
D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. GCPII Activity Assay
2.3. Metabolic Stability Assays
2.4. Pharmacokinetic Studies in Mice
2.5. Bioanalysis in Plasma and Brain
2.6. D-DOPA Target Engagement Studies
2.7. D-DOPA Mode of Inhibition Studies
3. Results and Discussion
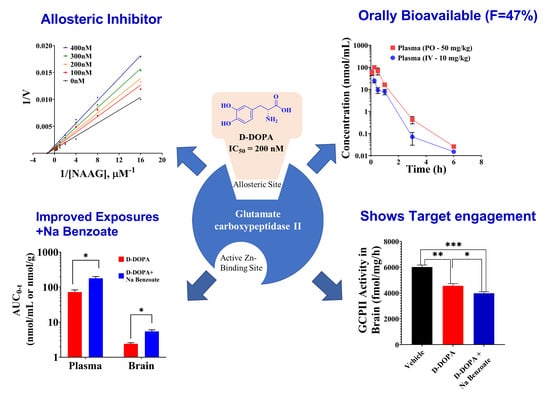
3.1. IC50 of Catechol-Based Scaffolds
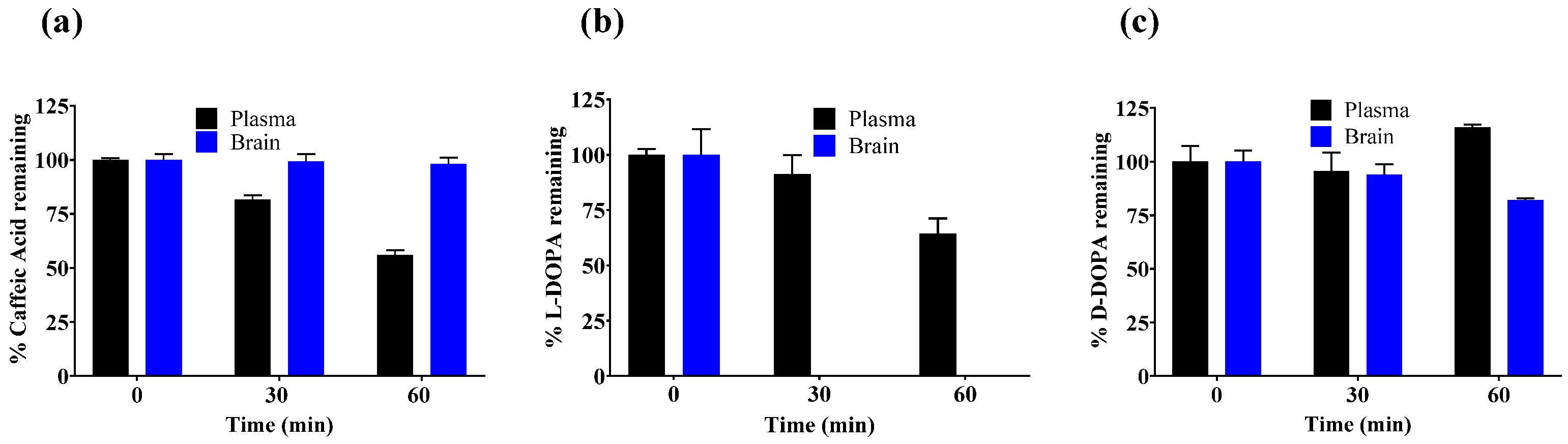
3.2. In Vitro Metabolic Stability of Caffeic Acid, L-DOPA, and D-DOPA
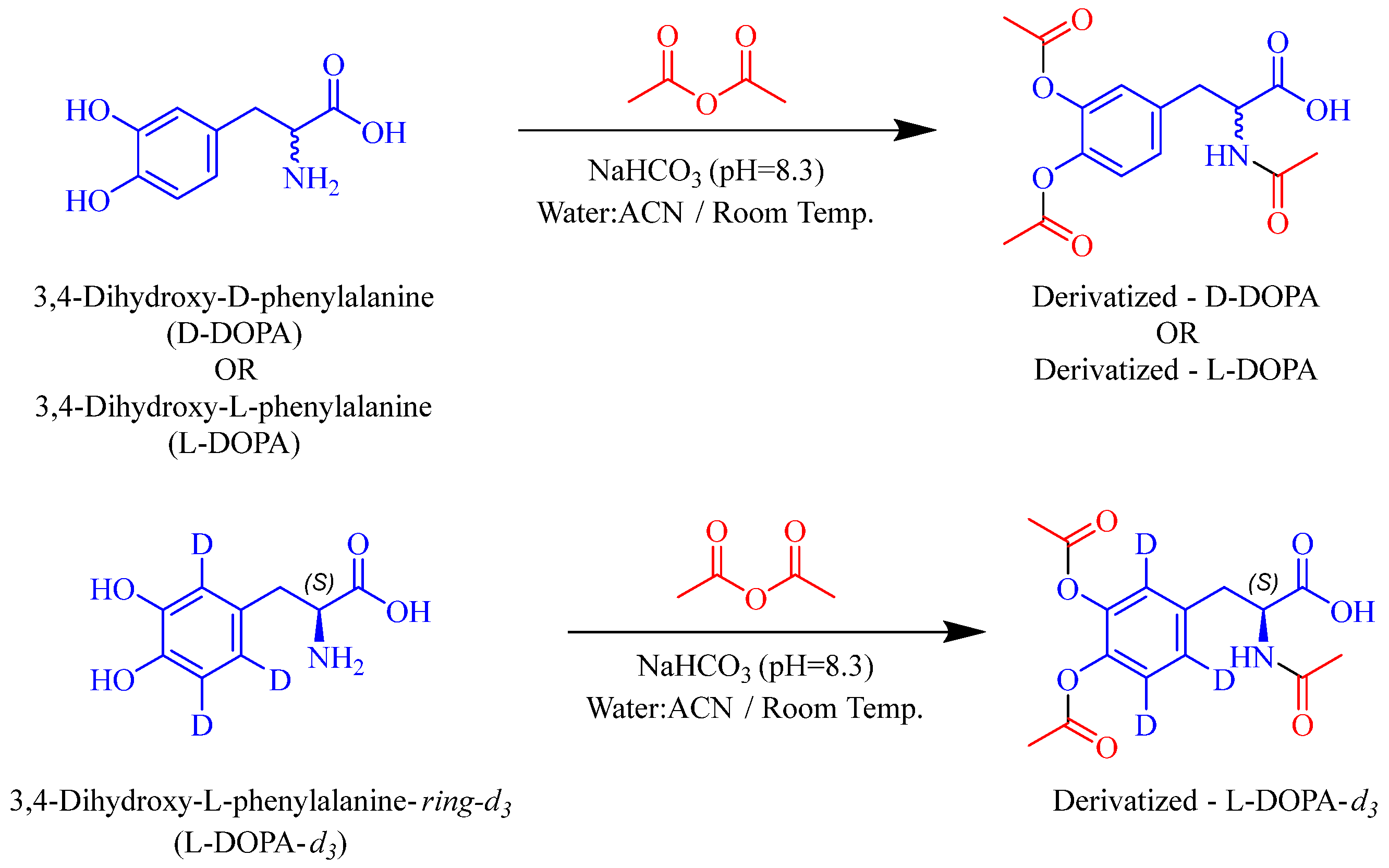
3.3. Bioanalytical Methods for Caffeic Acid, L-DOPA, and D-DOPA
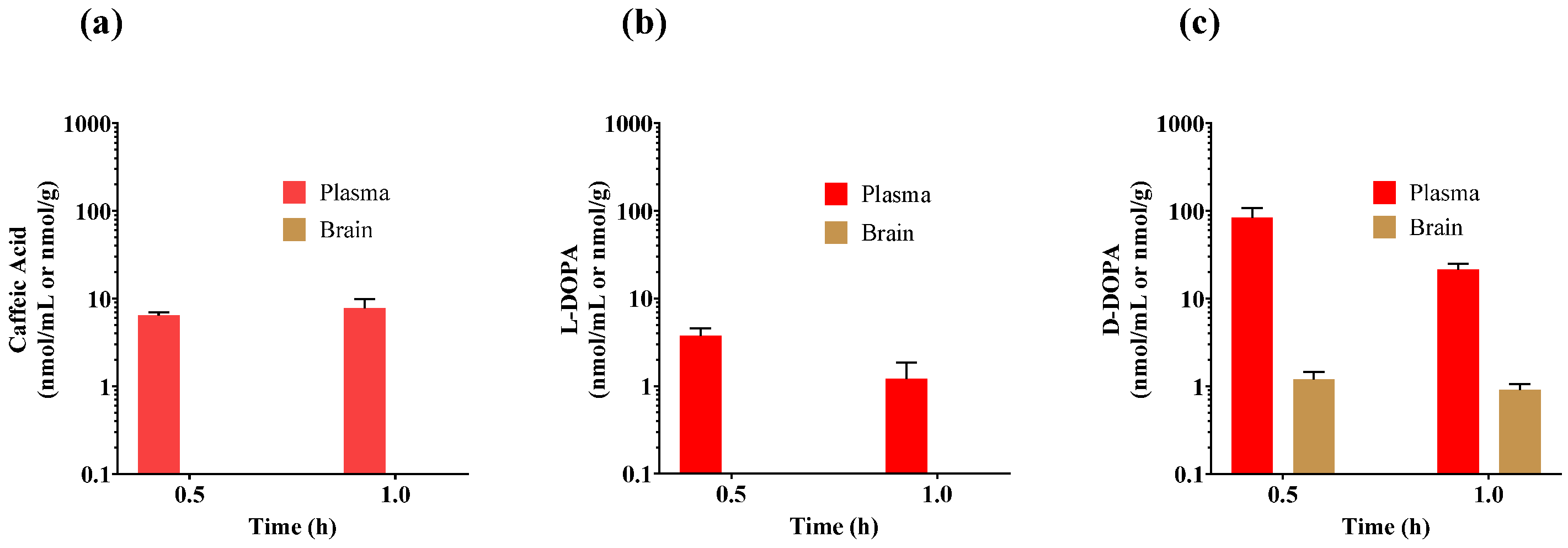
3.4. Initial Pharmacokinetic Studies of Caffeic Acid, L-DOPA, and D-DOPA in Mice
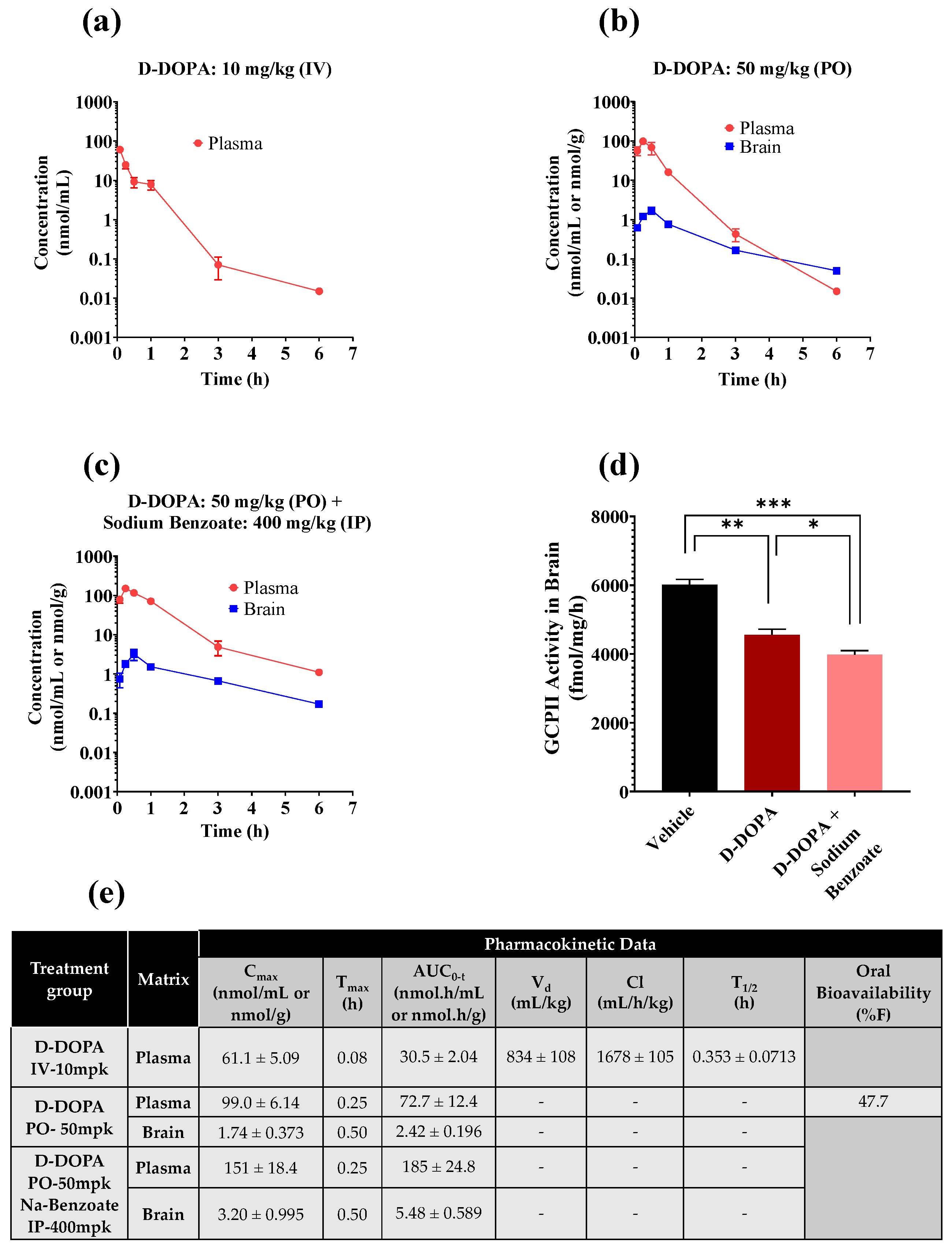
3.5. Pharmacokinetic and Target Engagement Studies of D-DOPA + Sodium Benzoate in Mice
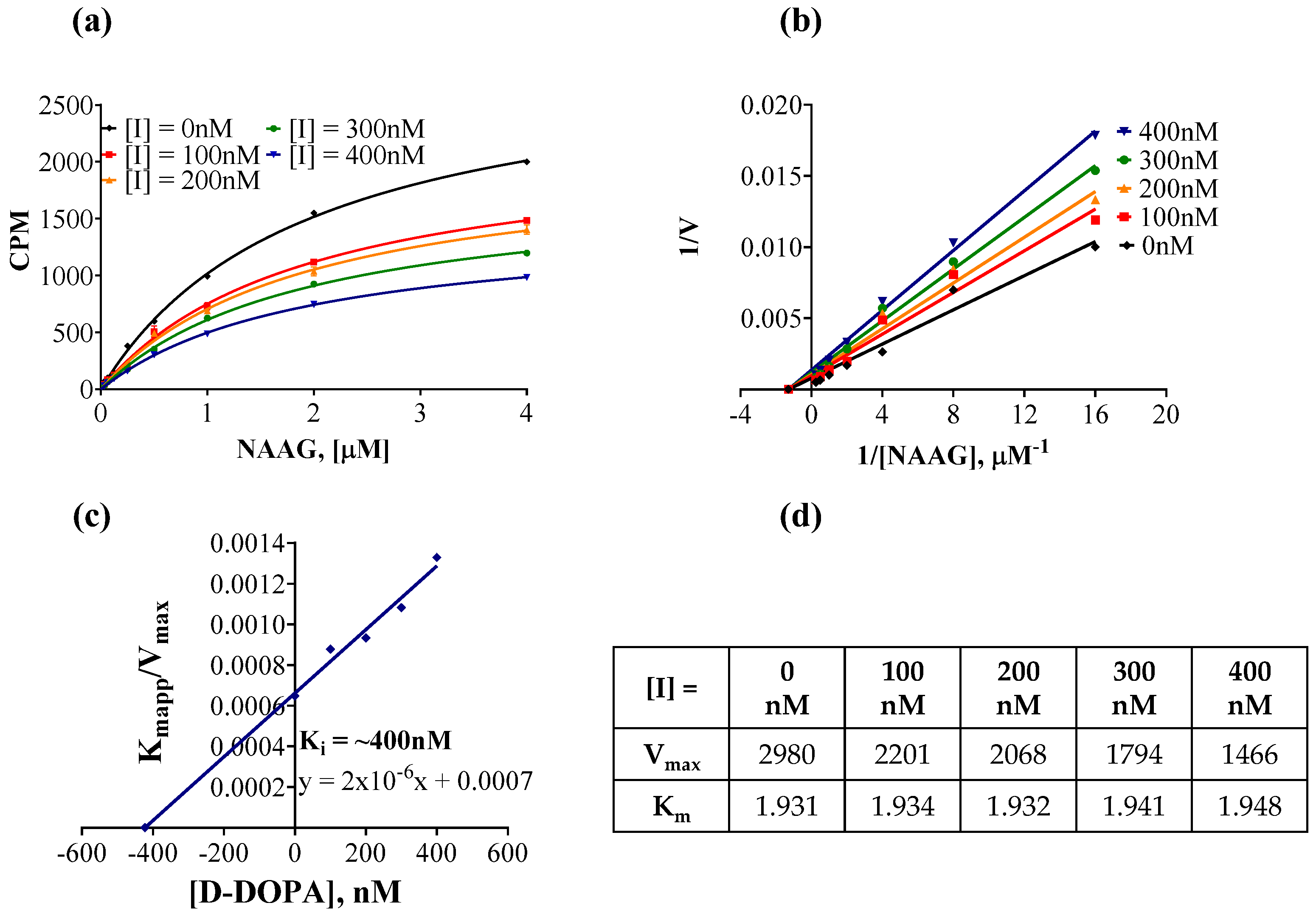
3.6. Characterization of D-DOPA’s Mode of GCPII Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Keefe, D.S.; Su, S.L.; Bacich, D.J.; Horiguchi, Y.; Luo, Y.; Powell, C.T.; Zandvliet, D.; Russell, P.J.; Molloy, P.L.; Nowak, N.J.; et al. Mapping, genomic organization and promoter analysis of the human prostate-specific membrane antigen gene. Biochim. Biophys. Acta 1998, 1443, 113–127. [Google Scholar] [CrossRef]
- Robinson, M.B.; Blakely, R.D.; Couto, R.; Coyle, J.T. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate. Identification and characterization of a novel N-acetylated alpha-linked acidic dipeptidase activity from rat brain. J. Biol. Chem. 1987, 262, 14498–14506. [Google Scholar] [CrossRef]
- Chang, S.S.; Gaudin, P.B.; Reuter, V.E.; Heston, W.D. Prostate-specific membrane antigen: Present and future applications. Urology 2000, 55, 622–629. [Google Scholar] [CrossRef]
- Pinto, J.T.; Suffoletto, B.P.; Berzin, T.M.; Qiao, C.H.; Lin, S.; Tong, W.P.; May, F.; Mukherjee, B.; Heston, W.D. Prostate-specific membrane antigen: A novel folate hydrolase in human prostatic carcinoma cells. Clin. Cancer Res. 1996, 2, 1445–1451. [Google Scholar]
- Zhao, R.; Matherly, L.H.; Goldman, I.D. Membrane transporters and folate homeostasis: Intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med. 2009, 11, e4. [Google Scholar] [CrossRef]
- Barinka, C.; Rojas, C.; Slusher, B.; Pomper, M. Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr. Med. Chem. 2012, 19, 856–870. [Google Scholar] [CrossRef]
- Guilarte, T.R.; Hammoud, D.A.; McGlothan, J.L.; Caffo, B.S.; Foss, C.A.; Kozikowski, A.P.; Pomper, M.G. Dysregulation of glutamate carboxypeptidase II in psychiatric disease. Schizophr. Res. 2008, 99, 324–332. [Google Scholar] [CrossRef]
- Yang, S.; Datta, D.; Elizabeth, W.; Duque, A.; Morozov, Y.M.; Arellano, J.; Slusher, B.S.; Wang, M.; Arnsten, A.F.T. Inhibition of glutamate-carboxypeptidase-II in dorsolateral prefrontal cortex: Potential therapeutic target for neuroinflammatory cognitive disorders. Mol. Psychiatry 2022, 1–12. [Google Scholar] [CrossRef]
- Ferraris, D.V.; Shukla, K.; Tsukamoto, T. Structure-activity relationships of glutamate carboxypeptidase II (GCPII) inhibitors. Curr. Med. Chem. 2012, 19, 1282–1294. [Google Scholar] [CrossRef]
- Schmidt, L.H.; Heitkotter, B.; Schulze, A.B.; Schliemann, C.; Steinestel, K.; Trautmann, M.; Marra, A.; Hillejan, L.; Mohr, M.; Evers, G.; et al. Prostate specific membrane antigen (PSMA) expression in non-small cell lung cancer. PLoS ONE 2017, 12, e0186280. [Google Scholar] [CrossRef]
- Conway, R.E.; Petrovic, N.; Li, Z.; Heston, W.; Wu, D.; Shapiro, L.H. Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol. Cell Biol. 2006, 26, 5310–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Song, B.; Zhu, W.; Xu, X.; Gong, Q.Q.; Morando, C.; Dassopoulos, T.; Newberry, R.D.; Hunt, S.R.; Li, E. An ileal Crohn’s disease gene signature based on whole human genome expression profiles of disease unaffected ileal mucosal biopsies. PLoS ONE 2012, 7, e37139. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Yanai, H.; Baram, L.; Elad, H.; Meirovithz, E.; Ofer, A.; Brazowski, E.; Tulchinsky, H.; Pasmanik-Chor, M.; Dotan, I. Gene expression profiles of ileal inflammatory bowel disease correlate with disease phenotype and advance understanding of its immunopathogenesis. Inflamm Bowel. Dis. 2013, 19, 2509–2521. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.L.; Abbas, A.R.; Lees, C.W.; Cornelius, J.; Toy, K.; Modrusan, Z.; Clark, H.F.; Arnott, I.D.; Penman, I.D.; Satsangi, J.; et al. Characterization of intestinal gene expression profiles in Crohn’s disease by genome-wide microarray analysis. Inflamm Bowel. Dis. 2010, 16, 1717–1728. [Google Scholar] [CrossRef]
- Rais, R.; Jiang, W.; Zhai, H.; Wozniak, K.M.; Stathis, M.; Hollinger, K.R.; Thomas, A.G.; Rojas, C.; Vornov, J.J.; Marohn, M.; et al. FOLH1/GCPII is elevated in IBD patients, and its inhibition ameliorates murine IBD abnormalities. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Evans, J.C.; Malhotra, M.; Cryan, J.F.; O’Driscoll, C.M. The therapeutic and diagnostic potential of the prostate specific membrane antigen/glutamate carboxypeptidase II (PSMA/GCPII) in cancer and neurological disease. Br. J. Pharmacol. 2016, 173, 3041–3079. [Google Scholar] [CrossRef]
- Vornov, J.J.; Hollinger, K.R.; Jackson, P.F.; Wozniak, K.M.; Farah, M.H.; Majer, P.; Rais, R.; Slusher, B.S. Chapter Nine-Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. In Advances in Pharmacology; Schwarcz, R., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 76, pp. 215–255. [Google Scholar]
- Majer, P.; Jancarik, A.; Krecmerova, M.; Tichy, T.; Tenora, L.; Wozniak, K.; Wu, Y.; Pommier, E.; Ferraris, D.; Rais, R.; et al. Discovery of Orally Available Prodrugs of the Glutamate Carboxypeptidase II (GCPII) Inhibitor 2-Phosphonomethylpentanedioic Acid (2-PMPA). J. Med. Chem. 2016, 59, 2810–2819. [Google Scholar] [CrossRef]
- Ferraris, D.V.; Majer, P.; Ni, C.; Slusher, C.E.; Rais, R.; Wu, Y.; Wozniak, K.M.; Alt, J.; Rojas, C.; Slusher, B.S.; et al. delta-Thiolactones as prodrugs of thiol-based glutamate carboxypeptidase II (GCPII) inhibitors. J. Med. Chem. 2014, 57, 243–247. [Google Scholar] [CrossRef]
- Rais, R.; Vavra, J.; Tichy, T.; Dash, R.P.; Gadiano, A.J.; Tenora, L.; Monincova, L.; Barinka, C.; Alt, J.; Zimmermann, S.C.; et al. Discovery of a para-Acetoxy-benzyl Ester Prodrug of a Hydroxamate-Based Glutamate Carboxypeptidase II Inhibitor as Oral Therapy for Neuropathic Pain. J. Med. Chem. 2017, 60, 7799–7809. [Google Scholar] [CrossRef]
- Dash, R.P.; Tichy, T.; Veeravalli, V.; Lam, J.; Alt, J.; Wu, Y.; Tenora, L.; Majer, P.; Slusher, B.S.; Rais, R. Enhanced Oral Bioavailability of 2-(Phosphonomethyl)-pentanedioic Acid (2-PMPA) from its (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-Based Prodrugs. Mol. Pharm. 2019, 16, 4292–4301. [Google Scholar] [CrossRef]
- Nedelcovych, M.; Dash, R.P.; Tenora, L.; Zimmermann, S.C.; Gadiano, A.J.; Garrett, C.; Alt, J.; Hollinger, K.R.; Pommier, E.; Jancarik, A.; et al. Enhanced Brain Delivery of 2-(Phosphonomethyl)pentanedioic Acid Following Intranasal Administration of Its gamma-Substituted Ester Prodrugs. Mol. Pharm. 2017, 14, 3248–3257. [Google Scholar] [CrossRef]
- Rais, R.; Wozniak, K.; Wu, Y.; Niwa, M.; Stathis, M.; Alt, J.; Giroux, M.; Sawa, A.; Rojas, C.; Slusher, B.S. Selective CNS Uptake of the GCP-II Inhibitor 2-PMPA following Intranasal Administration. PLoS ONE 2015, 10, e0131861. [Google Scholar] [CrossRef]
- Hollinger, K.R.; Sharma, A.; Tallon, C.; Lovell, L.; Thomas, A.G.; Zhu, X.; Wiseman, R.; Wu, Y.; Kambhampati, S.P.; Liaw, K.; et al. Dendrimer-2PMPA selectively blocks upregulated microglial GCPII activity and improves cognition in a mouse model of multiple sclerosis. Nanotheranostics 2022, 6, 126–142. [Google Scholar] [CrossRef]
- Arteaga Cabeza, O.; Zhang, Z.; Smith Khoury, E.; Sheldon, R.A.; Sharma, A.; Zhang, F.; Slusher, B.S.; Kannan, R.M.; Kannan, S.; Ferriero, D.M. Neuroprotective effects of a dendrimer-based glutamate carboxypeptidase inhibitor on superoxide dismutase transgenic mice after neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2021, 148, 105201. [Google Scholar] [CrossRef]
- Veldkamp, K.L.; Tubergen, P.J.; Swartz, M.A.; DeVries, J.T.; Tatko, C.D. Zinc binding with L-dopa peptides. Inorg. Chim. Acta 2017, 461, 120–126. [Google Scholar] [CrossRef]
- Rahman, F.; Nguyen, T.M.; Adekoya, O.A.; Campestre, C.; Tortorella, P.; Sylte, I.; Winberg, J.O. Inhibition of bacterial and human zinc-metalloproteases by bisphosphonate- and catechol-containing compounds. J. Enzym. Inhib. Med. Chem. 2021, 36, 819–830. [Google Scholar] [CrossRef]
- Kyriakou, S.; Mitsiogianni, M.; Mantso, T.; Cheung, W.; Todryk, S.; Veuger, S.; Pappa, A.; Tetard, D.; Panayiotidis, M.I. Anticancer activity of a novel methylated analogue of L-mimosine against an in vitro model of human malignant melanoma. Invest. New Drugs 2020, 38, 621–633. [Google Scholar] [CrossRef]
- Shchepin, R.; Moller, M.N.; Kim, H.Y.; Hatch, D.M.; Bartesaghi, S.; Kalyanaraman, B.; Radi, R.; Porter, N.A. Tyrosine-lipid peroxide adducts from radical termination: Para coupling and intramolecular Diels-Alder cyclization. J. Am. Chem. Soc. 2010, 132, 17490–17500. [Google Scholar] [CrossRef]
- Rojas, C.; Frazier, S.T.; Flanary, J.; Slusher, B.S. Kinetics and inhibition of glutamate carboxypeptidase II using a microplate assay. Anal. Biochem. 2002, 310, 50–54. [Google Scholar] [CrossRef]
- Zimmermann, S.C.; Tichy, T.; Vavra, J.; Dash, R.P.; Slusher, C.E.; Gadiano, A.J.; Wu, Y.; Jancarik, A.; Tenora, L.; Monincova, L.; et al. N-Substituted Prodrugs of Mebendazole Provide Improved Aqueous Solubility and Oral Bioavailability in Mice and Dogs. J. Med. Chem. 2018, 61, 3918–3929. [Google Scholar] [CrossRef]
- van Faassen, M.; Bischoff, R.; Eijkelenkamp, K.; de Jong, W.H.A.; van der Ley, C.P.; Kema, I.P. In Matrix Derivatization Combined with LC-MS/MS Results in Ultrasensitive Quantification of Plasma Free Metanephrines and Catecholamines. Anal. Chem. 2020, 92, 9072–9078. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Ma, X.; Chu, Y.; Li, S.; Guo, J.; Jia, Y.; Zhou, S.; Zhu, Y.; Liu, C. Simultaneous determination of caffeic acid and its major pharmacologically active metabolites in rat plasma by LC-MS/MS and its application in pharmacokinetic study. Biomed. Chromatogr. 2015, 29, 552–559. [Google Scholar] [CrossRef]
- Rahn, K.A.; Watkins, C.C.; Alt, J.; Rais, R.; Stathis, M.; Grishkan, I.; Crainiceau, C.M.; Pomper, M.G.; Rojas, C.; Pletnikov, M.V.; et al. Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20101–20106. [Google Scholar] [CrossRef]
- Maruyama, W.; Naoi, M.; Narabayashi, H. The metabolism of L-DOPA and L-threo-3,4-dihydroxyphenylserine and their effects on monoamines in the human brain: Analysis of the intraventricular fluid from parkinsonian patients. J. Neurol. Sci. 1996, 139, 141–148. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed]
- Peaston, R.T.; Weinkove, C. Measurement of catecholamines and their metabolites. Ann. Clin. Biochem. 2004, 41, 17–38. [Google Scholar] [CrossRef]
- Li, W.L.; Rossi, D.T.; Fountain, S.T. Development and validation of a semi-automated method for L-dopa and dopamine in rat plasma using electrospray LC/MS/MS. J. Pharm. Biomed. 2000, 24, 325–333. [Google Scholar] [CrossRef]
- Igarashi, K.; Hotta, K.; Kasuya, F.; Abe, K.; Sakoda, S. Determination of cabergoline and L-dopa in human plasma using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003, 792, 55–61. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Alany, R.G.; Chuang, V.; Wen, J.Y. Studies of the Rate Constant of L-DOPA Oxidation and Decarboxylation by HPLC. Chromatographia 2012, 75, 597–606. [Google Scholar] [CrossRef]
- Cho, Y.A.; Park, S.; Seo, O.N.; Jeong, S.W.; Lee, W.-K.; Kim, C.Y.; Kim, S.T.; Cho, M.J.; Shin, S.C. Development and validation of an LC–ESI–MS/MS method for simultaneous determination of levodopa, dopamine, L-α-methyldopa and 3-O-methyldopa in rat plasma. J. Pharm. Investig. 2012, 42, 361–368. [Google Scholar] [CrossRef]
- Lv, L.; Jiang, W.Z.; Zhou, S.Y.; Huang, X.Z.; Shi, X.X.; Lv, C.; Wu, L.L.; Xu, C.Y. LC-MS-MS Simultaneous Determination of l-Dopa and Its Prodrug l-Dopa n-Pentyl Ester Hydrochloride in Rat Plasma. Chromatographia 2010, 72, 239–243. [Google Scholar] [CrossRef]
- Wang, S.J.; Zeng, J.; Yang, B.K.; Zhong, Y.M. Bioavailability of caffeic acid in rats and its absorption properties in the Caco-2 cell model. Pharm. Biol. 2014, 52, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- de Vries, M.H.; Hamelijnck, M.A.; Hofman, G.A.; Koster, A.S.; Noordhoek, J. Decarboxylation of L-dopa in the rat isolated vascularly perfused small intestine: Contribution to systemic elimination and dose-dependent first pass effect. J. Pharm. Pharmacol. 1992, 44, 311–314. [Google Scholar] [CrossRef]
- Vieira-Coelho, M.A.; Soares-da-Silva, P. Dopamine formation, from its immediate precursor 3,4-dihydroxyphenylalanine, along the rat digestive tract. Fundam. Clin. Pharmacol. 1993, 7, 235–243. [Google Scholar] [CrossRef]
- Muller, T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Jenner, P.; Marsden, C.D. Peripheral pharmacokinetic handling and metabolism of L-dopa in the rat: The effect of route of administration and carbidopa pretreatment. J. Pharm. Pharmacol. 1991, 43, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Diederich, C.; Milakofsky, L.; Hare, T.A.; Hofford, J.M.; Dadmarz, M.; Vogel, W.H. Effects of L-DOPA/carbidopa administration on the levels of L-DOPA, other amino acids and related compounds in the plasma, brain and heart of the rat. Pharmacology 1997, 55, 109–116. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Vijayakumar, K.; Ramakrishnan, K.; Chakravarthy, V.S. A Multi-Scale Computational Model of Levodopa-Induced Toxicity in Parkinson’s Disease. Front. Neurosci. 2022, 16, 797127. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-induced neurotoxicity: The role of dopamine quinone formation and tyrosinase in a model of Parkinson’s disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Alexander, T.; Sortwell, C.E.; Sladek, C.D.; Roth, R.H.; Steece-Collier, K. Comparison of neurotoxicity following repeated administration of l-dopa, d-dopa and dopamine to embryonic mesencephalic dopamine neurons in cultures derived from Fisher 344 and Sprague-Dawley donors. Cell Transplant. 1997, 6, 309–315. [Google Scholar] [CrossRef]
- Abbott, A. Levodopa: The story so far. Nature 2010, 466, S6–S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoon, M.; Petzer, J.P.; Viljoen, F.; Petzer, A. The Design and Evaluation of an l-Dopa-Lazabemide Prodrug for the Treatment of Parkinson’s Disease. Molecules 2017, 22, 2076. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.; Siddiqui, A.; Silverman, P.B. Sodium benzoate differentially blocks circling induced by D-and L-dopa in the hemi-parkinsonian rat. Neurosci. Lett. 1996, 218, 145–148. [Google Scholar] [CrossRef]
- Wu, M.; Zhou, X.J.; Konno, R.; Wang, Y.X. D-dopa is unidirectionally converted to L-dopa by D-amino acid oxidase, followed by dopa transaminase. Clin. Exp. Pharmacol. Physiol. 2006, 33, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Karoum, F.; Freed, W.J.; Chuang, L.W.; Cannon-Spoor, E.; Wyatt, R.J.; Costa, E. D-dopa and L-dopa similarly elevate brain dopamine and produce turning behavior in rats. Brain Res. 1988, 440, 190–194. [Google Scholar] [CrossRef]
- Rais, R.; Thomas, A.G.; Wozniak, K.; Wu, Y.; Jaaro-Peled, H.; Sawa, A.; Strick, C.A.; Engle, S.J.; Brandon, N.J.; Rojas, C.; et al. Pharmacokinetics of oral D-serine in D-amino acid oxidase knockout mice. Drug Metab. Dispos. 2012, 40, 2067–2073. [Google Scholar] [CrossRef]
- Ferraris, D.; Duvall, B.; Ko, Y.S.; Thomas, A.G.; Rojas, C.; Majer, P.; Hashimoto, K.; Tsukamoto, T. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J. Med. Chem. 2008, 51, 3357–3359. [Google Scholar] [CrossRef]
- van der Post, J.P.; de Visser, S.J.; de Kam, M.L.; Woelfler, M.; Hilt, D.C.; Vornov, J.; Burak, E.S.; Bortey, E.; Slusher, B.S.; Limsakun, T.; et al. The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br. J. Clin. Pharmacol. 2005, 60, 128–136. [Google Scholar] [CrossRef]
- Vornov, J.J.; Wozniak, K.M.; Wu, Y.; Rojas, C.; Rais, R.; Slusher, B.S. Pharmacokinetics and pharmacodynamics of the glutamate carboxypeptidase II inhibitor 2-MPPA show prolonged alleviation of neuropathic pain through an indirect mechanism. J. Pharmacol. Exp. Ther. 2013, 346, 406–413. [Google Scholar] [CrossRef]
- Rais, R.; Hoover, R.; Wozniak, K.; Rudek, M.A.; Tsukamoto, T.; Alt, J.; Rojas, C.; Slusher, B.S. Reversible disulfide formation of the glutamate carboxypeptidase II inhibitor E2072 results in prolonged systemic exposures in vivo. Drug Metab. Dispos. 2012, 40, 2315–2323. [Google Scholar] [CrossRef]
- Sala, M.; Hollinger, K.R.; Thomas, A.G.; Dash, R.P.; Tallon, C.; Veeravalli, V.; Lovell, L.; Kogler, M.; Hrebabecky, H.; Prochazkova, E.; et al. Novel Human Neutral Sphingomyelinase 2 Inhibitors as Potential Therapeutics for Alzheimer’s Disease. J. Med. Chem. 2020, 63, 6028–6056. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.F.; Tays, K.L.; Maclin, K.M.; Ko, Y.S.; Li, W.; Vitharana, D.; Tsukamoto, T.; Stoermer, D.; Lu, X.C.; Wozniak, K.; et al. Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J. Med. Chem. 2001, 44, 4170–4175. [Google Scholar] [CrossRef]
- Jackson, P.F.; Cole, D.C.; Slusher, B.S.; Stetz, S.L.; Ross, L.E.; Donzanti, B.A.; Trainor, D.A. Design, synthesis, and biological activity of a potent inhibitor of the neuropeptidase N-acetylated alpha-linked acidic dipeptidase. J. Med. Chem. 1996, 39, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Majer, P.; Jackson, P.F.; Delahanty, G.; Grella, B.S.; Ko, Y.S.; Li, W.; Liu, Q.; Maclin, K.M.; Polakova, J.; Shaffer, K.A.; et al. Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: Discovery of an orally active GCP II inhibitor. J. Med. Chem. 2003, 46, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Zhang, J.; Nan, F.; Petukhov, P.A.; Grajkowska, E.; Wroblewski, J.T.; Yamamoto, T.; Bzdega, T.; Wroblewska, B.; Neale, J.H. Synthesis of urea-based inhibitors as active site probes of glutamate carboxypeptidase II: Efficacy as analgesic agents. J. Med. Chem. 2004, 47, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Stoermer, D.; Liu, Q.; Hall, M.R.; Flanary, J.M.; Thomas, A.G.; Rojas, C.; Slusher, B.S.; Tsukamoto, T. Synthesis and biological evaluation of hydroxamate-Based inhibitors of glutamate carboxypeptidase II. Bioorg Med. Chem. Lett. 2003, 13, 2097–2100. [Google Scholar] [CrossRef]
- Parellada, J.; Suarez, G.; Guinea, M. Inhibition of zinc metallopeptidases by flavonoids and related phenolic compounds: Structure-activity relationships. J. Enzym. Inhib. 1998, 13, 347–359. [Google Scholar] [CrossRef]
- Rahn, K.A.; Slusher, B.S.; Kaplin, A.I. Glutamate in CNS neurodegeneration and cognition and its regulation by GCPII inhibition. Curr Med. Chem. 2012, 19, 1335–1345. [Google Scholar] [CrossRef]
- Slusher, B.S.; Vornov, J.J.; Thomas, A.G.; Hurn, P.D.; Harukuni, I.; Bhardwaj, A.; Traystman, R.J.; Robinson, M.B.; Britton, P.; Lu, X.C.; et al. Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat. Med. 1999, 5, 1396–1402. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y.; Inagaki, T. Inhibition of spinal N-acetylated-alpha-linked acidic dipeptidase produces an antinociceptive effect in the rat formalin test. Neuroscience 2001, 102, 473–479. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nozaki-Taguchi, N.; Sakashita, Y. Spinal N-acetyl-alpha-linked acidic dipeptidase (NAALADase) inhibition attenuates mechanical allodynia induced by paw carrageenan injection in the rat. Brain Res. 2001, 909, 138–144. [Google Scholar] [CrossRef]
- Chen, S.R.; Wozniak, K.M.; Slusher, B.S.; Pan, H.L. Effect of 2-(phosphono-methyl)-pentanedioic acid on allodynia and afferent ectopic discharges in a rat model of neuropathic pain. J. Pharmacol. Exp. Ther. 2002, 300, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.J.; Sen, S.; Matthews, E.A.; Flatters, S.L.; Wozniak, K.M.; Slusher, B.S.; Dickenson, A.H. Effects of GCP-II inhibition on responses of dorsal horn neurones after inflammation and neuropathy: An electrophysiological study in the rat. Neuropeptides 2003, 37, 298–306. [Google Scholar] [CrossRef]
- Ghadge, G.D.; Slusher, B.S.; Bodner, A.; Canto, M.D.; Wozniak, K.; Thomas, A.G.; Rojas, C.; Tsukamoto, T.; Majer, P.; Miller, R.J.; et al. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc. Natl. Acad Sci. USA 2003, 100, 9554–9559. [Google Scholar] [CrossRef] [PubMed]
- Ghose, S.; Chin, R.; Gallegos, A.; Roberts, R.; Coyle, J.; Tamminga, C. Localization of NAAG-related gene expression deficits to the anterior hippocampus in schizophrenia. Schizophr. Res. 2009, 111, 131–137. [Google Scholar] [CrossRef]
- Vornov, J.J.; Peters, D.; Nedelcovych, M.; Hollinger, K.; Rais, R.; Slusher, B.S. Looking for Drugs in All the Wrong Places: Use of GCPII Inhibitors Outside the Brain. Neurochem. Res. 2020, 45, 1256–1267. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | IC50 (μM) | Compound | Structure | IC50 (μM) |
|---|---|---|---|---|---|
| 1 (L-DOPA) |  | 0.6 | 6 |  | 20 |
| 2 (Tyr) |  | 100 | 7 (Dopamine) |  | >100 |
| 3 |  | >100 | 8 |  | 4 |
| 4 |  | >100 | 9 (Caffeic Acid) |  | 0.3 |
| 5 |  | >100 | 10 (D-DOPA) |  | 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gori, S.S.; Thomas, A.G.; Pal, A.; Wiseman, R.; Ferraris, D.V.; Gao, R.-d.; Wu, Y.; Alt, J.; Tsukamoto, T.; Slusher, B.S.; et al. D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics 2022, 14, 2018. https://doi.org/10.3390/pharmaceutics14102018
Gori SS, Thomas AG, Pal A, Wiseman R, Ferraris DV, Gao R-d, Wu Y, Alt J, Tsukamoto T, Slusher BS, et al. D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics. 2022; 14(10):2018. https://doi.org/10.3390/pharmaceutics14102018
Chicago/Turabian StyleGori, Sadakatali S., Ajit G. Thomas, Arindom Pal, Robyn Wiseman, Dana V. Ferraris, Run-duo Gao, Ying Wu, Jesse Alt, Takashi Tsukamoto, Barbara S. Slusher, and et al. 2022. "D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II" Pharmaceutics 14, no. 10: 2018. https://doi.org/10.3390/pharmaceutics14102018
APA StyleGori, S. S., Thomas, A. G., Pal, A., Wiseman, R., Ferraris, D. V., Gao, R. -d., Wu, Y., Alt, J., Tsukamoto, T., Slusher, B. S., & Rais, R. (2022). D-DOPA Is a Potent, Orally Bioavailable, Allosteric Inhibitor of Glutamate Carboxypeptidase II. Pharmaceutics, 14(10), 2018. https://doi.org/10.3390/pharmaceutics14102018