
Albumin–Methotrexate Prodrug Analogues That Undergo Intracellular Reactivation Following Entrance into Cancerous Glioma Cells


Abstract
:1. Introduction
2. Experimental Procedures
2.1. Materials
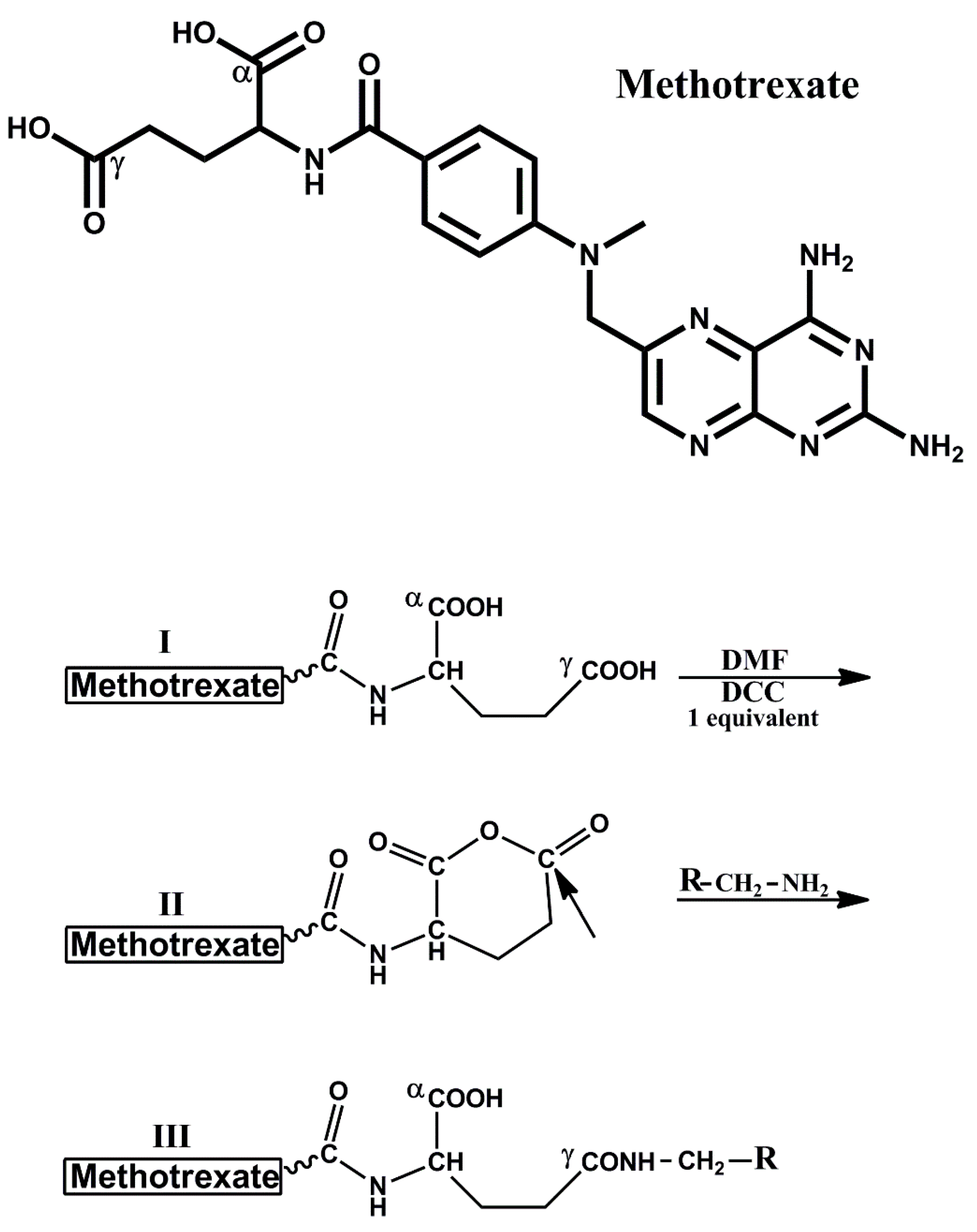
2.2. Synthesis of MTX–Anhydride
2.3. Synthesis of MTX–Dicystamine [MTX–COγNH–(CH2)2–S–S–(CH2)2–NH2]
2.4. Synthesis of MTX–Hexamethyl-Amine and MTX–Glutathione
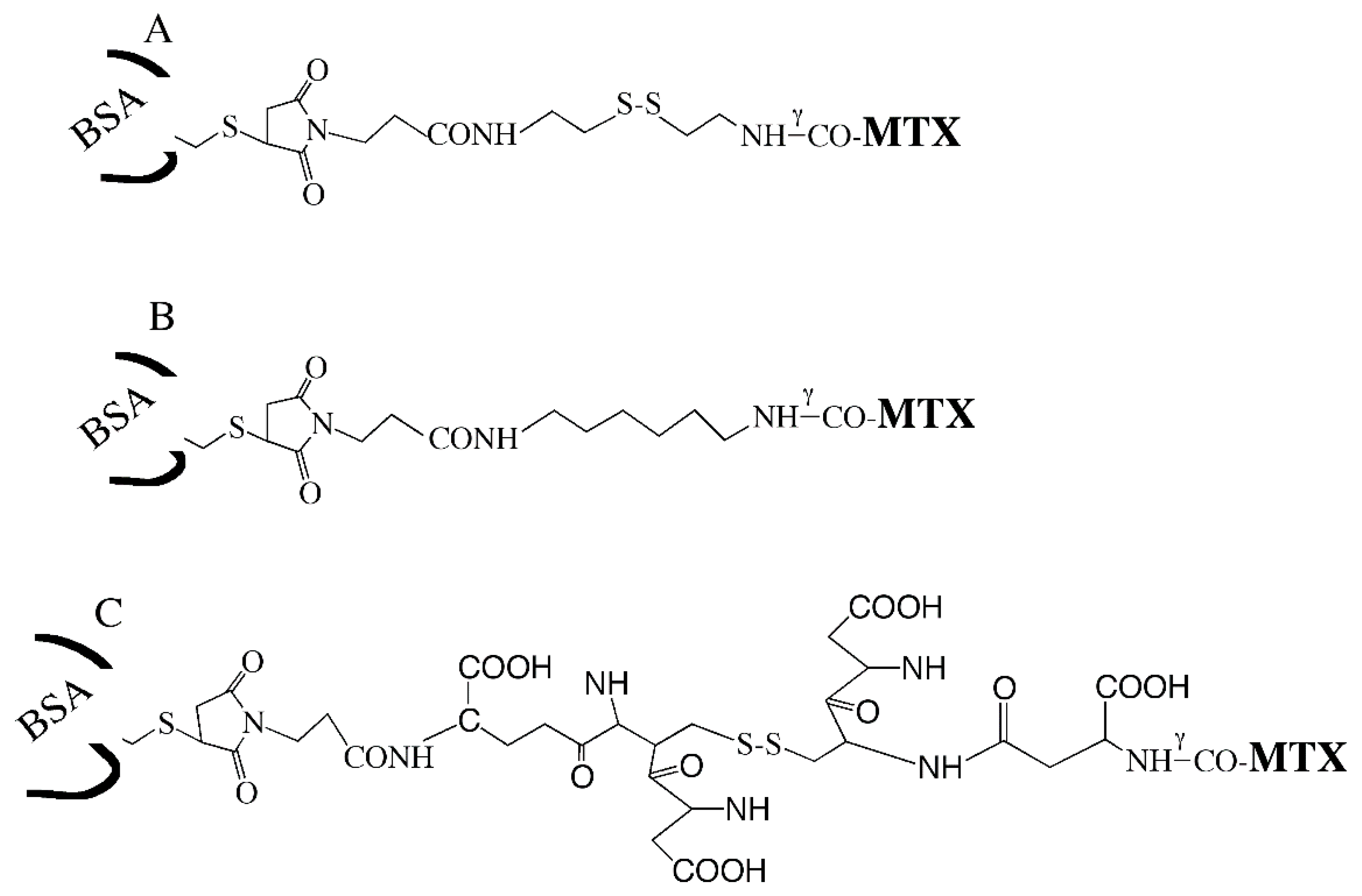
2.5. Preparation of BSA–MTX Conjugates
2.6. Cleavage of BSA–(CH2)6–MTX with Cyanogen Bromide
2.7. Preparation of HSA(MTX)40
2.8. Partial Purification of DHFR from Chicken Liver
2.9. Cell-Free Enzymatic Assay for DHFR
2.10. Maleimide–Thiol Exchange Assay
2.11. Cancer Cell Lines
2.12. Growth Inhibition Effects of BSA–MTX Analogues
2.13. Statistical Analysis
3. Results
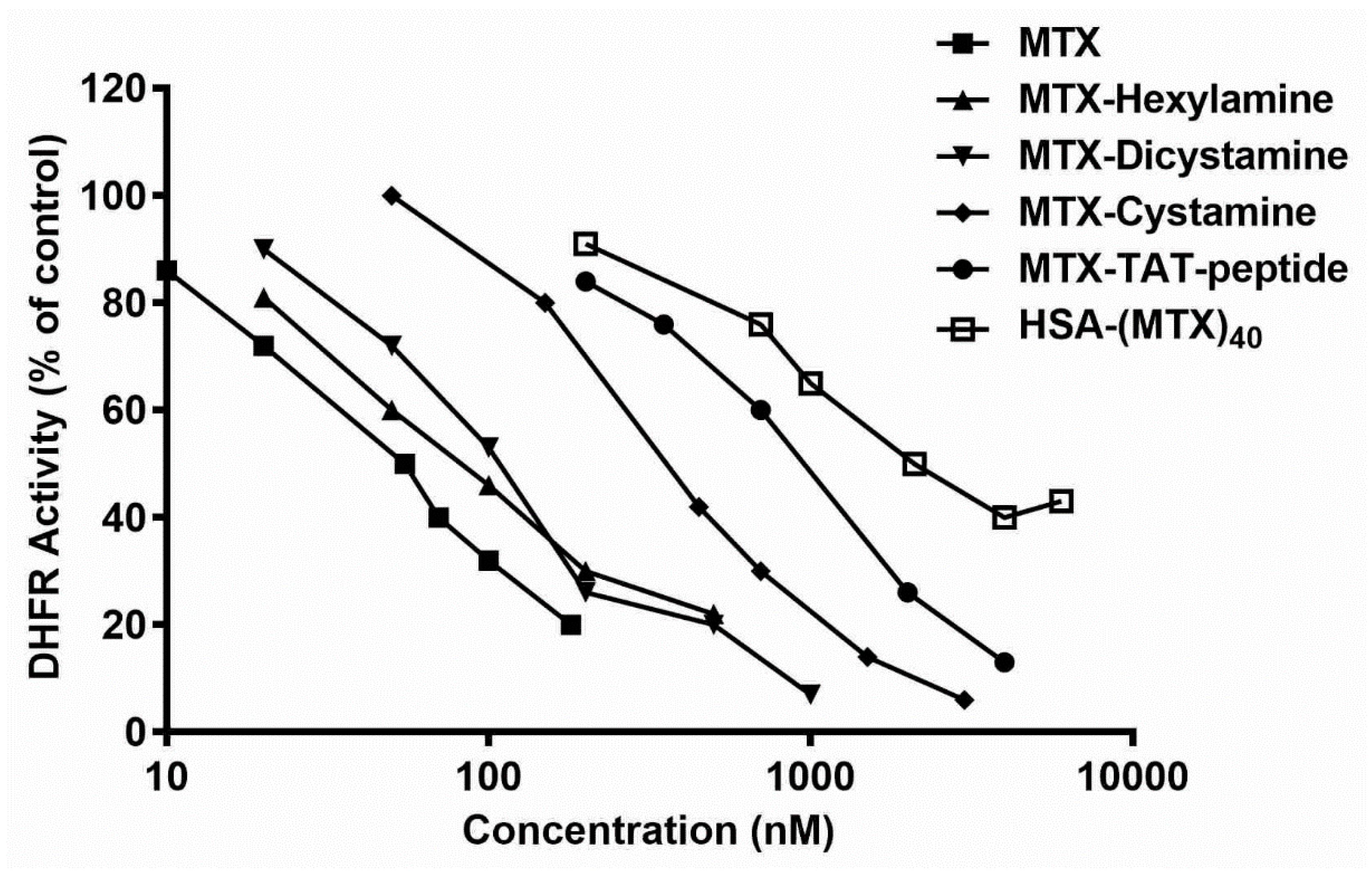
3.1. Searching for a Chemical Procedure to Obtain Active MTX Derivatives Following the Covalent Introduction of Amino-Containing Nucleophiles
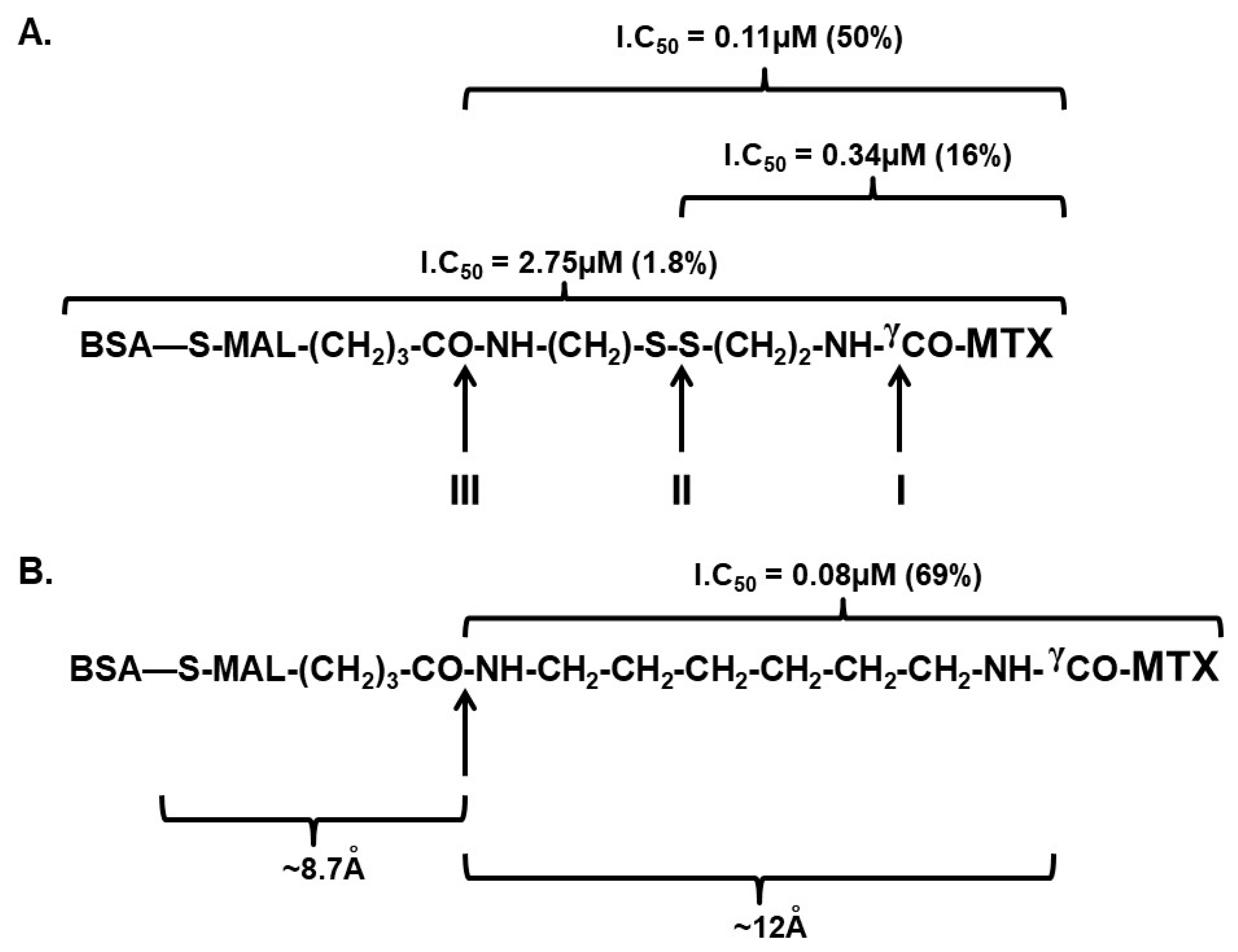
3.2. Preparing BSA–MTX Prodrug Analogues
3.3. BSA–MTX Analogues Are Prodrugs
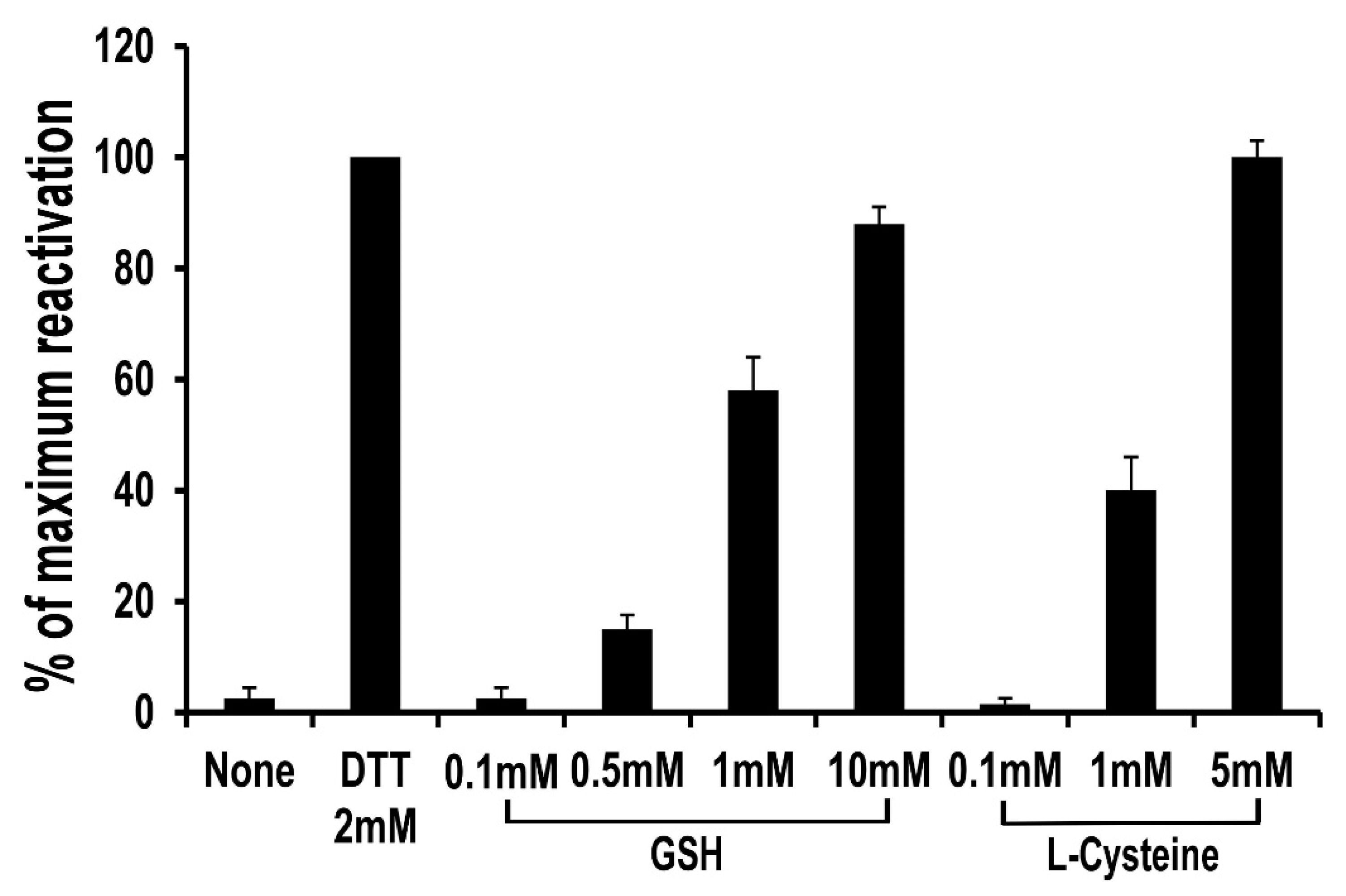
3.4. Lack of Maleimide–Thiol Exchange of BSA–(CH2)6–MTX following Treatment with Reduced Glutathione (GSH)
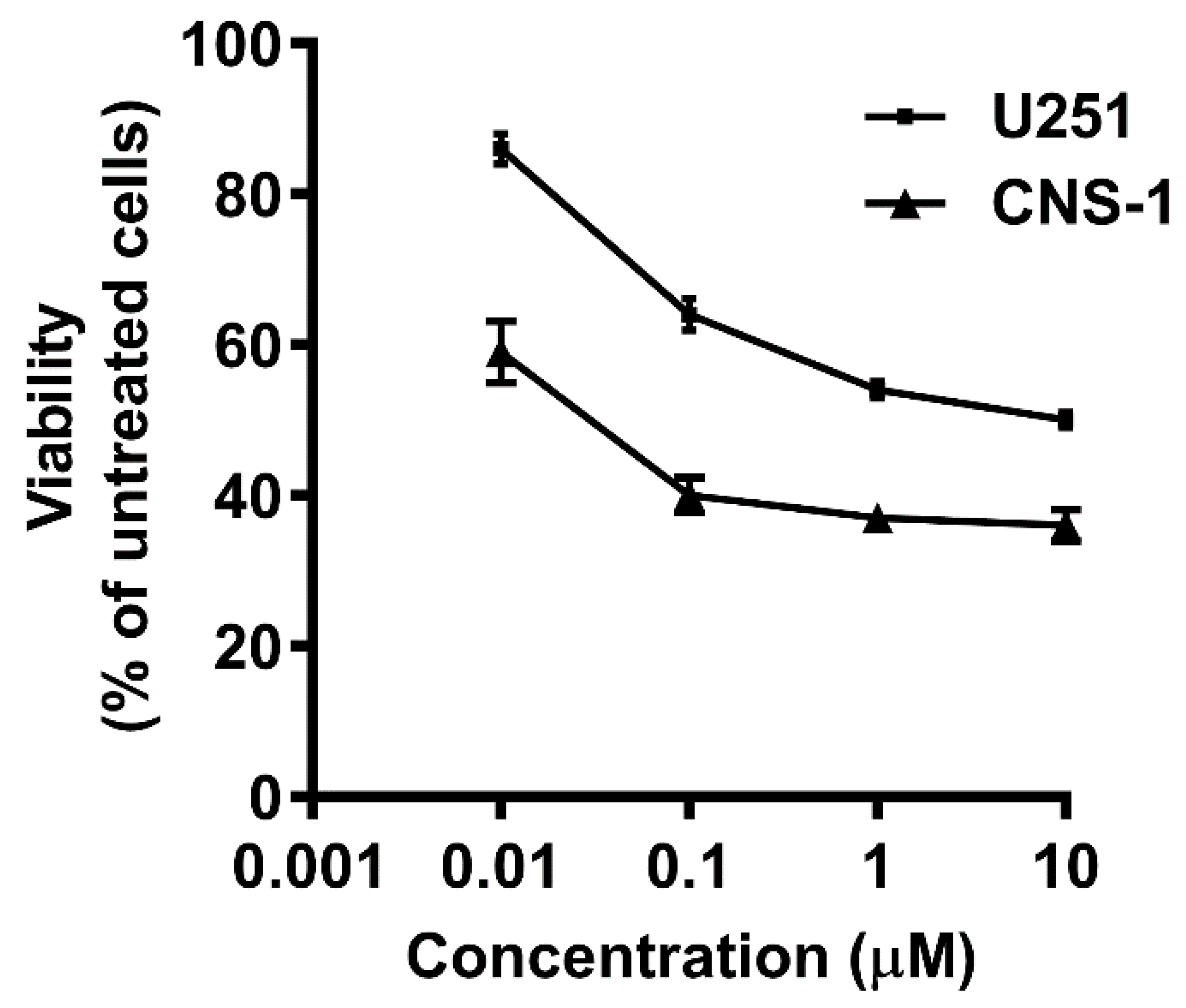
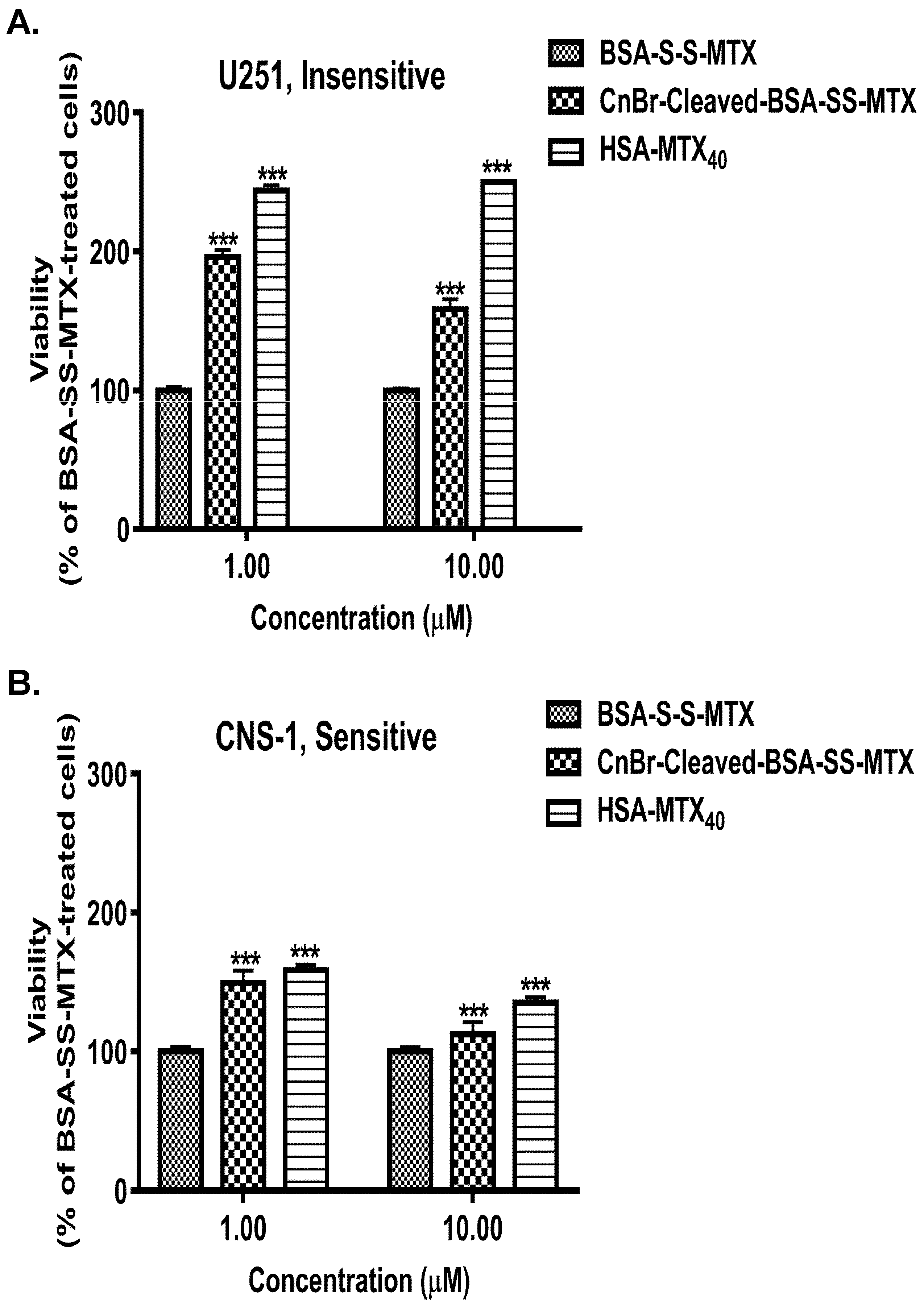
3.5. Selecting MTX-Sensitive and MTX-Insensitive Glioma Cell Lines for Studying the Antiproliferative Effects of BSA–MTX Analogues
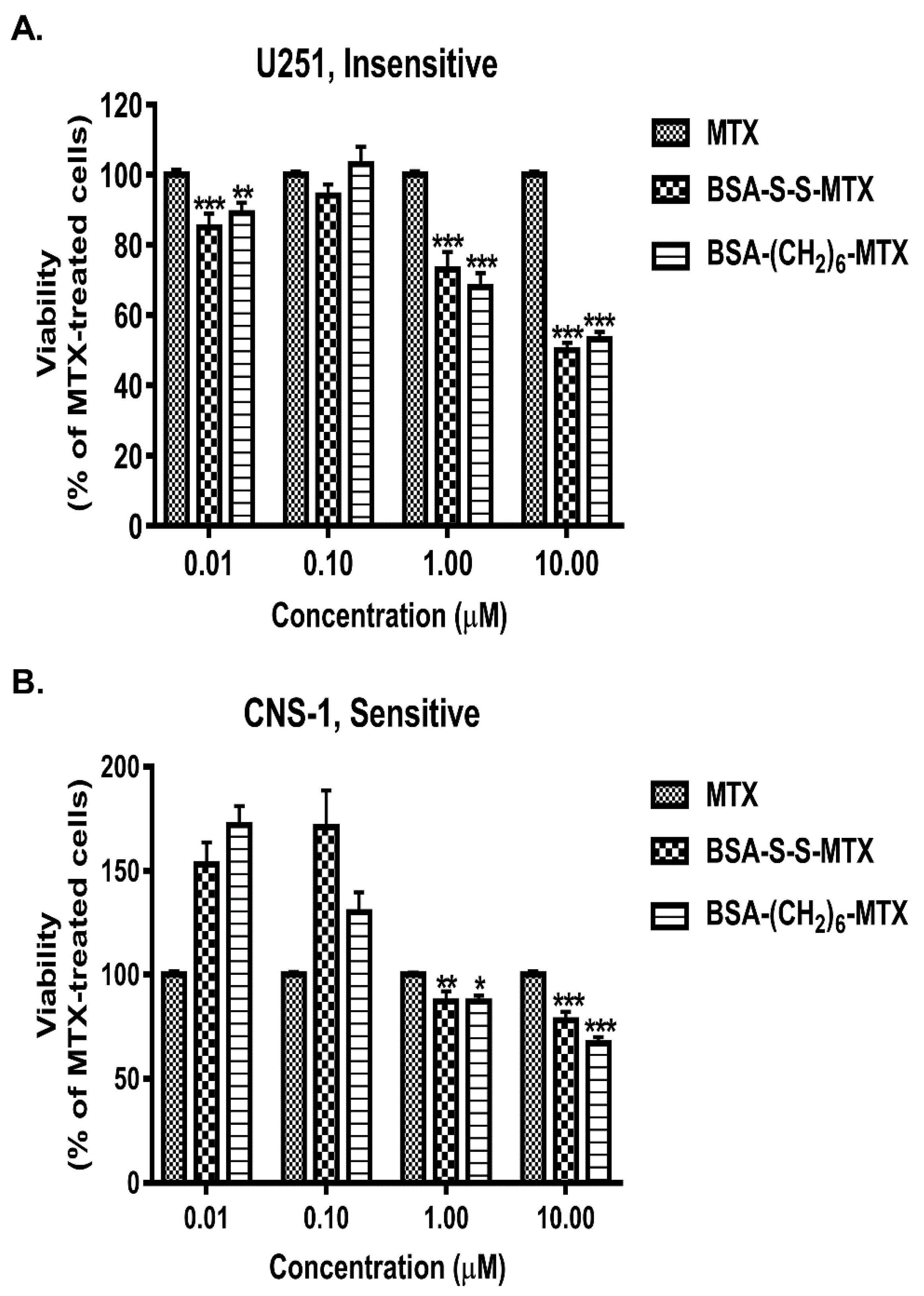
3.6. Comparison of the Antiproliferative Potencies of the BSA–MTX Conjugates in the Insensitive and Sensitive Glioma Cell Lines
3.7. Analyzing the Antiproliferative Potencies of Albumin–MTX Conjugates That Lost the Native Three-Dimensional Structure of the Protein
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | bovine serum albumin |
| MTX | methotrexate |
| DHF | dihydrofolic acid |
| DHFR | dihydrofolate reductase |
| MAL–(CH2)3–COOSu | maleimidopropionic acid-N-hydroxysuccinimide ester |
| GSSG | oxidized glutathione |
| BSA–S–S–MTX | BSA–S–MAL–(CH2)3–CONH–(CH2)2–S–S–(CH2)2–NHγCO–MTX |
| BSA–GSSG–MTX | BSA–S–MAL– (CH2)3–CONH–GSSG–NHγCO–MTX |
| BSA–(CH2)6–MTX | BSA–S–MAL–(CH2)3–CONH–(CH2)6–NHγCO–MTX |
| MALDI-TOF | matrix-assisted laser desorption ionization time of flight |
| ESMS | electrospray single quadruple mass spectroscopy |
| DCC | N,N′ dicyclohexylcarbodiimide |
| GSH | reduced glutathione |
| DMF | dimethylformamide |
| DIPEA | N,N′ diisopropylethylamine |
References
- Abolmaali, S.S.; Tamaddon, A.M.; Dinarvand, R. A review of therapeutic challenges and achievements of methotrexate de-livery systems for treatment of cancer and rheumatoid arthritis. Cancer Chemother. Pharmacol. 2013, 71, 1115–1130. [Google Scholar] [CrossRef] [PubMed]
- Genestier, L.; Paillot, R.; Quemeneur, L.; Izeradjene, K.; Revillard, J.-P. Mechanisms of action of methotrexate. Immunopharmacology 2000, 47, 247–257. [Google Scholar] [CrossRef]
- Sirotnak, F.M.; Moccio, D.M. Pharmacokinetic basis for differences in methotrexate sensitivity of normal proliferative tissues in the mouse. Cancer Res. 1980, 40, 1230–1234. [Google Scholar] [PubMed]
- Sirotnak, F.M.; Kurita, S.; Hutchison, D.J. On the nature of a transport alteration determining resistance to amethopterin in the L1210 leukemia. Cancer Res. 1968, 28, 75–80. [Google Scholar]
- Hill, B.T.; Bailey, B.D.; White, J.C.; Goldman, I.D. Characteristics of transport of 4-amino antifolates and folate compounds by two lines of L5178Y lymphoblasts, one with impaired transport of methotrexate. Cancer Res. 1979, 39, 2440–2446. [Google Scholar] [PubMed]
- Neumann, E.; Frei, E.; Funk, D.; Becker, M.D.; Schrenk, H.-H.; Müller-Ladner, U.; Fiehn, C. Native albumin for targeted drug delivery. Expert Opin. Drug Deliv. 2010, 7, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Levick, J.R. Permeability of rheumatoid and normal human synovium to specific plasma proteins. Arthritis Rheum. 1981, 24, 1550–1560. [Google Scholar] [CrossRef]
- Fitzpatrick, J.J.; Garnett, M.C. Design, synthesis and in vitro testing of methotrexate carrier conjugates linked via oligopeptide spacers. Anticancer Drug Des. 1995, 10, 1–9. [Google Scholar]
- Matthews, D.A.; Alden, R.A.; Bolin, J.T.; Filman, D.J.; Freer, S.T.; Hamlin, R.; Hol, W.G.; Kisliuk, R.L.; Pastore, E.J.; Plante, L.T.; et al. Dihydrofolate reductase from Lactobacillus casei. X-ray structure of the enzyme methotrexate.NADPH complex. J. Biol. Chem. 1978, 253, 6946–6954. [Google Scholar] [CrossRef]
- Rosowsky, A.; Forsch, R.; Uren, J.; Wick, M. Methotrexate analogues. Synthesis of new gamma-substituted derivatives as dihydrofolate reductase inhibitors and potential anticancer agents. J. Med. Chem. 1981, 24, 1450–1455. [Google Scholar]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef] [Green Version]
- Austin, C.D.; Wen, X.; Gazzard, L.; Nelson, C.; Scheller, R.H.; Scales, S.J. Oxidizing potential of endosomes and lysosomes limits intracellular cleavage of disulfide-based antibody-drug conjugates. Proc. Natl. Acad. Sci. USA 2005, 102, 17987–17992. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, S.; Luo, T.; Wang, C.; Zhao, J. Disulfide Linkage: A Potent Strategy in Tumor-Targeting Drug Discovery. Curr. Med. Chem. 2012, 19, 2976–2983. [Google Scholar] [CrossRef]
- Cooper, I.; Sasson, K.; Teichberg, V.I.; Schnaider-Beeri, M.; Fridkin, M.; Shechter, Y. Peptide derived from HIV-1 TAT protein de-stabilizes a monolayer of endothelial cells in an in vitro model of the blood-brain barrier and allows permeation of high molecular weight proteins. J. Biol. Chem. 2012, 287, 44676–44683. [Google Scholar] [CrossRef] [Green Version]
- Sharabi, S.; Guez, D.; Daniels, D.; Cooper, I.; Atrakchi, D.; Liraz-Zaltsman, S.; Last, D.; Mardor, Y. The application of point source electroporation and chemotherapy for the treatment of glioma: A randomized controlled rat study. Sci. Rep. 2020, 10, 2178. [Google Scholar] [CrossRef]
- Sharabi, S.; Bresler, Y.; Ravid, O.; Shemesh, C.; Atrakchi, D.; Schnaider-Beeri, M.; Gosselet, F.; Dehouck, L.; Last, D.; Guez, D.; et al. Transient blood–brain barrier disruption is induced by low pulsed electrical fields in vitro: An analysis of permeability and trans-endothelial electric resistivity. Drug Deliv. 2019, 26, 459–469. [Google Scholar] [CrossRef] [Green Version]
- Sharabi, S.; Last, D.; Daniels, D.; Fabian, I.D.; Atrakchi, D.; Bresler, Y.; Liraz-Zaltsman, S.; Cooper, I.; Mardor, Y. Non-Invasive Low Pulsed Electrical Fields for Inducing BBB Disruption in Mice-Feasibility Demonstration. Pharmaceutics 2021, 13, 169. [Google Scholar] [CrossRef]
- Cooper, I.; Last, D.; Guez, D.; Sharabi, S.; Goldman, S.E.; Lubitz, I.; Daniels, D.; Salomon, S.; Tamar, G.; Tamir, T.; et al. Combined Local Blood–Brain Barrier Opening and Systemic Methotrexate for the Treatment of Brain Tumors. Br. J. Pharmacol. 2015, 35, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.T.; Kemerer, V.F. Purification and characterization of beef liver dihydrofolate reductase. Arch. Biochem. Biophys. 1976, 172, 289–300. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kempton, R.J.; Anstead, G.M.; Freisheim, J.H. Fluorescent analogues of methotrexate: Characterization and interaction with dihydrofolate reductase. Biochemistry 1983, 22, 390–395. [Google Scholar] [CrossRef]
- Cooper, I.; Malina, K.C.-K.; Cagnotto, A.; Bazzoni, G.; Salmona, M.; Teichberg, V.I. Interactions of the prion peptide (PrP 106-126) with brain capillary endothelial cells: Coordinated cell killing and remodeling of intercellular junctions. J. Neurochem. 2011, 116, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Piper, J.R.; Montgomery, J.A.; Sirotnak, F.M.; Chello, P.L. Syntheses of alpha- and gamma-substituted amides, peptides, and esters of methotrexate and their evaluation as inhibitors of folate metabolism. J. Med. Chem. 1982, 25, 182–187. [Google Scholar] [PubMed]
- Miyachi, H.; Takemura, Y.; Kobayashi, H.; Ando, Y. Expression of variant dihydrofolate reductase with decreased binding affinity to antifolates in MOLT-3 human leukemia cell lines resistant to trimetrexate. Cancer Lett. 1995, 88, 93–99. [Google Scholar] [CrossRef]
- Huang, X.; Day, N.; Luo, X.; Roupioz, Y.; Seid, M.; Keillor, J.W. Synthesis and characterization of a series of novel glutamic gamma-15N-anilide dipeptides. J. Pept. Res. 1999, 53, 126–133. [Google Scholar] [CrossRef]
- Shen, B.-Q.; Xu, K.; Liu, L.; Raab, H.; Bhakta, S.; Kenrick, M.; Parsons-Reponte, K.L.; Tien, J.; Yu, S.-F.; Mai, E.; et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 2012, 30, 184–189. [Google Scholar] [CrossRef]
- Wolff, J.E.A.; Trilling, T.; Mölenkamp, G.; Egeler, R.M.; Jürgens, H. Chemosensitivity of glioma cells in vitro: A meta analysis. J. Cancer Res. Clin. Oncol. 1999, 125, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in Cancer Cell Death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ames, B.N.; Gold, L.S.; Willett, W.C. The causes and prevention of cancer. Proc. Natl. Acad. Sci. USA 1995, 92, 5258–5265. [Google Scholar] [CrossRef] [Green Version]
- Go, Y.-M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta (BBA)–Gen. Subj. 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [Green Version]
- Spinella, M.J.; Brigle, K.E.; Sierra, E.E.; Goldman, I.D. Distinguishing between folate receptor-alpha-mediated transport and reduced folate carrier-mediated transport in L1210 leukemia cells. J. Biol. Chem. 1995, 270, 7842–7849. [Google Scholar] [CrossRef] [Green Version]
- Wosikowski, K.; Biedermann, E.; Rattel, B.; Breiter, N.; Jank, P.; Löser, R.; Jansen, G.; Peters, G.J. In vitro and in vivo antitumor activity of methotrexate conjugated to human serum albumin in human cancer cells. Clin. Cancer Res. 2003, 9, 1917–1926. [Google Scholar] [PubMed]
- Ayusawa, D.; Koyama, H.; Seno, T. Resistance to methotrexate in thymidylate synthetase-deficient mutants of cultured mouse mammary tumor FM3A cells. Cancer Res. 1981, 41, 1497–1501. [Google Scholar] [PubMed]
- Banerjee, D.; Mayer-Kuckuk, P.; Capiaux, G.; Budak-Alpdogan, T.; Gorlick, R.; Bertino, J.R. Novel aspects of resistance to drugs targeted to dihydrofolate reductase and thymidylate synthase. Biochim. Biophys. Acta (BBA)–Mol. Basis Dis. 2002, 1587, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Cowan, K.H.; Jolivet, J. A methotrexate-resistant human breast cancer cell line with multiple defects, including diminished formation of methotrexate polyglutamates. J. Biol. Chem. 1984, 259, 10793–10800. [Google Scholar] [CrossRef]
- Ravasco, J.M.J.M.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with Maleimides: A Useful Tool for Chemical Biology. Chem. A Eur. J. 2019, 25, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Tunable Degradation of Maleimide–Thiol Adducts in Reducing Environments. Bioconjugate Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [Green Version]
- Carter, D.C.; Ho, J.X. Structure of Serum Albumin. In Lipoproteins, Apolipoproteins, and Lipases; Elsevier: Amsterdam, The Netherlands, 1994; pp. 153–203. [Google Scholar]
- Assaraf, Y.G. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007, 26, 153–181. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.N.; Poe, M.; Greenfield, N.J.; Hirshfield, J.M.; Hoogsteen, K. Methotrexate binding to dihydrofolate reductase from a methotrexate-resistant strain of Escherichia coli. J. Biol. Chem. 1973, 248, 6375–6379. [Google Scholar] [CrossRef]
- Cooper, I.; Fridkin, M.; Shechter, Y. Conjugation of Methotrexate-Amino Derivatives to Macromolecules through Carboxylate Moieties Is Superior over Conventional Linkage to Amino Residues: Chemical, Cell-Free and In Vitro Characterizations. PLoS ONE 2016, 11, e0158352. [Google Scholar] [CrossRef]
- Stehle, G.; Sinn, H.; Wunder, A.; Schrenk, H.H.; Schütt, S.; Maier-Borst, W.; Heene, D.L. The loading rate determines tumor targeting properties of methotrexate-albumin conjugates in rats. Anti-Cancer Drugs 1995, 8, 677–685. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative Designation | Abbreviated Structure | IC50 nM | % Inhibition relative to MTX |
|---|---|---|---|
| Methotrexate | MTX | 55 ± 4 (a) | 100 |
| MTX–hexylamine | MTX–CONH–(CH2)6–NH2 | 80 ± 4 | 69 |
| MTX–dicystamine | MTX–CONH–(CH2)2–S–S–(CH2)2–NH2 | 110 ± 12 | 50 |
| MTX–cystamine | MTX–CONH–(CH2)2–SH | 340 ± 11 | 16.2 |
| MTX–TAT–peptide | MTX–AYGRKKRRQRRR | 1000 ± 200 | 5.5 |
| HSA–(MTX)40 | HSA–(NH–CO–MTX)40 | 2100 ± 150 | 2.6 |
| Derivative Designation | DHFR Inhibitory Potency (IC50, nM) | % Activity Relative to MTX | Fold Reactivation by DTT |
|---|---|---|---|
| BSA–(CH2)6–MTX | 790 ± 30 | 7.0 | |
| BSA–S–S–MTX | 2750 ± 200 | 2.0 | |
| BSA–S–S–MTX + 2 mM DTT | 390 ± 30 | 14.1 | 7.05 |
| BSA–GSSG–MTX | 1750 ± 120 | 3.14 | |
| BSA–GSSG–MTX + 2 mM DTT | 340 ± 20 | 16.2 | 5.16 |
| Treatment | Absorbance at 372 nm per mg Protein |
|---|---|
| Control | 0.479 ± 0.03 |
| GSH (a) | 0.487 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, I.; Schnaider-Beeri, M.; Fridkin, M.; Shechter, Y. Albumin–Methotrexate Prodrug Analogues That Undergo Intracellular Reactivation Following Entrance into Cancerous Glioma Cells. Pharmaceutics 2022, 14, 71. https://doi.org/10.3390/pharmaceutics14010071
Cooper I, Schnaider-Beeri M, Fridkin M, Shechter Y. Albumin–Methotrexate Prodrug Analogues That Undergo Intracellular Reactivation Following Entrance into Cancerous Glioma Cells. Pharmaceutics. 2022; 14(1):71. https://doi.org/10.3390/pharmaceutics14010071
Chicago/Turabian StyleCooper, Itzik, Michal Schnaider-Beeri, Mati Fridkin, and Yoram Shechter. 2022. "Albumin–Methotrexate Prodrug Analogues That Undergo Intracellular Reactivation Following Entrance into Cancerous Glioma Cells" Pharmaceutics 14, no. 1: 71. https://doi.org/10.3390/pharmaceutics14010071
APA StyleCooper, I., Schnaider-Beeri, M., Fridkin, M., & Shechter, Y. (2022). Albumin–Methotrexate Prodrug Analogues That Undergo Intracellular Reactivation Following Entrance into Cancerous Glioma Cells. Pharmaceutics, 14(1), 71. https://doi.org/10.3390/pharmaceutics14010071