
Optimisation of a Microfluidic Method for the Delivery of a Small Peptide

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
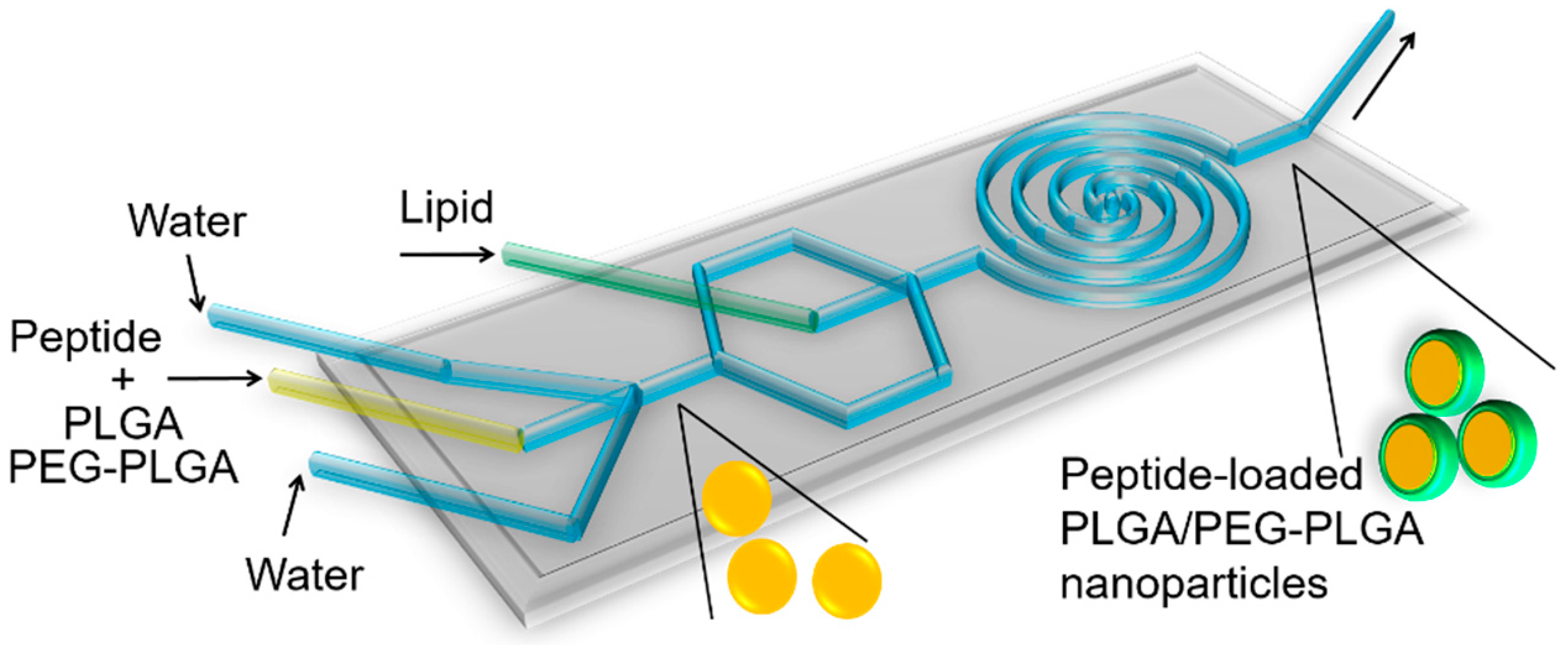
2.2. Preparation of Peptide-Loaded Nanoparticles
2.3. Particle Size, Polydispersity Index (PDI) and Zeta Potential
2.4. Incorporation Efficiency
2.5. In Vitro Release Profile
2.6. Measurement of Peptide by Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
2.7. Morphology
2.8. Pharmacokinetics Study and Biodistribution
2.9. Data Analysis
3. Results and Discussion
3.1. Optimisation of Particle Size
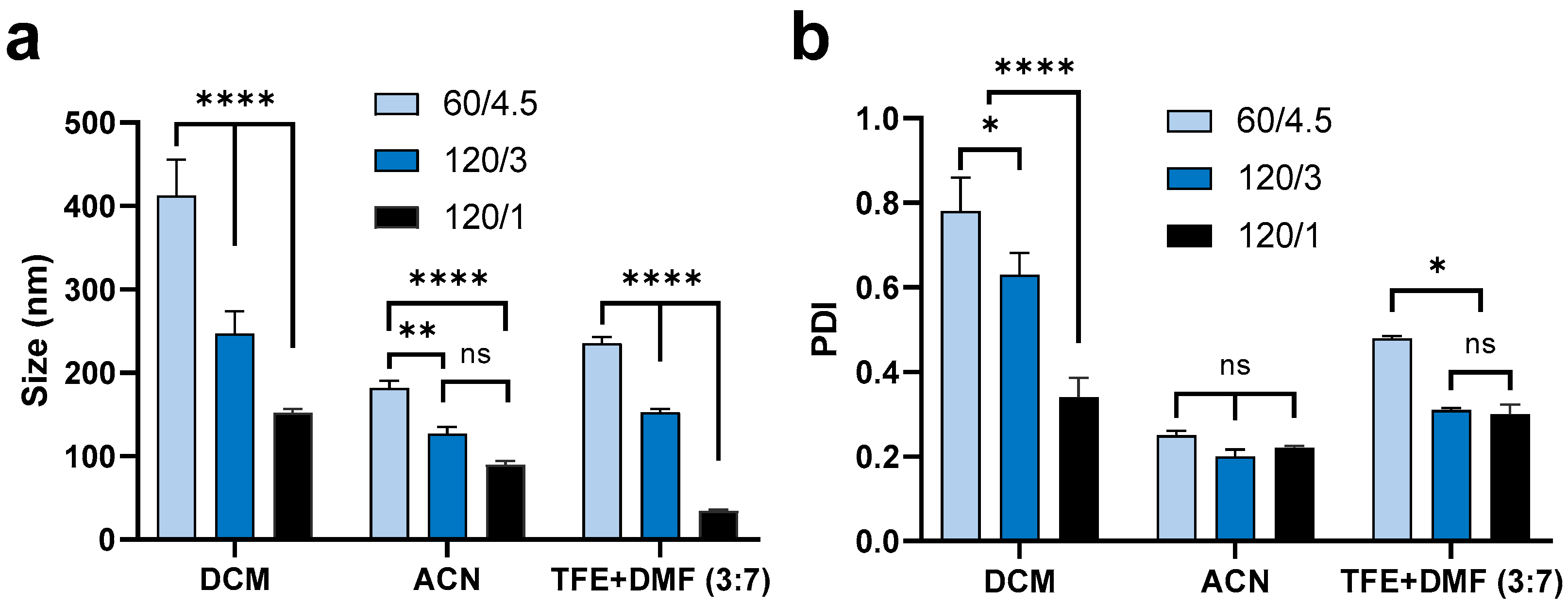
3.1.1. Effect of Flow Rate (Side/Centre)
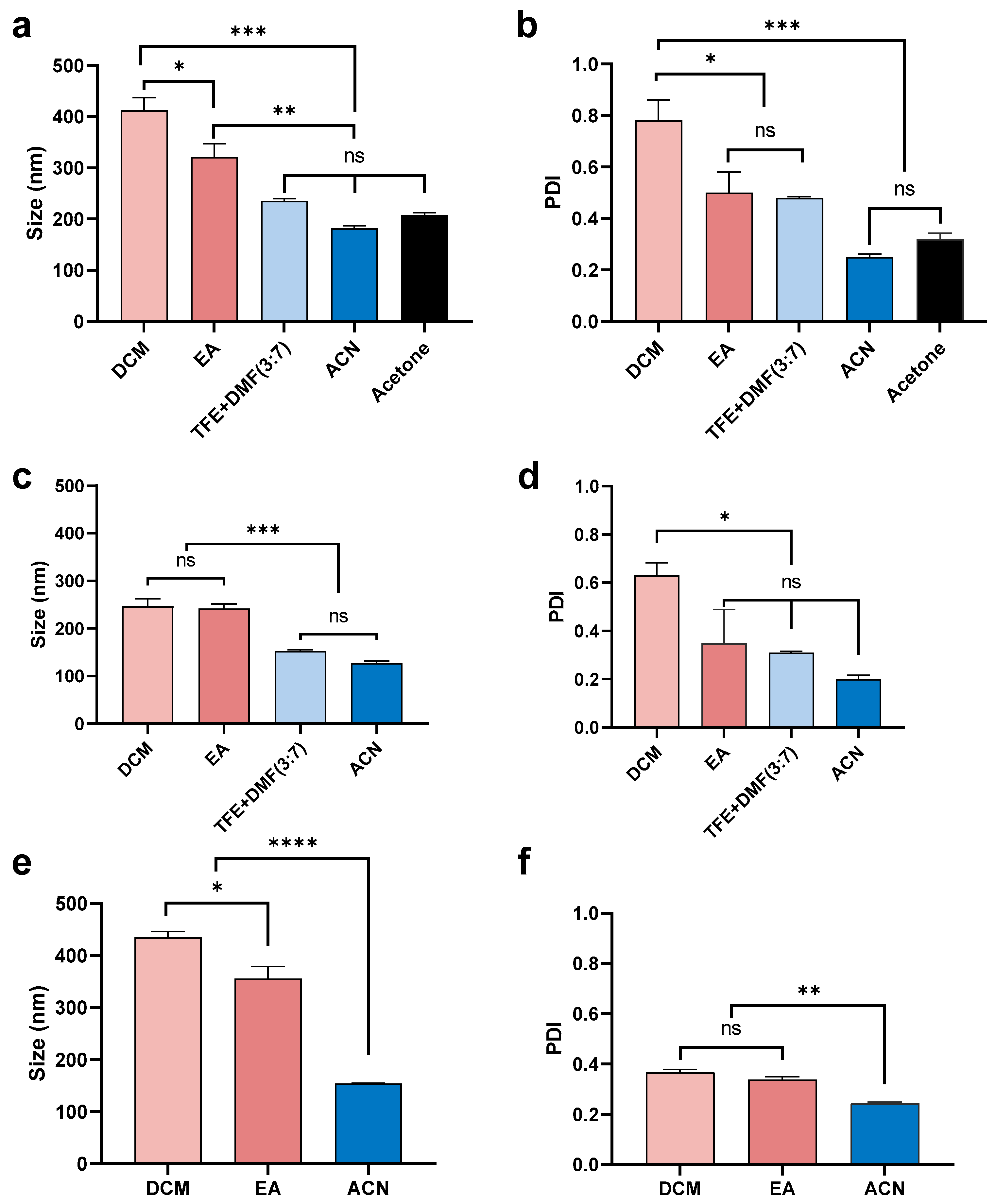
3.1.2. Effect of Organic Solvent
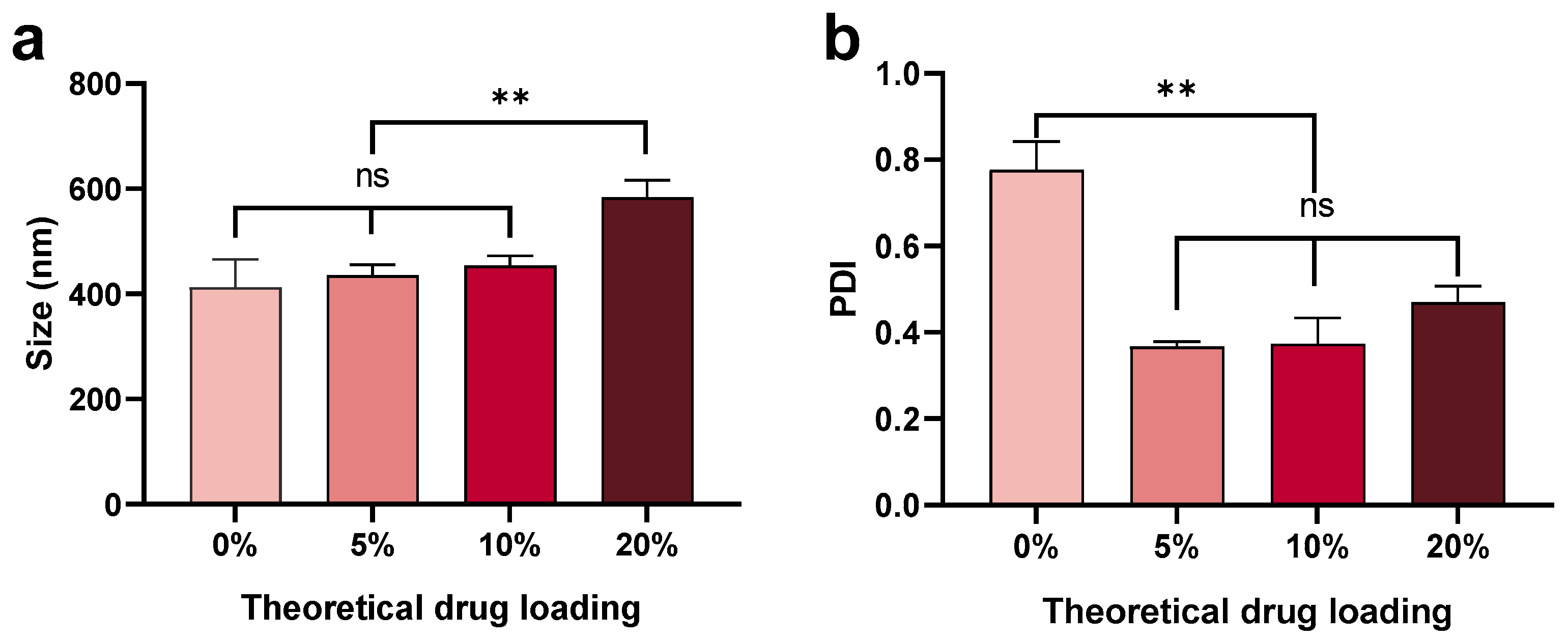
3.1.3. Effect of Theoretical Drug Loading
3.1.4. Effect of Polymer
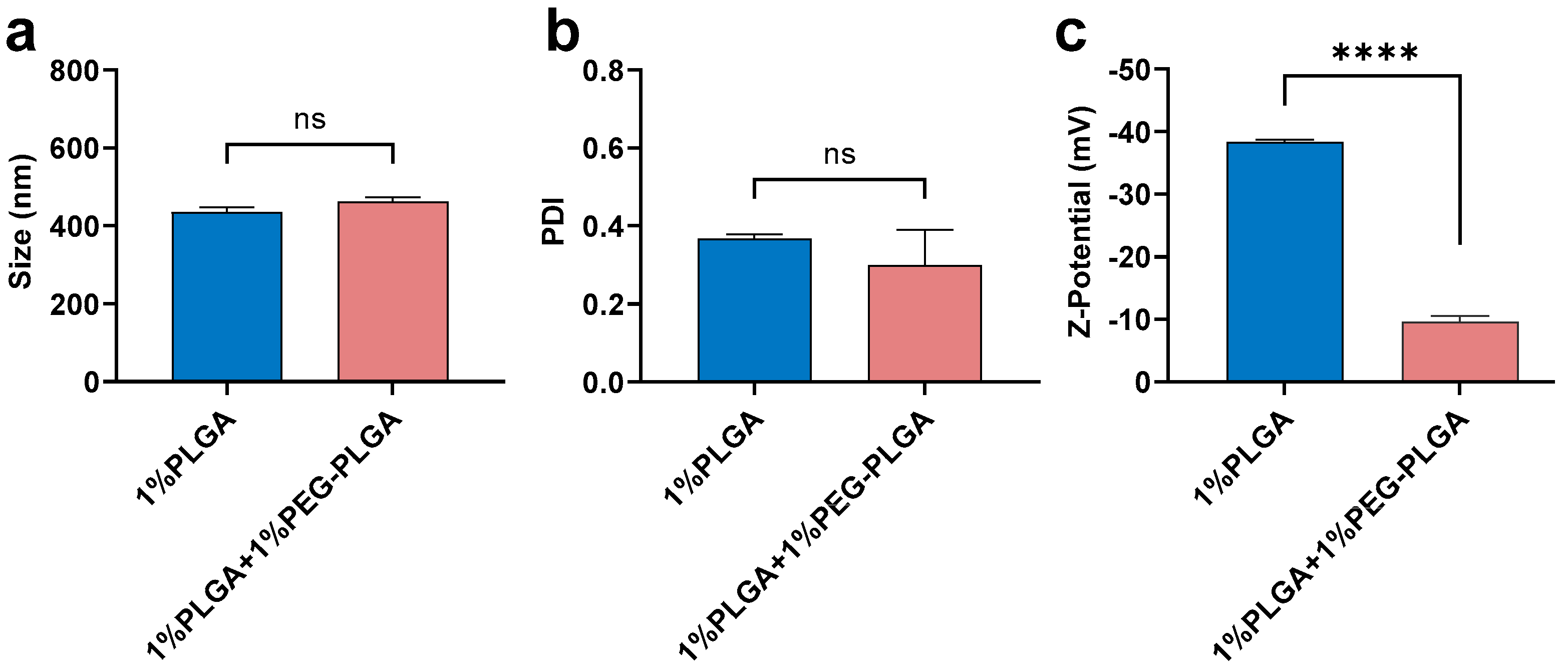
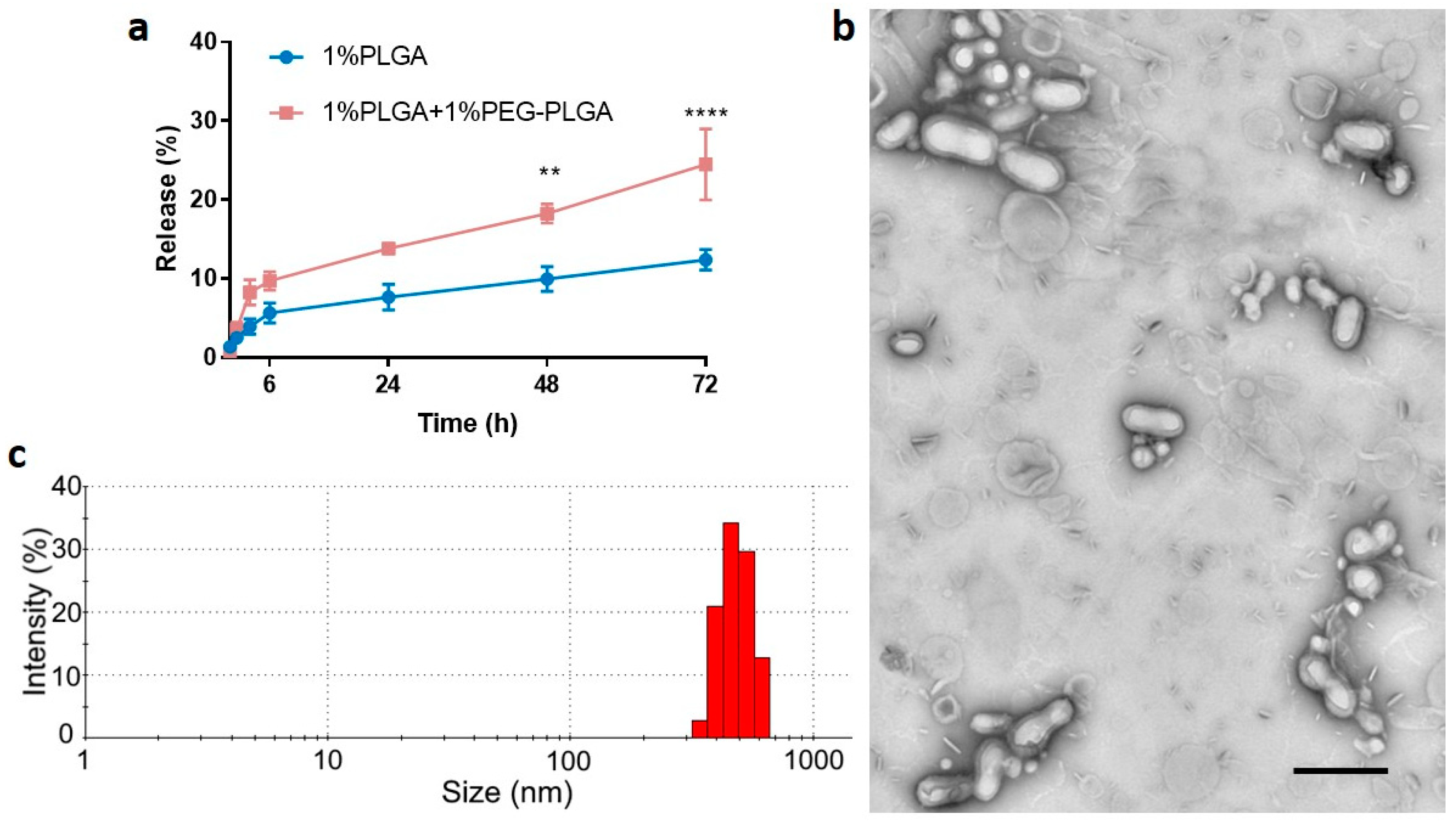
3.2. Optimisation of In Vitro Release Profile and Further Characterization
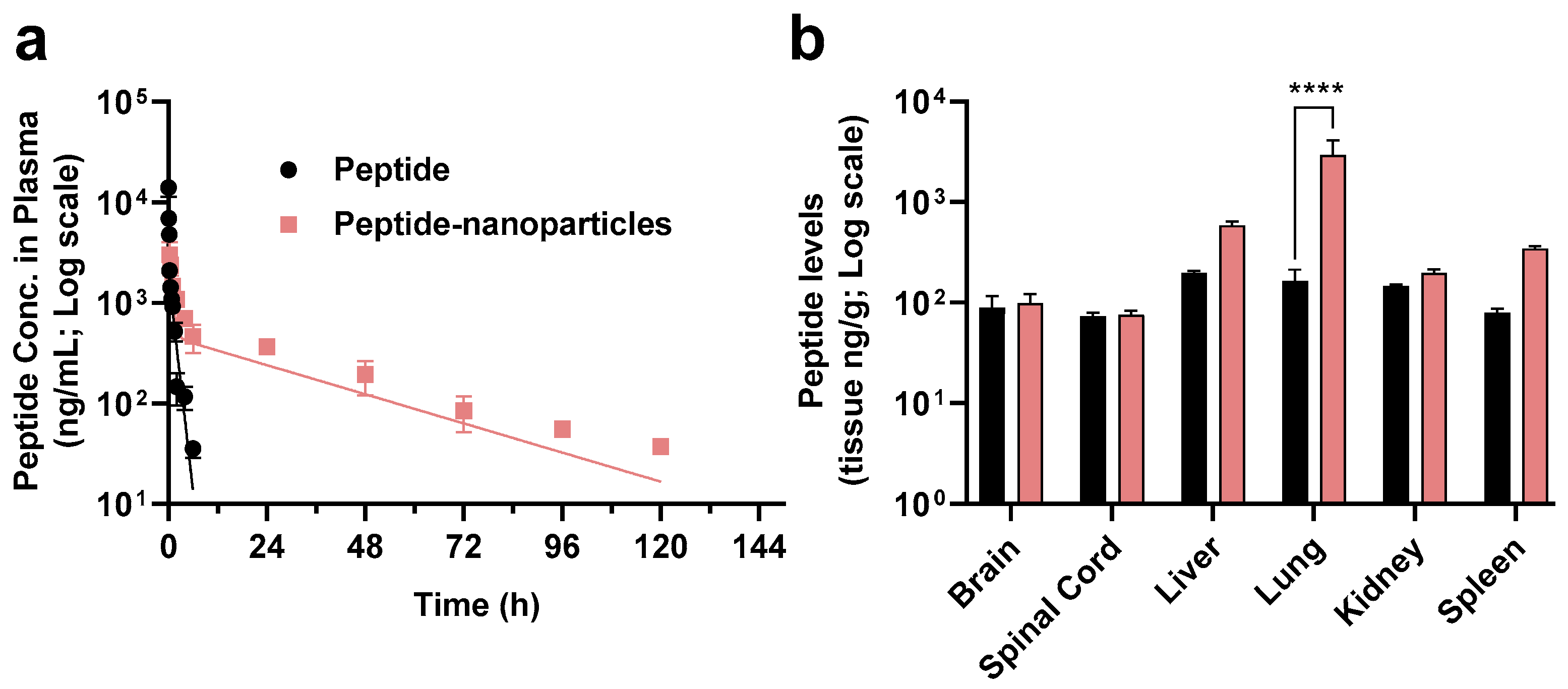
3.3. In Vivo Pharmacokinetics Study and Bio-Distribution
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Allahyari, M.; Mohit, E. Peptide/protein vaccine delivery system based on PLGA particles. Hum. Vaccin Immunother. 2016, 12, 806–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Operti, M.C.; Bernhardt, A.; Grimm, S.; Engel, A.; Figdor, C.G.; Tagit, O. PLGA-based nanomedicines manufacturing: Technologies overview and challenges in industrial scale-up. Int. J. Pharm. 2021, 605, 120807. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Han, F.Y.; Thurecht, K.J.; Whittaker, A.K.; Smith, M.T. Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading. Front. Pharmacol. 2016, 7, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Maincent, P.; Dellacherie, E.; Alonso, M.J.; Labrude, P.; Vigneron, C. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J. Control Release 1998, 50, 31–40. [Google Scholar] [CrossRef]
- Bisht, R.; Rupenthal, I.D. PLGA nanoparticles for intravitreal peptide delivery: Statistical optimization, characterization and toxicity evaluation. Pharm. Dev. Technol. 2018, 23, 324–333. [Google Scholar] [CrossRef]
- Han, F.Y.; Whittaker, A.; Howdle, S.M.; Naylor, A.; Shabir-Ahmed, A.; Smith, M.T. Sustained-Release Hydromorphone Microparticles Produced by Supercritical Fluid Polymer Encapsulation. J. Pharm. Sci. 2019, 108, 811–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, F.; Yang, M. Design of PLGA-based depot delivery systems for biopharmaceuticals prepared by spray drying. Int. J. Pharm. 2016, 498, 82–95. [Google Scholar] [CrossRef]
- Morikawa, Y.; Tagami, T.; Hoshikawa, A.; Ozeki, T. The Use of an Efficient Microfluidic Mixing System for Generating Stabilized Polymeric Nanoparticles for Controlled Drug Release. Biol. Pharm. Bull. 2018, 41, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Operti, M.C.; Dölen, Y.; Keulen, J.; van Dinther, E.A.W.; Figdor, C.G.; Tagit, O. Microfluidics-Assisted Size Tuning and Biological Evaluation of PLGA Particles. Pharmaceutics 2019, 11, 590. [Google Scholar] [CrossRef] [Green Version]
- Operti, M.C.; Fecher, D.; van Dinther, E.A.W.; Grimm, S.; Jaber, R.; Figdor, C.G.; Tagit, O. A comparative assessment of continuous production techniques to generate sub-micron size PLGA particles. Int. J. Pharm. 2018, 550, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Hashimoto, M.; Dang, T.T.; Hoare, T.; Kohane, D.S.; Whitesides, G.M.; Langer, R.; Anderson, D.G. Preparation of monodisperse biodegradable polymer microparticles using a microfluidic flow-focusing device for controlled drug delivery. Small 2009, 5, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.H.; Teh, S.Y.; Jester, J.; Lee, A.P. PLGA micro/nanosphere synthesis by droplet microfluidic solvent evaporation and extraction approaches. Lab Chip 2010, 10, 1820–1825. [Google Scholar] [CrossRef]
- Valencia, P.M.; Basto, P.A.; Zhang, L.; Rhee, M.; Langer, R.; Farokhzad, O.C.; Karnik, R. Single-Step Assembly of Homogenous Lipid−Polymeric and Lipid−Quantum Dot Nanoparticles Enabled by Microfluidic Rapid Mixing. ACS Nano 2010, 4, 1671–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Zhang, L.; Liu, C.; Li, X.; Hu, G.; Sun, J.; Jiang, X. Microfluidic based high throughput synthesis of lipid-polymer hybrid nanoparticles with tunable diameters. Biomicrofluidics 2015, 9, 052604. [Google Scholar] [CrossRef] [Green Version]
- Valencia, P.M.; Pridgen, E.M.; Rhee, M.; Langer, R.; Farokhzad, O.C.; Karnik, R. Microfluidic platform for combinatorial synthesis and optimization of targeted nanoparticles for cancer therapy. ACS Nano 2013, 7, 10671–10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, Q.; Ma, Y.; Sun, J. Microfluidic Methods for Fabrication and Engineering of Nanoparticle Drug Delivery Systems. ACS Appl. Bio Mater. 2020, 3, 107–120. [Google Scholar] [CrossRef]
- Bose, R.J.; Lee, S.-H.; Park, H. Lipid-based surface engineering of PLGA nanoparticles for drug and gene delivery applications. Biomater. Res. 2016, 20, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Feng, Q.; Wang, J.; Zhang, S.; Ding, B.; Wei, Y.; Dong, M.; Ryu, J.Y.; Yoon, T.Y.; Shi, X.; et al. Microfluidic Synthesis of Hybrid Nanoparticles with Controlled Lipid Layers: Understanding Flexibility-Regulated Cell-Nanoparticle Interaction. ACS Nano 2015, 9, 9912–9921. [Google Scholar] [CrossRef] [Green Version]
- Faraji, A.H.; Wipf, P. Nanoparticles in cellular drug delivery. Bioorganic Med. Chem. 2009, 17, 2950–2962. [Google Scholar] [CrossRef] [PubMed]
- Hoshyar, N.; Gray, S.; Han, H.; Bao, G. The effect of nanoparticle size on in vivo pharmacokinetics and cellular interaction. Nanomedicine 2016, 11, 673–692. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popović, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Al-Jamal, K.T.; Kostarelos, K.; Reineke, J. Physiologically based pharmacokinetic modeling of nanoparticles. ACS Nano 2010, 4, 6303–6317. [Google Scholar] [CrossRef]
- Ferguson, R.M.; Minard, K.R.; Krishnan, K.M. Optimization of nanoparticle core size for magnetic particle imaging. J. Magn. Magn. Mater. 2009, 321, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhang, L.; Wang, J.; Feng, Q.; Liu, D.; Yin, Q.; Xu, D.; Wei, Y.; Ding, B.; Shi, X.; et al. Tunable rigidity of (polymeric core)-(lipid shell) nanoparticles for regulated cellular uptake. Adv. Mater. 2015, 27, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, W.; Sun, J.; Liu, C.; Yin, Q.; Zhang, L.; Xianyu, Y.; Shi, X.; Hu, G.; Jiang, X. A microfluidic tubing method and its application for controlled synthesis of polymeric nanoparticles. Lab Chip 2014, 14, 1673–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, A.L.; Rosalia, R.A.; Sazak, A.; Carstens, M.G.; Ossendorp, F.; Oostendorp, J.; Jiskoot, W. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: Low-burst release is crucial for efficient CD8+ T cell activation. Eur. J. Pharm. Biopharm. 2013, 83, 338–345. [Google Scholar] [CrossRef]
- Kanai, T.; Tsuchiya, M. Microfluidic devices fabricated using stereolithography for preparation of monodisperse double emulsions. Chem. Eng. J. 2016, 290, 400–404. [Google Scholar] [CrossRef]
- Zhou, Z.; Kong, T.; Mkaouar, H.; Salama, K.N.; Zhang, J.M. A hybrid modular microfluidic device for emulsion generation. Sens. Actuators A Phys. 2018, 280, 422–428. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Hensel, A.; Brandner, J.J.; Zhang, K.; Du, X.; Yang, Y. A review on emulsification via microfluidic processes. Front. Chem. Sci. Eng. 2020, 14, 350–364. [Google Scholar] [CrossRef]
- March, D.R.; Proctor, L.M.; Stoermer, M.J.; Sbaglia, R.; Abbenante, G.; Reid, R.C.; Woodruff, T.M.; Wadi, K.; Paczkowski, N.; Tyndall, J.D.; et al. Potent cyclic antagonists of the complement C5a receptor on human polymorphonuclear leukocytes. Relationships between structures and activity. Mol. Pharm. 2004, 65, 868–879. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Pollitt, S.; Proctor, L.M.; Stocks, S.Z.; Manthey, H.D.; Williams, H.M.; Mahadevan, I.B.; Shiels, I.A.; Taylor, S.M. Increased potency of a novel complement factor 5a receptor antagonist in a rat model of inflammatory bowel disease. J. Pharm. Exp. 2005, 314, 811–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, U.; Woodruff, T.M.; Stadnyk, A.W. The C5a receptor antagonist PMX205 ameliorates experimentally induced colitis associated with increased IL-4 and IL-10. Br. J. Pharmacol. 2013, 168, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Brennan, F.H.; Gordon, R.; Lao, H.W.; Biggins, P.J.; Taylor, S.M.; Franklin, R.J.; Woodruff, T.M.; Ruitenberg, M.J. The Complement Receptor C5aR Controls Acute Inflammation and Astrogliosis following Spinal Cord Injury. J. Neurosci. 2015, 35, 6517–6531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staab, E.B.; Sanderson, S.D.; Wells, S.M.; Poole, J.A. Treatment with the C5a receptor/CD88 antagonist PMX205 reduces inflammation in a murine model of allergic asthma. Int. Immunopharmacol. 2014, 21, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.D.; Kumar, V.; Fung, J.N.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1(G93A) mouse model of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2017, 174, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Lee, J.D.; Clark, R.J.; Woodruff, T.M. Development and validation of a LC-MS/MS assay for pharmacokinetic studies of complement C5a receptor antagonists PMX53 and PMX205 in mice. Sci. Rep. 2018, 8, 8101. [Google Scholar] [CrossRef]
- Strachan, A.J.; Shiels, I.A.; Reid, R.C.; Fairlie, D.P.; Taylor, S.M. Inhibition of immune-complex mediated dermal inflammation in rats following either oral or topical administration of a small molecule C5a receptor antagonist (vol 134, pg 579, 2001). Brit. J. Pharm. 2002, 135, 579–580. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Lee, J.D.; Clark, R.J.; Noakes, P.G.; Taylor, S.M.; Woodruff, T.M. Preclinical Pharmacokinetics of Complement C5a Receptor Antagonists PMX53 and PMX205 in Mice. ACS Omega 2020, 5, 2345–2354. [Google Scholar] [CrossRef] [Green Version]
- Li, X.X.; Lee, J.D.; Massey, N.L.; Guan, C.; Robertson, A.A.B.; Clark, R.J.; Woodruff, T.M. Pharmacological characterisation of small molecule C5aR1 inhibitors in human cells reveals biased activities for signalling and function. Biochem. Pharm. 2020, 180, 114156. [Google Scholar] [CrossRef]
- Feng, Q.; Sun, J.S.; Jiang, X.Y. Microfluidics-mediated assembly of functional nanoparticles for cancer-related pharmaceutical applications. Nanoscale 2016, 8, 12430–12443. [Google Scholar] [CrossRef]
- Zhu, M.; Whittaker, A.K.; Jiang, X.; Tang, R.; Li, X.; Xu, W.; Fu, C.; Smith, M.T.; Han, F.Y. Use of Microfluidics to Fabricate Bioerodable Lipid Hybrid Nanoparticles Containing Hydromorphone or Ketamine for the Relief of Intractable Pain. Pharm. Res. 2020, 37, 211. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chan, J.M.; Gu, F.X.; Rhee, J.-W.; Wang, A.Z.; Radovic-Moreno, A.F.; Alexis, F.; Langer, R.; Farokhzad, O.C. Self-Assembled Lipid−Polymer Hybrid Nanoparticles: A Robust Drug Delivery Platform. ACS Nano 2008, 2, 1696–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.C.; Lee, H.S.; Choung, I.Y.; Cho, K.I.; Ahn, Y.; Choi, E.J. The effect of type of organic phase solvents on the particle size of poly (d, l-lactide-co-glycolide) nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2006, 276, 162–167. [Google Scholar] [CrossRef]
- Sahana, D.; Mittal, G.; Bhardwaj, V.; Kumar, M.R. PLGA nanoparticles for oral delivery of hydrophobic drugs: Influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J. Pharm. Sci. 2008, 97, 1530–1542. [Google Scholar] [CrossRef]
- Martins, J.P.; Torrieri, G.; Santos, H.A. The importance of microfluidics for the preparation of nanoparticles as advanced drug delivery systems. Expert Opin. Drug Deliv. 2018, 15, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Utada, A.S.; Fernandez-Nieves, A.; Stone, H.A.; Weitz, D.A. Dripping to jetting transitions in coflowing liquid streams. Phys. Rev. Lett. 2007, 99, 094502. [Google Scholar] [CrossRef]
- Lince, F.; Marchisio, D.L.; Barresi, A.A. Strategies to control the particle size distribution of poly-epsilon-caprolactone nanoparticles for pharmaceutical applications. J. Colloid Interface Sci. 2008, 322, 505–515. [Google Scholar] [CrossRef]
- Proctor, L.M.; Woodruff, T.M.; Sharma, P.; Shiels, I.A.; Taylor, S.M. Transdermal pharmacology of small molecule cyclic C5a antagonists. Adv. Exp. Med. Biol. 2006, 586, 329–345. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.G.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.P.; Pei, Y.Y.; Zhang, X.Y.; Gu, Z.H.; Zhou, Z.H.; Yuan, W.F.; Zhou, J.J.; Zhu, J.H.; Gao, X.J. PEGylated PLGA nanoparticles as protein carriers: Synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Garinot, M.; Fievez, V.; Pourcelle, V.; Stoffelbach, F.; des Rieux, A.; Plapied, L.; Theate, I.; Freichels, H.; Jerome, C.; Marchand-Brynaert, J.; et al. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J. Control. Release 2007, 120, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xianyu, Y.; Li, M.; Liu, W.; Zhang, L.; Liu, D.; Liu, C.; Hu, G.; Jiang, X. A microfluidic origami chip for synthesis of functionalized polymeric nanoparticles. Nanoscale 2013, 5, 5262–5265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cheng, C.; Prud’Homme, R.K.; Fox, R. Mixing in a multi-inlet vortex mixer (MIVM) for flash nano-precipitation. Chem. Eng. Sci. 2008, 63, 2829–2842. [Google Scholar] [CrossRef]
- Johnson, B.K.; Prud’homme, R.K. Chemical processing and micromixing in confined impinging jets. Aiche J. 2003, 49, 2264–2282. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef]
- Zhang, J.; Woodruff, T.M.; Clark, R.J.; Martin, D.J.; Minchin, R.F. Release of bioactive peptides from polyurethane films in vitro and in vivo: Effect of polymer composition. Acta Biomater. 2016, 41, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, N.; Li, P.; Jiang, Y.; Sun, H.; Cui, J.; Zhao, G.; Li, D.; Guo, Y.; Chen, Y.; Gao, J.; et al. C5a receptor1 inhibition alleviates influenza virus-induced acute lung injury. Int. Immunopharmacol. 2018, 59, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Carvelli, J.; Demaria, O.; Vely, F.; Batista, L.; Chouaki Benmansour, N.; Fares, J.; Carpentier, S.; Thibult, M.L.; Morel, A.; Remark, R.; et al. Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature 2020, 588, 146–150. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.Y.; Xu, W.; Kumar, V.; Cui, C.S.; Li, X.; Jiang, X.; Woodruff, T.M.; Whittaker, A.K.; Smith, M.T. Optimisation of a Microfluidic Method for the Delivery of a Small Peptide. Pharmaceutics 2021, 13, 1505. https://doi.org/10.3390/pharmaceutics13091505
Han FY, Xu W, Kumar V, Cui CS, Li X, Jiang X, Woodruff TM, Whittaker AK, Smith MT. Optimisation of a Microfluidic Method for the Delivery of a Small Peptide. Pharmaceutics. 2021; 13(9):1505. https://doi.org/10.3390/pharmaceutics13091505
Chicago/Turabian StyleHan, Felicity Y., Weizhi Xu, Vinod Kumar, Cedric S. Cui, Xaria Li, Xingyu Jiang, Trent M. Woodruff, Andrew K. Whittaker, and Maree T. Smith. 2021. "Optimisation of a Microfluidic Method for the Delivery of a Small Peptide" Pharmaceutics 13, no. 9: 1505. https://doi.org/10.3390/pharmaceutics13091505
APA StyleHan, F. Y., Xu, W., Kumar, V., Cui, C. S., Li, X., Jiang, X., Woodruff, T. M., Whittaker, A. K., & Smith, M. T. (2021). Optimisation of a Microfluidic Method for the Delivery of a Small Peptide. Pharmaceutics, 13(9), 1505. https://doi.org/10.3390/pharmaceutics13091505