
The Physicochemical, Biopharmaceutical, and In Vitro Efficacy Properties of Freeze-Dried Dexamethasone-Loaded Lipomers

 ,
,  , and
, and 
Abstract
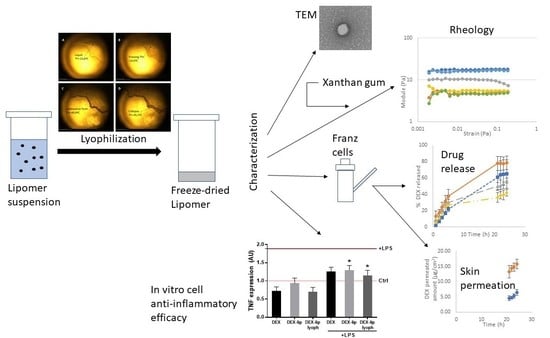
:
1. Introduction
2. Materials and Methods
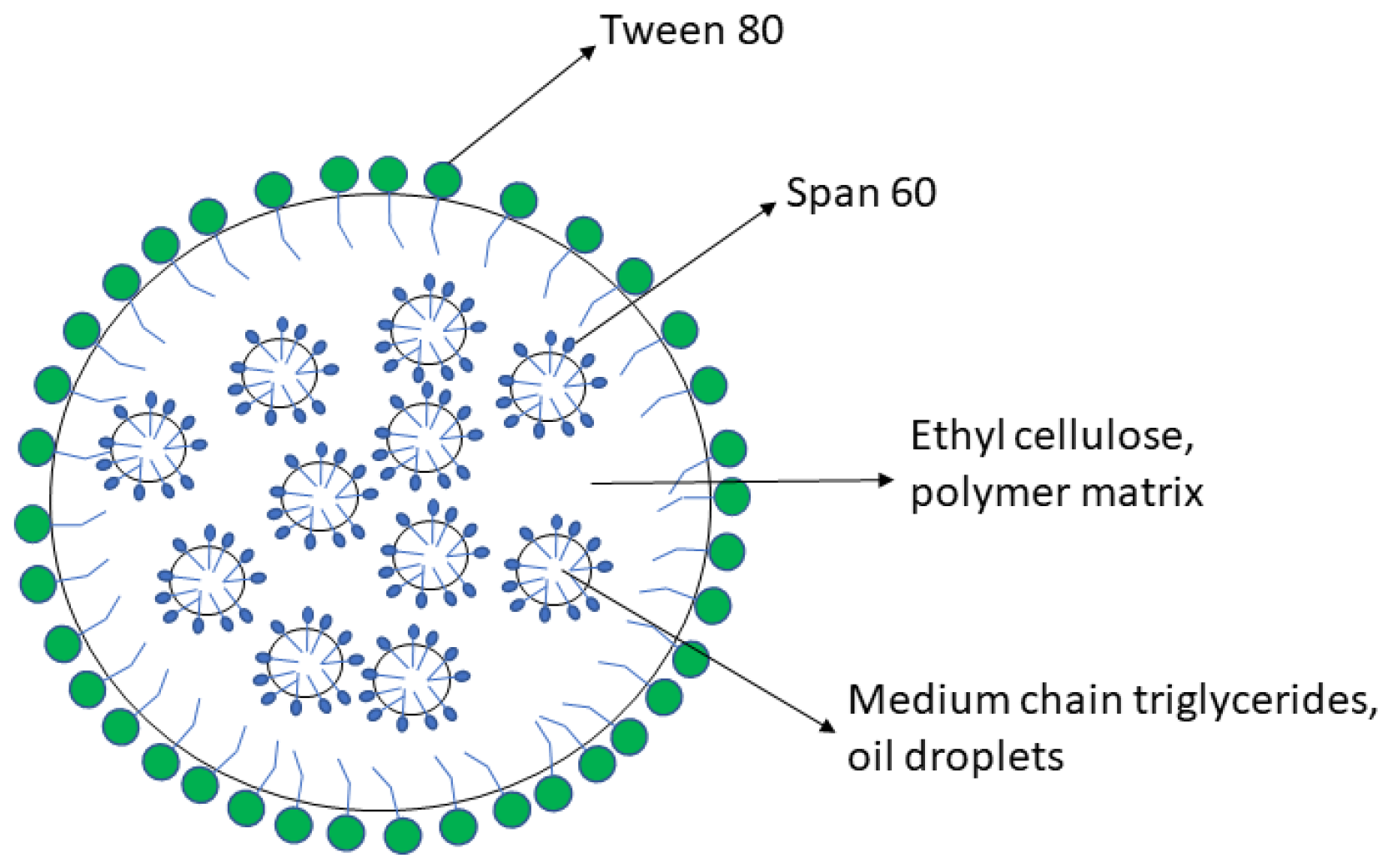
2.1. Materials and Lipomer Formulation
2.2. Freeze Drying
2.3. Physicochemical Characterization
2.3.1. Z-Average, PdI, and Z-Potential before and after Freeze Drying
2.3.2. Transmission Electron Microscopy before and after Freeze Drying
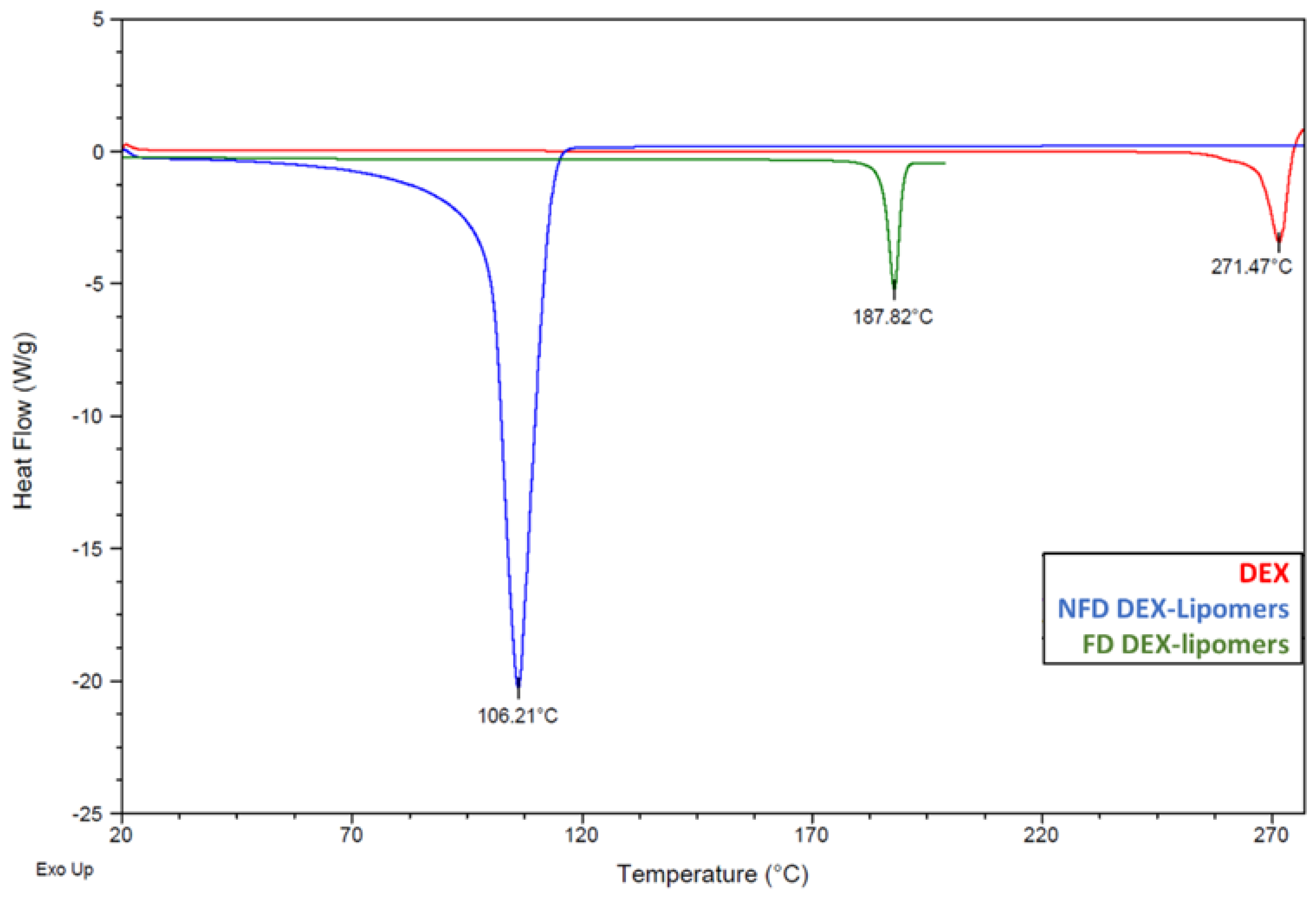
2.3.3. Differential Scanning Calorimetry before and after Freeze Drying
2.4. Gel Formulations
2.5. High-Performance Liquid Chromatography (HPLC)
2.6. In Vitro Release Tests
2.7. Pig Skin In Vitro Permeation Tests
2.8. In Vitro Cytotoxicity/Anti-TNFα Efficacy
2.9. Statistical Analysis
3. Results and Discussion
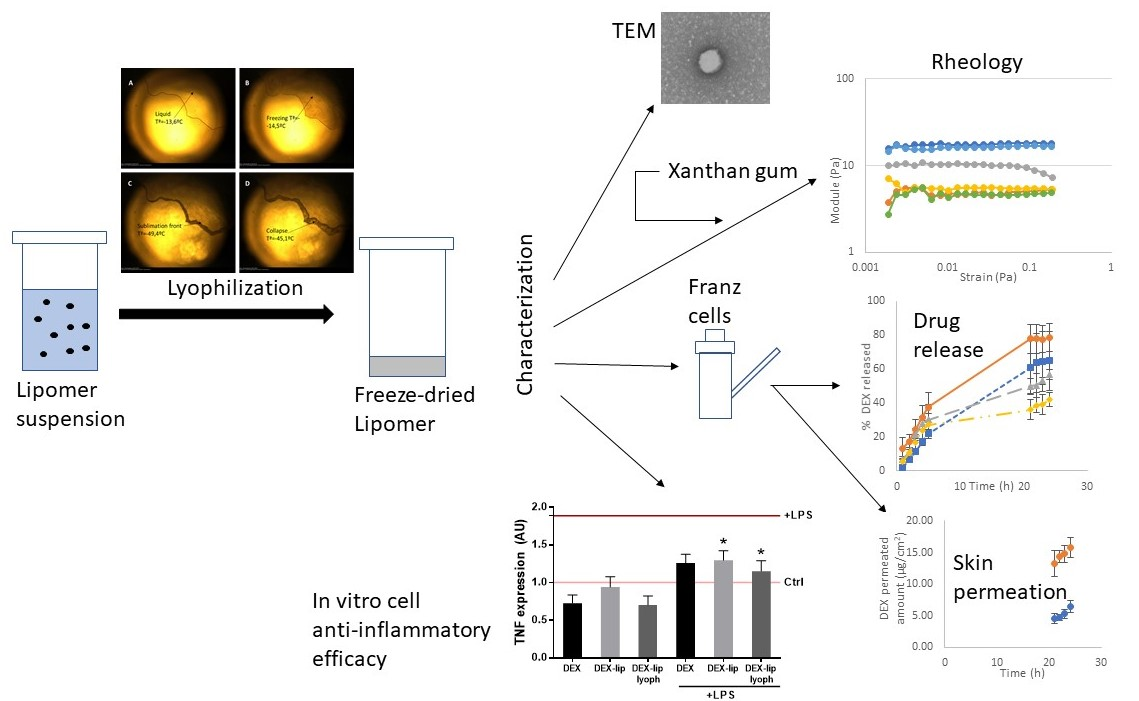
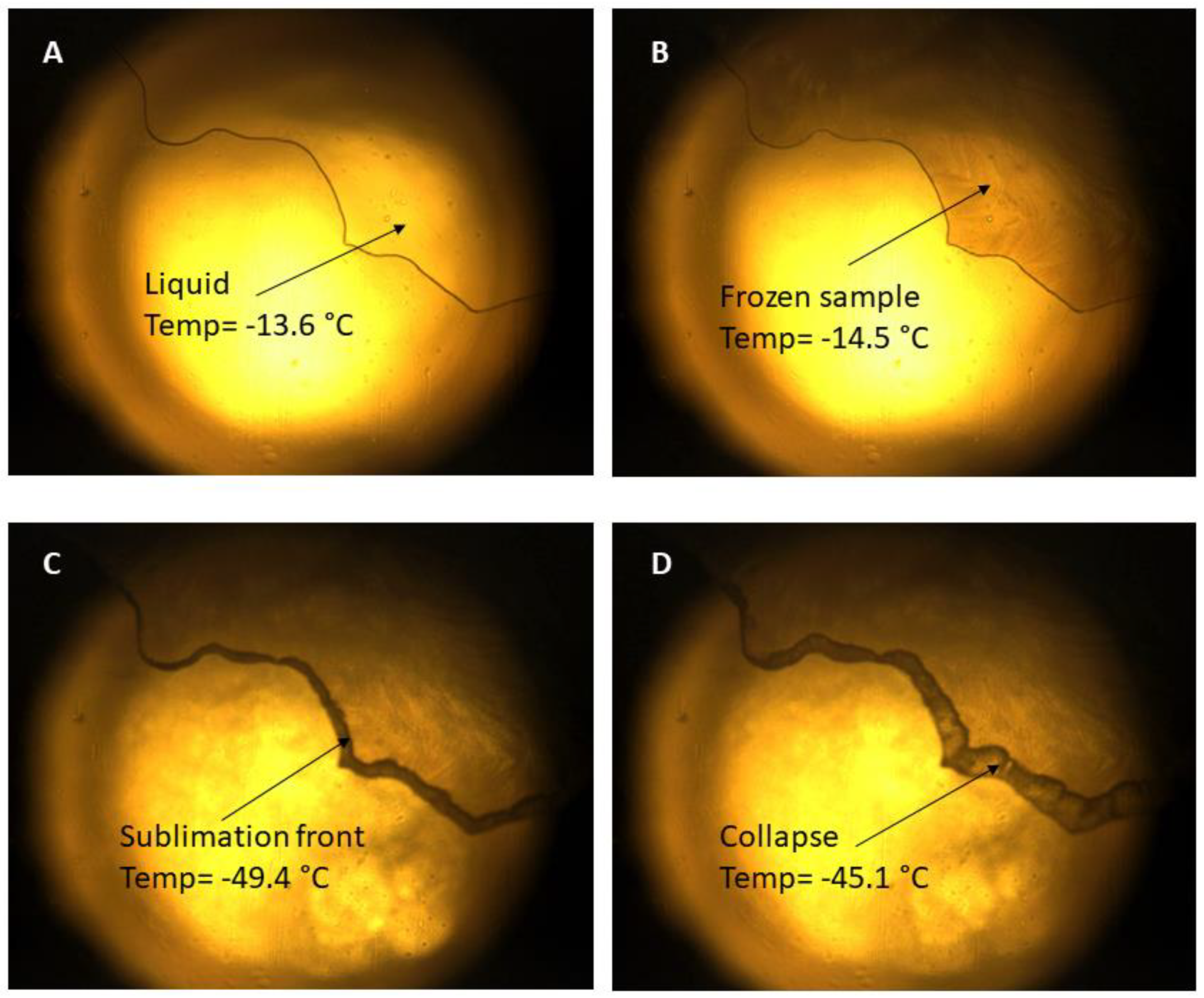
3.1. Freeze Drying
3.2. Physicochemical Characterization before and after Freeze Drying
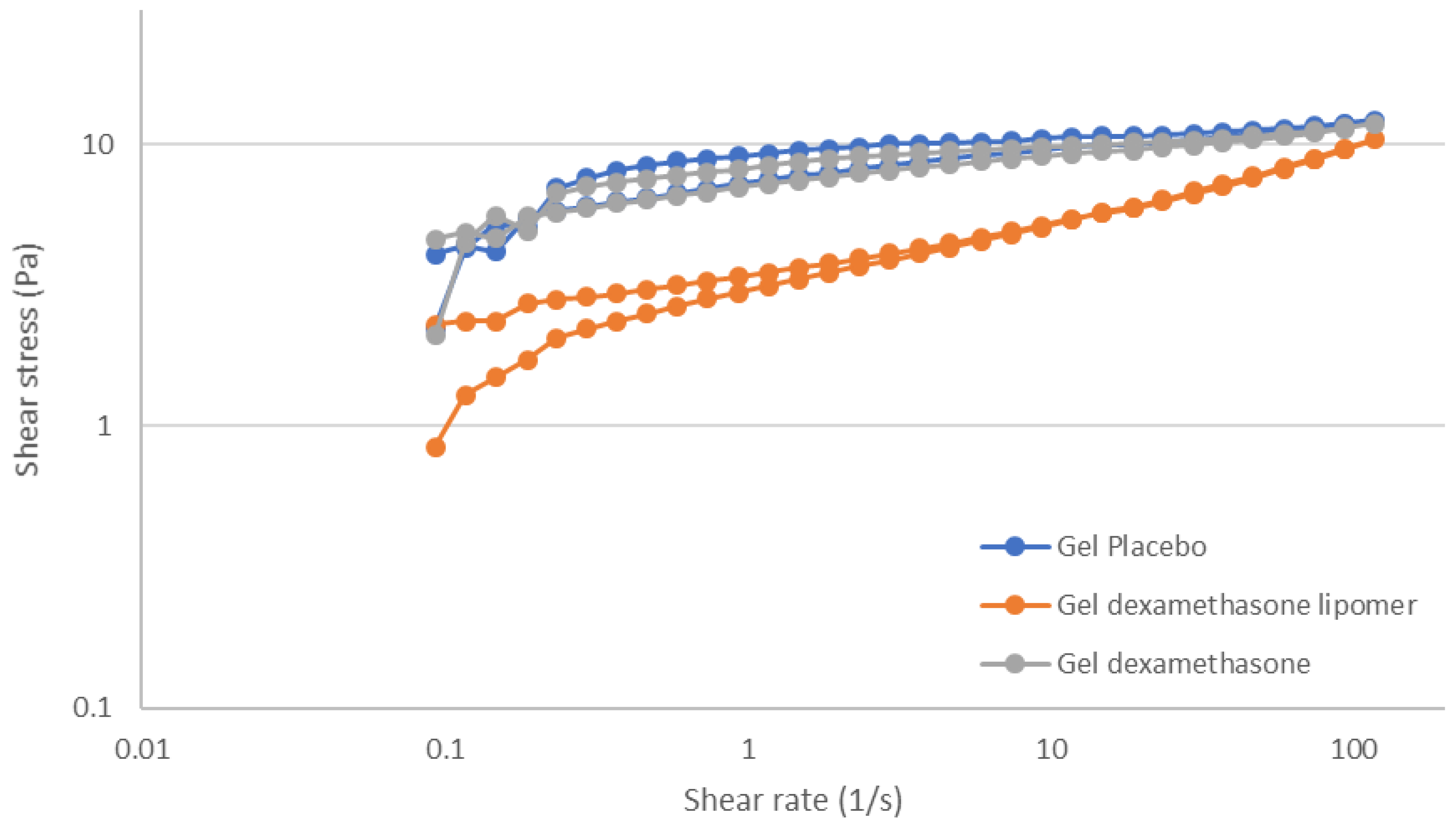
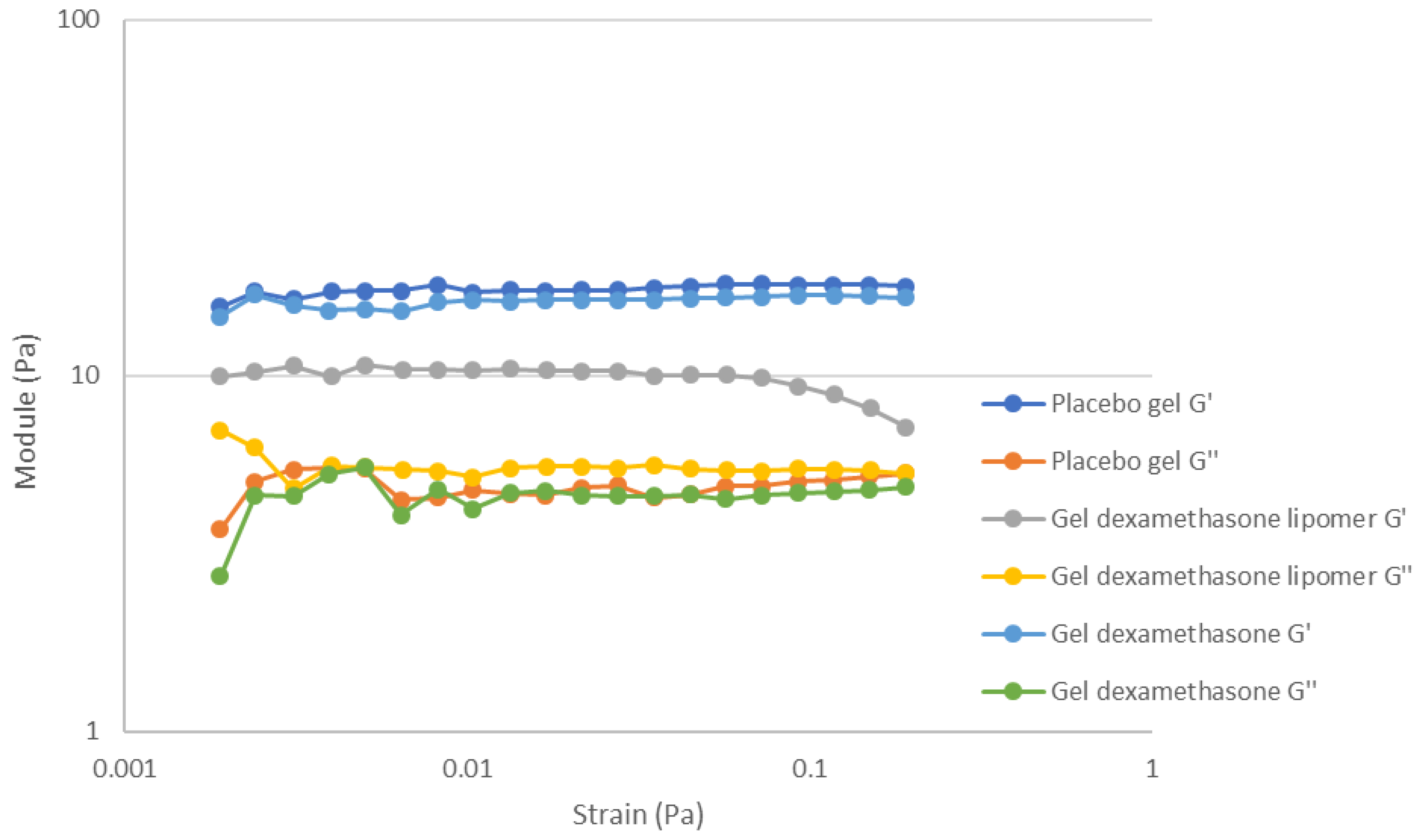
3.3. Gel Formulations Rheology Studies
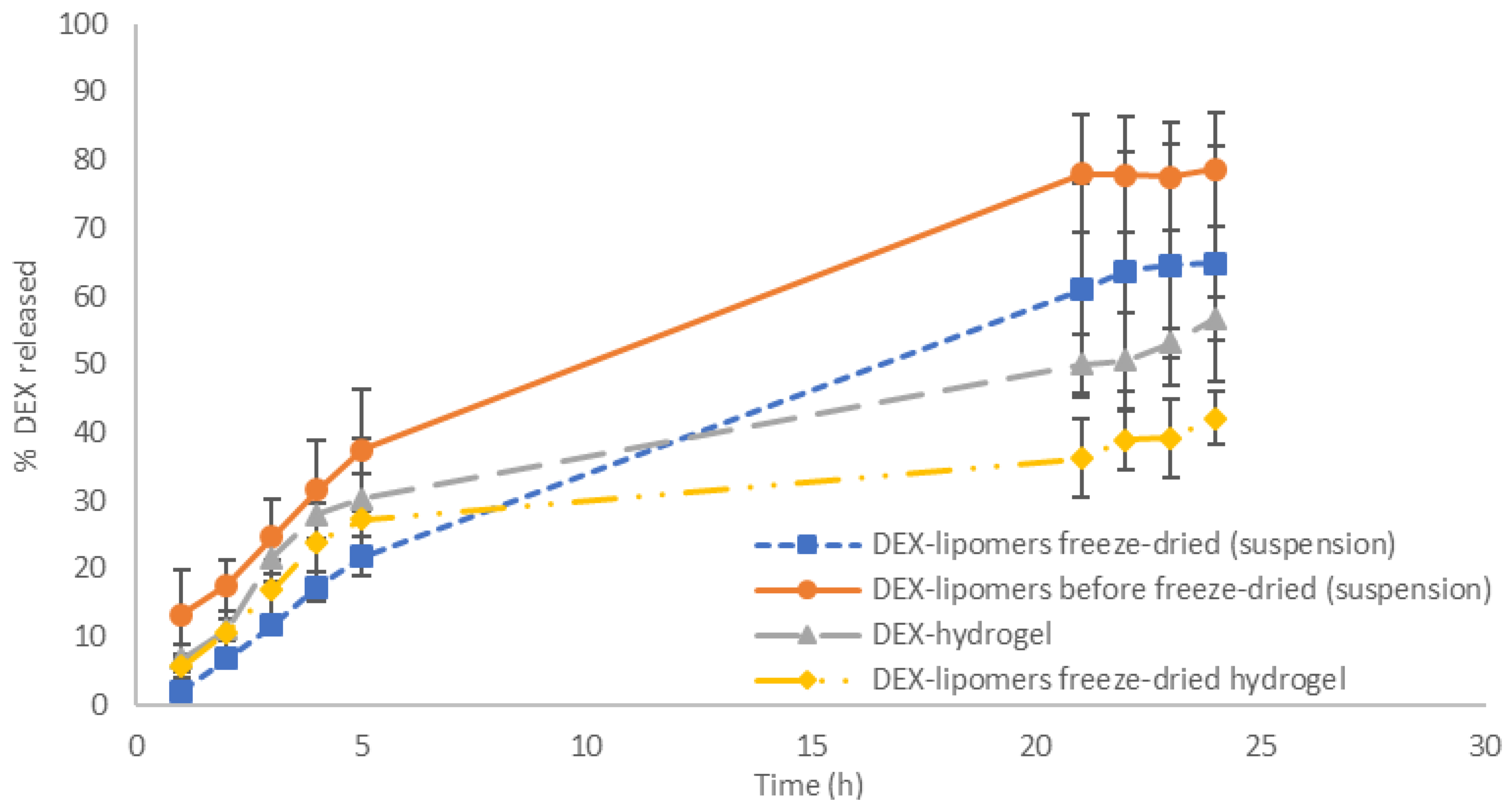
3.4. In Vitro Release Tests
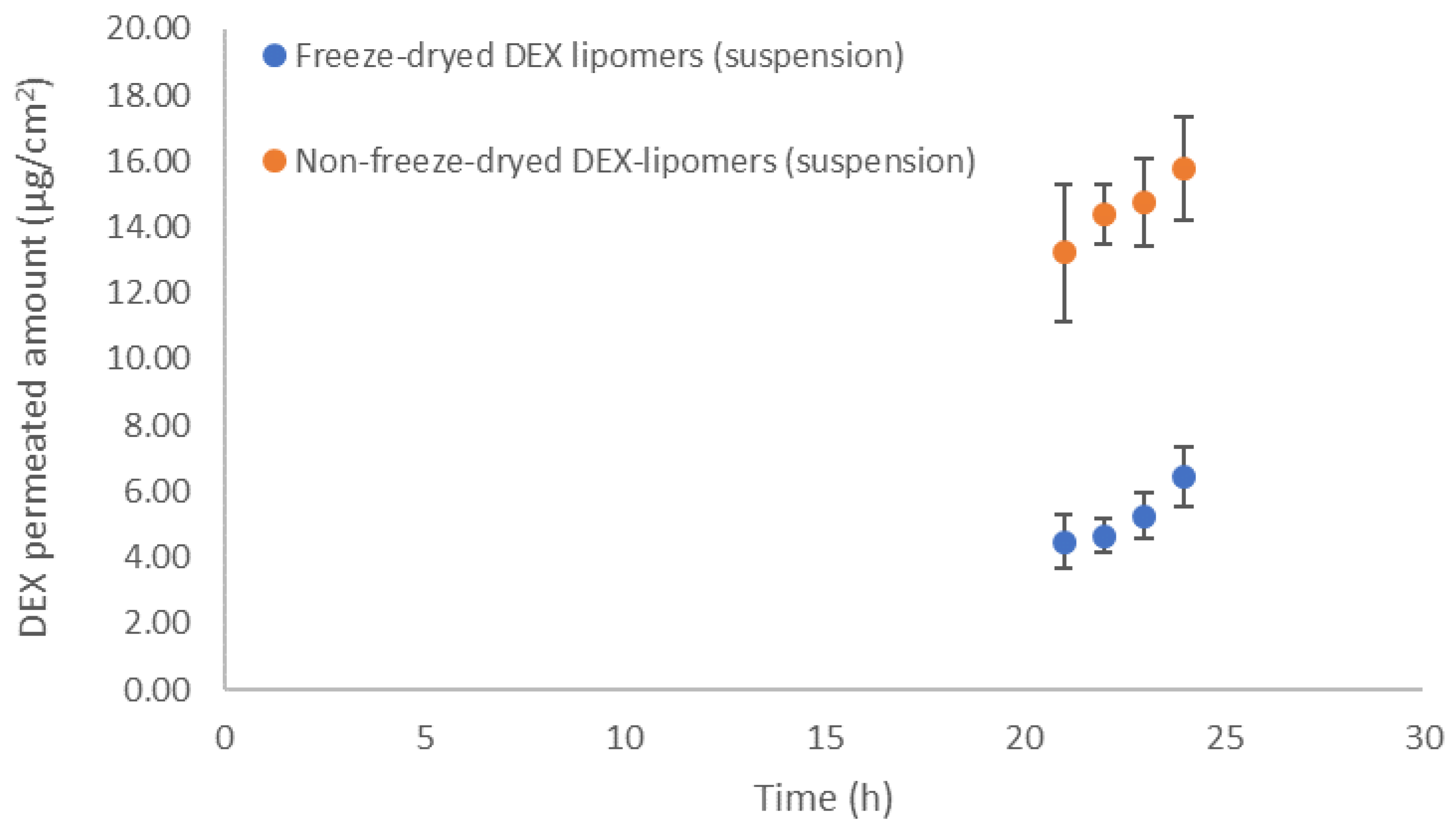
3.5. In Vitro Permeation Tests
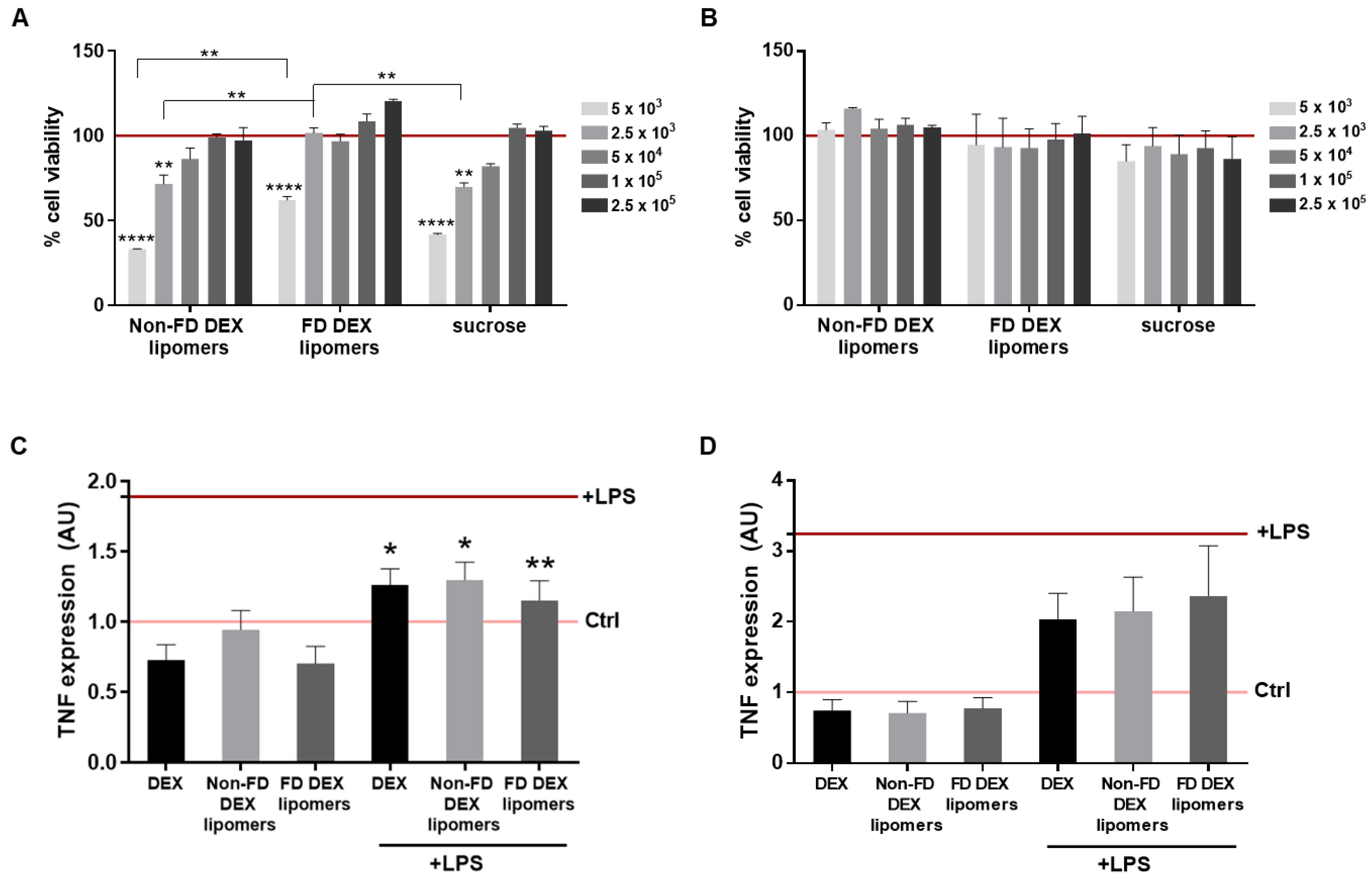
3.6. Cytotoxicity and Anti-TNFα Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaul, S.; Gulati, N.; Verma, D.; Mukherjee, S.; Nagaich, U. Role of Nanotechnology in Cosmeceuticals: A Review of Recent Advances. J. Pharm. 2018, 2018, 420204. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.-L.; Aljuffali, I.A.; Li, Y.-C.; Fang, J.-Y. Delivery and targeting of nanoparticles into hair follicles. Ther. Deliv. 2014, 5, 991–1006. [Google Scholar] [CrossRef]
- Wosicka, H.; Cal, K. Targeting to the hair follicles: Current status and potential. J. Dermatol. Sci. 2010, 57, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Juárez-Rendón, K.J.; Rivera Sánchez, G.; Reyes-López, M.Á.; García-Ortiz, J.E.; Bocanegra-García, V.; Guardiola-Avila, I.; Altamirano-García, M.L. Alopecia areata. Actualidad y perspectivas. Arch. Argent. Pediatr. 2017, 115, e404–e411. [Google Scholar] [PubMed]
- Hordinsky, M.K. Overview of Alopecia Areata. J. Investig. Dermatol. Symp. Proc. 2013, 16, S13–S15. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, W.-S. Management of alopecia areata: Updates and algorithmic approach. J. Dermatol. 2017, 44, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Pena-Rodríguez, E.; Lajarin-Reinares, M.; Mata-Ventosa, A.; Pérez-Torras, S.; Fernández-Campos, F. Dexamethasone-Loaded Lipomers: Development, Characterization, and Skin Biodistribution Studies. Pharmaceutics 2021, 13, 533. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-drying of nanoparticles: Formulation, process and storage considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef] [PubMed]
- Mohammady, M.; Mohammadi, Y.; Yousefi, G. Freeze-Drying of Pharmaceutical and Nutraceutical Nanoparticles: The Effects of Formulation and Technique Parameters on Nanoparticles Characteristics. J. Pharm. Sci. 2020, 109, 3235–3247. [Google Scholar] [CrossRef]
- Holzer, M.; Vogel, V.; Mäntele, W.; Schwartz, D.; Haase, W.; Langer, K. Physico-chemical characterisation of PLGA nanoparticles after freeze-drying and storage. Eur. J. Pharm. Biopharm. 2009, 72, 428–437. [Google Scholar] [CrossRef]
- Fonte, P.; Soares, S.; Sousa, F.; Costa, A.M.M.M.; Seabra, V.; Reis, S.; Sarmento, B. Stability Study Perspective of the Effect of Freeze-Drying Using Cryoprotectants on the Structure of Insulin Loaded into PLGA Nanoparticles. Biomacromolecules 2014, 15, 3753–3765. [Google Scholar] [CrossRef] [PubMed]
- Srihaphon, K.; Lamlertthon, S.; Pitaksuteepong, T. Influence of stabilizers and cryoprotectants on the characteristics of freeze-dried plga nanoparticles containing morus alba stem extract. Songklanakarin J. Sci. Technol. 2021, 43, 72–79. [Google Scholar]
- Gajra, B.; Dalwadi, C.; Patel, R. Formulation and optimization of itraconazole polymeric lipid hybrid nanoparticles (Lipomer) using box behnken design. DARU J. Pharm. Sci. 2015, 23, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, G.; Sathesh Kumar, S. Lyophilization and stability study of noscapine-loaded polycaprolactone nanoparticles. Drug Invent. Today 2019, 12, 2516–2521. [Google Scholar]
- Choi, M.J.; Briancon, S.; Andrieu, J.; Min, S.G.; Fessi, H. Effect of Freeze-Drying Process Conditions on the Stability of Nanoparticles. Dry. Technol. 2004, 22, 335–346. [Google Scholar] [CrossRef]
- Pan-In, P.; Banlunara, W.; Chaichanawongsaroj, N.; Wanichwecharungruang, S. Ethyl cellulose nanoparticles: Clarithomycin encapsulation and eradication of H. pylori. Carbohydr. Polym. 2014, 109, 22–27. [Google Scholar] [CrossRef]
- Mirabedini, S.; Dutil, I.; Farnood, R. Preparation and characterization of ethyl cellulose-based core–shell microcapsules containing plant oils. Colloids Surf. A Physicochem. Eng. Asp. 2012, 394, 74–84. [Google Scholar] [CrossRef]
- Abd-Elrahman, M.I.; Ahmed, S.M. Thermal Degradation Kinetics and Geometrical Stability of D-Sucrose. Int. J. Polym. Mater. 2009, 58, 322–335. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An Add-In Program for Modeling and Comparison of Drug Dissolution Profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Starciuc, T.; Malfait, B.; Danede, F.; Paccou, L.; Guinet, Y.; Correia, N.T.; Hedoux, A. Trehalose or Sucrose: Which of the Two Should be Used for Stabilizing Proteins in the Solid State? A Dilemma Investigated by In Situ Micro-Raman and Dielectric Relaxation Spectroscopies During and After Freeze-Drying. J. Pharm. Sci. 2020, 109, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.N.; Mallik, S.; Sarkar, K. Role of freeze-drying in the presence of mannitol on the echogenicity of echogenic liposomes. J. Acoust. Soc. Am. 2017, 142, 3670–3676. [Google Scholar] [CrossRef]
- Almalik, A.; Alradwan, I.; Kalam, M.A.; Alshamsan, A. Effect of cryoprotection on particle size stability and preservation of chitosan nanoparticles with and without hyaluronate or alginate coating. Saudi Pharm. J. 2017, 25, 861–867. [Google Scholar] [CrossRef]
- Bonaccorso, A.; Musumeci, T.; Carbone, C.; Vicari, L.; Lauro, M.R.; Puglisi, G. Revisiting the role of sucrose in PLGA-PEG nanocarrier for potential intranasal delivery. Pharm. Dev. Technol. 2017, 23, 265–274. [Google Scholar] [CrossRef]
- Kannan, V.; Balabathula, P.; Thoma, L.A.; Wood, G.C. Effect of sucrose as a lyoprotectant on the integrity of paclitaxel-loaded liposomes during lyophilization. J. Liposome Res. 2014, 25, 270–278. [Google Scholar] [CrossRef]
- Nail, S.L.; Jiang, S.; Chongprasert, S.; Knopp, S.A. Fundamentals of Freeze-Drying. Pharm. Biotechnol. 2002, 14, 281–360. [Google Scholar] [CrossRef]
- Meister, E.; Sasić, S.; Gieseler, H. Freeze-Dry Microscopy: Impact of Nucleation Temperature and Excipient Concentration on Collapse Temperature Data. AAPS PharmSciTech 2009, 10, 582–588. [Google Scholar] [CrossRef] [Green Version]
- Ward, K.R.; Matejtschuk, P. The use of microscopy, thermal analysis, and impedance measurements to establish critical formulation parameters for freeze-drying cycle development. In Freeze-Drying/Lyophilization of Pharmaceutical and Biological Products, 3rd ed.; CRC Press: London, UK, 2016; pp. 112–135. [Google Scholar] [CrossRef]
- Meister, E.; Gieseler, H. Freeze-Dry Microscopy of Protein/Sugar Mixtures: Drying Behavior, Interpretation of Collapse Temperatures and a Comparison to Corresponding Glass Transition Data. J. Pharm. Sci. 2009, 98, 3072–3087. [Google Scholar] [CrossRef] [PubMed]
- Pikal, M.J.; Shah, S. The collapse temperature in freeze drying: Dependence on measurement methodology and rate of water removal from the glassy phase. Int. J. Pharm. 1990, 62, 165–186. [Google Scholar] [CrossRef]
- Pauline, M.D. Chapter 11—Unit Operations, Bioprocess Engineering Principles, 2nd ed.; Academic Press: Amsterdam, The Netherlands, 2013; pp. 445–595. ISBN 9780122208515. [Google Scholar] [CrossRef]
- Talik, P.; Piwowarczyk, J.; Muszyńska, B.; Hubicka, U. DSC study of hydration and water-holding behaviour of cultured in vitro mycelium and naturally grown fruiting bodies of freeze-dried Boletus badius, Agaricus bisporus and Cantharellus cibarius. J. Therm. Anal. Calorim. 2021, 143, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Osswald, T.; Rudolph, N. Generalized Newtonian Fluid (GNF) Models. In Polymer Rheology: Fundamentals and Applications; Strohm, C., Ed.; Hanser Publications: München, Germany, 2014; pp. 59–99. [Google Scholar]
- Fernández-Campos, F.; Obach, M.; Moreno, M.; García, A.; González, J. Pharmaceutical development of a generic corticoid semisolid formulation. J. Drug Deliv. Sci. Technol. 2017, 42, 227–236. [Google Scholar] [CrossRef]
- Tauber, M.; Buche, S.; Reygagne, P.; Berthelot, J.-M.; Aubin, F.; Ghislain, P.-D.; Cohen, J.-D.; Coquerelle, P.; Goujon, E.; Jullien, D.; et al. Alopecia areata occurring during anti-TNF therapy: A national multicenter prospective study. J. Am. Acad. Dermatol. 2014, 70, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Pratt, C.H.; King, L.E.; Messenger, A.G.; Christiano, A.M.; Sundberg, J.P. Alopecia areata. Nat. Rev. Dis. Primers 2017, 3, 17011. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rheological Model | Equation | |
|---|---|---|
| Newton | (1) | |
| Bingham | (2) | |
| Ostwald–de Waele | (3) | |
| Herschel–Bulkley | (4) | |
| Casson | (5) | |
| Cross | (6) |
| Kinetic Model | Equation | |
|---|---|---|
| First Order | (9) | |
| Higuchi | (10) | |
| Korsmeyer–Peppas | (11) | |
| Weibull | (12) |
| FD Cycle | Temperature | Time |
|---|---|---|
| Soak | 10 °C | 1 h |
| Freezing | −55 °C | 4 h |
| Primary Drying | −30 °C | 72 h |
| Secondary Drying ramp | −30 °C to 30 °C | 4 h |
| Secondary Drying | 30 °C | 4 h |
| Formulation | Before Freeze Drying | After Freeze Drying | ||||||
|---|---|---|---|---|---|---|---|---|
| Hydrodynamic Diameter (nm) | PDI | Z-Pot (mV) | EE (%) | Hydrodynamic Diameter (nm) | PDI | Z-Pot (mV) | EE (%) | |
| DEX-lipomers (no cryo) | 185.23 ± 5.24 | 0.360 ± 0.019 | 39.0 ± 0.1 | 98.60 ± 0.01 | 1850.00 ± 188.75 | 0.313 ± 0.051 | 35.9 ± 2.0 | 98.94 ± 0.01 |
| DEX-lipomers (trehalose 6%) | 186.87 ± 2.68 | 0.361 ± 0.015 | 36.3 ± 0.4 | 446.70 ± 3.21 | 0.355 ± 0.013 | 34.3 ± 0.5 | 98.97 ± 0.01 | |
| DEX-lipomers (sucrose 6%) | 185.67 ± 4.92 | 0.349 ± 0.016 | 37.3 ± 0.5 | 374.33 ± 7.60 | 0.229 ± 0.011 | 34.7 ± 0.4 | 98.87 ± 0.01 | |
| DEX-lipomers (mannitol 6%) | 183.97 ± 1.27 | 0.334 ± 0.008 | 37.4 ± 0.4 | 749.53 ± 26.49 | 0.435 ± 0.013 | 34.9 ± 1.7 | 94.63 ± 6.22 | |
| Rheological Model | Hydrogel Placebo (Cost) | FD-DEX-Lipomers Hydrogel (Cost) | DEX Hydrogel (Cost) |
|---|---|---|---|
| Newton | 904.563 | 137.743 | 768.095 |
| Bingham | 59.985 | 19.360 | 44.060 |
| Ostwald–de Waele | 23.138 | 0.665 | 13.814 |
| Herschel–Bulkley | 2.721 | 0.656 | 3.372 |
| Casson | 4.952 | 0.791 | 4.883 |
| Cross | 2.080 | 0.142 | 2.529 |
| Herschel–Bulkley Equation Parameter | Hydrogel Placebo | DEX-Lipomers Hydrogel | DEX Hydrogel |
|---|---|---|---|
| (Pa) | 11.312 ± 1.990 | −0.120 ± 0.250 * | 11.283 ± 0.189 |
| K | −2.040 ± 0.957 | 2.971 ± 0.523 * | −2.935 ± 0.196 |
| n | −0.611 ± 0.076 * | 0.259 ± 0.019 * | −0.411 ± 0.025 * |
| Parameter | Formulation | Mean ± SD |
|---|---|---|
| G′ | Placebo gel | 17.48 ± 0.44 Pa |
| Gel DEX-lipomer | 9.97 ± 0.35 Pa (*) | |
| Gel DEX | 16.19 ± 0.13 Pa (*) | |
| G″ | Placebo gel | 4.88 ± 0.09 Pa |
| Gel DEX-lipomer | 5.55 ± 0.24 Pa (*) | |
| Gel DEX | 4.60 ± 0.03 Pa | |
| G* | Placebo gel | 18.16 ± 0.43 Pa |
| Gel DEX-lipomer | 11.35 ± 0.42 Pa (*) | |
| Gel DEX | 16.84 ± 0.12 Pa | |
| tan δ | Placebo gel | 15.6 ± 0.64° |
| Gel DEX-lipomer | 29.4 ± 0.09° (*) | |
| Gel DEX | 15.8 ± 0.25° |
| Formulation | Model | AIC | Parameters | Value |
|---|---|---|---|---|
| Freeze-dried DEX-lipomers | First order | 28.59 | k (h−1) | 0.046 |
| Higuchi | 57.87 | kH (%h−1/2) | 12.579 | |
| Korsmeyer–Peppas | 35.48 | n | 0.800 | |
| kKP (%h−n) | 5.261 | |||
| Weibull | 28.57 | td (h) | 22.75 | |
| β | 1.008 | |||
| Freeze-dried DEX-lipomers hydrogel | First order | 44.988 | k (h−1) | 0.155 |
| Higuchi | 46.539 | kH (%h−1/2) | 44.196 | |
| Korsmeyer–Peppas | 49.847 | n | 0.383 | |
| kKP (%h−n) | 10.960 | |||
| Weibull | 48.870 | td (h) | 11.960 | |
| β | 0.601 | |||
| DEX hydrogel | First order | 48.877 | k (h−1) | 0.133 |
| Higuchi | 44.710 | kH (%h−1/2) | 11.277 | |
| Korsmeyer–Peppas | 52.096 | n | 0.590 | |
| kKP (%h−n) | 9.060 | |||
| Weibull | 48.484 | td (h) | 10.965 | |
| β | 0.700 |
| Parameter | Non-Freeze-Drying DEX-Lipomers (Mean ± SD) | Freeze-Drying DEX-Lipomers (Mean ± SD) |
|---|---|---|
| Jsup (µg/h·cm2) | 0.4759 ± 0.1123 | 0.3789 ± 0.2093 |
| Kp (cm/h) | 7.9316 × 10−5 ± 1.900 × 10−5 | 6.3148 × 10−5 ± 5.156 × 10−5 |
| P2 (1/h) | 0.0385 ± 0.0011 | 0.0117 ± 0.0039 (*) |
| P1 (cm) | 0.0020 ± 0.0006 | 0.0073 ± 0.0067 |
| tlag (h) | 4.213 ± 0.064 | 11.978 ± 4.776 (*) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pena-Rodríguez, E.; Mata-Ventosa, A.; Garcia-Vega, L.; Pérez-Torras, S.; Fernández-Campos, F. The Physicochemical, Biopharmaceutical, and In Vitro Efficacy Properties of Freeze-Dried Dexamethasone-Loaded Lipomers. Pharmaceutics 2021, 13, 1322. https://doi.org/10.3390/pharmaceutics13081322
Pena-Rodríguez E, Mata-Ventosa A, Garcia-Vega L, Pérez-Torras S, Fernández-Campos F. The Physicochemical, Biopharmaceutical, and In Vitro Efficacy Properties of Freeze-Dried Dexamethasone-Loaded Lipomers. Pharmaceutics. 2021; 13(8):1322. https://doi.org/10.3390/pharmaceutics13081322
Chicago/Turabian StylePena-Rodríguez, Eloy, Aida Mata-Ventosa, Laura Garcia-Vega, Sandra Pérez-Torras, and Francisco Fernández-Campos. 2021. "The Physicochemical, Biopharmaceutical, and In Vitro Efficacy Properties of Freeze-Dried Dexamethasone-Loaded Lipomers" Pharmaceutics 13, no. 8: 1322. https://doi.org/10.3390/pharmaceutics13081322
APA StylePena-Rodríguez, E., Mata-Ventosa, A., Garcia-Vega, L., Pérez-Torras, S., & Fernández-Campos, F. (2021). The Physicochemical, Biopharmaceutical, and In Vitro Efficacy Properties of Freeze-Dried Dexamethasone-Loaded Lipomers. Pharmaceutics, 13(8), 1322. https://doi.org/10.3390/pharmaceutics13081322