswire.com/news-releases/rcpe-awarded-two-fda-contracts-for-next-generation-pharma-technology-301163714.html (accessed on 2 July 2021).
- Garcia-Munoz, S. Two novel methods to analyze the combined effect of multiple raw-materials and processing conditions on the product’s final attributes: JRPLS and TPLS. Chemometr. Intell. Lab. Syst. 2014, 133, 49–62. [Google Scholar] [CrossRef]
- Wang, Y.; O’Connor, T.; Li, T.; Ashraf, M.; Cruz, C.N. Development and applications of a material library for pharmaceutical continuous manufacturing of solid dosage forms. Int. J. Pharm. 2019, 569, 118551. [Google Scholar] [CrossRef] [PubMed]
- Leane, M.; Pitt, K.; Reynolds, G. A proposal for a drug product Manufacturing Classification System (MCS) for oral solid dosage forms. Pharm. Dev. Technol. 2015, 20, 12–21. [Google Scholar] [CrossRef]
- Allenspach, C.; Timmins, P.; Lumay, G.; Holman, J.; Minko, T. Loss-in-weight feeding, powder flow and electrostatic evaluation for direct compression hydroxypropyl methylcellulose (HPMC) to support continuous manufacturing. Int. J. Pharm. 2021, 596, 120259. [Google Scholar] [CrossRef] [PubMed]
- Schenck, L.; Erdemir, D.; Gorka, L.S.; Merritt, J.M.; Marziano, I.; Ho, R.; Lee, M.; Bullard, J.; Boukerche, M.; Ferguson, S.; et al. Recent Advances in Co-processed APIs and Proposals for Enabling Commercialization of These Transformative Technologies. Mol. Pharm. 2020, 17, 2232–2244. [Google Scholar] [CrossRef]
- ISCMP 2014. Available online: https://iscmp.mit.edu/2014 (accessed on 2 July 2021).
- ISCMP 2016. Available online: https://iscmp.mit.edu/2016 (accessed on 2 July 2021).
- ISCMP 2018. Available online: https://iscmp.mit.edu/2018 (accessed on 2 July 2021).
- Dell ‘Orco, P.; Tix, M. The State of Continuous Processing. In Proceedings of the Third International Symposium on the Continuous Manufacturing of Pharmaceuticals (ISCMP), London, UK, 3–4 October 2018; Available online: https://issuu.com/luxevents2/docs/the_state_of_continuous_processing (accessed on 2 July 2021).
- Novartis-MIT Center for Continuous Manufacturing. Available online: https://novartis-mit.mit.edu/ (accessed on 2 July 2021).
- Continuus Pharmaceuticals. Available online: https://www.continuuspharma.com/ (accessed on 2 July 2021).
- CMAC. Future Manufacturing Research Hub. Available online: https://www.cmac.ac.uk/index.php (accessed on 2 July 2021).
- International Institute for Advanced Pharmaceutical Manufacturing—Center for Structured Organic Particulate System. I2APM-C-SOPS. Available online: https://www.i2apm.org/CSOPS.htm (accessed on 2 July 2021).
- Research Center Pharmaceutical Engineering (RCPE). Available online: https://www.rcpe.at/en/en_home/ (accessed on 2 July 2021).
- International Institute for Advanced Pharmaceutical Manufacturing. I2APM. Available online: https://www.i2apm.org/index.php (accessed on 2 July 2021).
- Medicines Manufacturing Innovation Centre (MMIC). Available online: https://www.uk-cpi.com/about/national-centres/medicines-manufacturing-innovation-centre (accessed on 2 July 2021).
- Tudor, D.; Accelerating Advanced Technology Translation. Presented at Virtual International Symposium on the Continuous Manufacture of Pharmaceuticals (ISCMP), 18 February 2021. Available online: https://www.youtube.com/watch?v=bekSNzLhZfQ (accessed on 2 July 2021).
- Science Foundation Ireland Pharmaceutical Research Centre (SSPC). Available online: https://sspc.ie/about-us/ (accessed on 2 July 2021).
- Ardill, L. SSPC to build €1.9m Pharma Manufacturing Facility at UL. Available online: https://www.siliconrepublic.com/innovation/sspc-sfi-infrastructure-award (accessed on 2 July 2021).
- National Institute for Innovation in Manufacturing Biopharmaceuticals (NIIMBL). Available online: https://niimbl.force.com/s/ (accessed on 2 July 2021).
- NIIMBL. NIIMBL and FDA Sign Agreement to Support Innovation in Biopharma Manufacturing. Available online: https://niimbl.force.com/s/news/a0a3u000004PnxiAAC/niimbl-and-fda-sign-agreement-to-support-innovation-in-biopharma-manufacturing (accessed on 2 July 2021).
- CPI. National Centres. Available online: https://www.uk-cpi.com/about/national-centres (accessed on 2 July 2021).
- Japan Agency for Medical Research and Development (AMED). Available online: https://www.amed.go.jp/en/index.html (accessed on 2 July 2021).
- United States Pharmacopeia (USP). Available online: https://www.usp.org/ (accessed on 2 July 2021).
- Phlow Corp. and USP Announce Strategic Alliance Focused on Pharmaceutical Continuous Manufacturing to Increase Supply of Essential Medicines for U.S. Patients, Phlow Corp, USP. 2021. Available online: https://www.businesswire.com/news/home/20210222005551/en/Phlow-Corp.-and-USP-Announce-Strategic-Alliance-Focused-on-Pharmaceutical-Continuous-Manufacturing-to-Increase-Supply-of-Essential-Medicines-for-U.S.-Patients (accessed on 28 July 2021).
- ASTM International. Available online: https://www.astm.org/ (accessed on 2 July 2021).
- ASTM. Standard Guide for Application of Continuous Processing in the Pharmaceutical Industry; ASTM: West Conshohocken, PA, USA, 2014; p. E2968-14. [Google Scholar]
- ASTM. Standard Guide for Application of Continuous Process Verification to Pharmaceutical and Biopharmaceutical Manufacturing; ASTM: West Conshohocken, PA, USA, 2016; p. E2537-16. [Google Scholar]
- International Society for Pharmaceutical Engineering (ISPE). Available online: https://ispe.org/ (accessed on 2 July 2021).
- International Society for Pharmaceutical Engineering. ISPE 2016 Continuous Manufacturing Conference Highlights. Pharm. Eng. 2016, 37, 35–42. [Google Scholar]
- Santos, P.D.L.; Hofer, J.; Jayjock, E.; Kramer, T.; Lief, K.; Otava, M. Sampling Considerations in Continuous Manufacturing. Pharm. Eng. 2019, 1–17. Available online: https://ispe.org/pharmaceutical-engineering/may-june-2019 (accessed on 2 July 2021).
- Wade, J.B.; Ievers, R.T. Process Validation—Small-Molecule Drug Substance & Drug Product Continuous Manufacturing Processes. Pharm. Eng. 2019. Available online: https://ispe.org/pharmaceutical-engineering/may-june-2019/process-validation-small-molecule-drug-substance-drug-product-continuous-manufacturing-processes (accessed on 2 July 2021).
- National Institute for Pharmaceutical Technology & Education (NIPTE). Available online: https://nipte.org/ (accessed on 2 July 2021).



{kind=link}
{kind=link}
| Drug Product | Indication | Company | Year of First Approval | Regulatory Body |
|---|---|---|---|---|
| Orkambi® | Cystic fibrosis | Vertex | 2015 | EMA, FDA |
| Prezista® * | HIV | Janssen (J&J) | 2016 | EMA, FDA |
| Verzenio® | Breast cancer | Eli Lilly | 2017 | EMA, FDA, PDMA |
| Lorbrena® | Lung cancer | Pfizer | 2018 | ** |
| Daurismo® | Myeloid leukaemia | Pfizer | 2018 | FDA |
| Symkevi®/Symdeko® | Cystic fibrosis | Vertex | 2018 | EMA/FDA |
| Tramacet® | Pain | J&J | ** | PMDA |
| Faster and leaner transition from development to commercial scale | Same equipment used during development, clinical supply manufacture and commercial production with batch sizes accommodated by changing run times Shorter cycle times More rapid development |
| Shorter supply chains | All operations conducted on one piece of equipment in one location without hold-ups and inter-site transfers between manufacturing stages Improved stability as no intermediate holding periods |
| Supply chain security | Enhanced domestic manufacture with reliance on high technology rather than low-cost labour Shorter manufacturing times meaning longer product shelf lives |
| Improved product quality | Domestic manufacture In-process monitoring, feedback and feed-forward controls. Maintenance of a state-of-control Reduced dependence on end-product testing Decreased regulatory oversight—frees resources for other higher-risk areas Fully aligned with principles of Quality by Design (QbD) |
| Cost benefits (after initial investment) | Lower production costs Improved utilisation of equipment Lower personnel requirements Smaller footprint |
| Supply chain responsiveness | Batch sizes tailored to requirements by adjusting run times Ability to respond rapidly to demands |
| Patient benefits | More suited to niche/personalised products Alternative manufacturing technologies (e.g., active pharmaceutical ingredient (API) printed or sprayed onto a dosage form) Lower risk of stock-outs |
| Societal benefits | Less environmental impact (less use of solvents, lower energy costs) Less waste/improved yields. Lowered risk that a whole batch will need to be rejected if it fails end-product testing Higher process intensification (less use of space, energy and raw materials) Improved safety (reduced handling and exposure to materials, easier cleaning) Source of high-tech jobs |
| Existing equipment and facilities are geared to batch processes | Initial investment cost to implement CM (albeit with lower subsequent costs) Existing equipment likely depreciated so may be less of an issue |
| Facilities not located to achieve end-to-end processing | API and drug product facilities may be in different countries (perhaps driven by tax benefits) or locations within a country |
| Different expertise requirements | CM requires experts in statistics, process control, modelling, QbD processes, PAT (Process Analytical Technology), etc. Requirement to better understand material attributes |
| CM process | Limited possibilities for re-work (but also less likely a need to do so) Limited opportunities to halt the process part way through |
| Maintenance | Control algorithms and models require adjustment to accommodate changes in raw materials. May have regulatory implications Sophisticated PAT equipment |
| Submission requirements | New submissions required to switch batch to CM No globally recognised harmonised CM approval process Concern that regulators will baulk at CM processes Limited expertise in non-FDA, EMA, MHRA, PMDA regulated countries Longer approval times for global registration |
| Equipment | Lack of appropriate bench and pilot-scale equipment Not all unit operations can currently be included in a CM process Handling of dry solids and solid laden fluids can be difficult |
| Experience | Lack of examples of end-to-end processes Limited company experience of CM submissions Limited experience in regulators (in particular EMA, MHRA and PMDA) |
| Number | Date | Title | Contents of Relevance to CM |
|---|---|---|---|
| ICH Q8(R2) | 2009 | Pharmaceutical Development | Control strategy, continuous process verification |
| ICH Q9 | 2015 | Quality Risk Management | Risk assessment and control |
| ICH Q10 | 2008 | Pharmaceutical Quality System | Continual improvement of process performance and product quality |
| Quality-IWG | 2012 | Points to consider for ICH Q8/Q9/Q10 guidelines | Models in QbD, continuous process verification |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahlich, J. Review: Continuous Manufacturing of Small Molecule Solid Oral Dosage Forms. Pharmaceutics 2021, 13, 1311. https://doi.org/10.3390/pharmaceutics13081311
Wahlich J. Review: Continuous Manufacturing of Small Molecule Solid Oral Dosage Forms. Pharmaceutics. 2021; 13(8):1311. https://doi.org/10.3390/pharmaceutics13081311
Chicago/Turabian StyleWahlich, John. 2021. "Review: Continuous Manufacturing of Small Molecule Solid Oral Dosage Forms" Pharmaceutics 13, no. 8: 1311. https://doi.org/10.3390/pharmaceutics13081311
APA StyleWahlich, J. (2021). Review: Continuous Manufacturing of Small Molecule Solid Oral Dosage Forms. Pharmaceutics, 13(8), 1311. https://doi.org/10.3390/pharmaceutics13081311