

Diallyl Disulfide Induces Chemosensitization to Sorafenib, Autophagy, and Cell Cycle Arrest and Inhibits Invasion in Hepatocellular Carcinoma

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Culture Conditions
2.3. Protocol Design
2.4. LIVE/DEAD Assay
2.5. Clonogenic Survival Assay
2.6. Autophagy-Induced Cell Death
2.7. Analysis of Cell Cycle Phase
2.8. Cell Migration
2.9. Cell Invasion
2.10. Gene Expression
2.11. Protein Expression
2.12. Analysis of the Compound–Gene–Protein Interaction Network
2.13. Statistical Analysis
3. Results
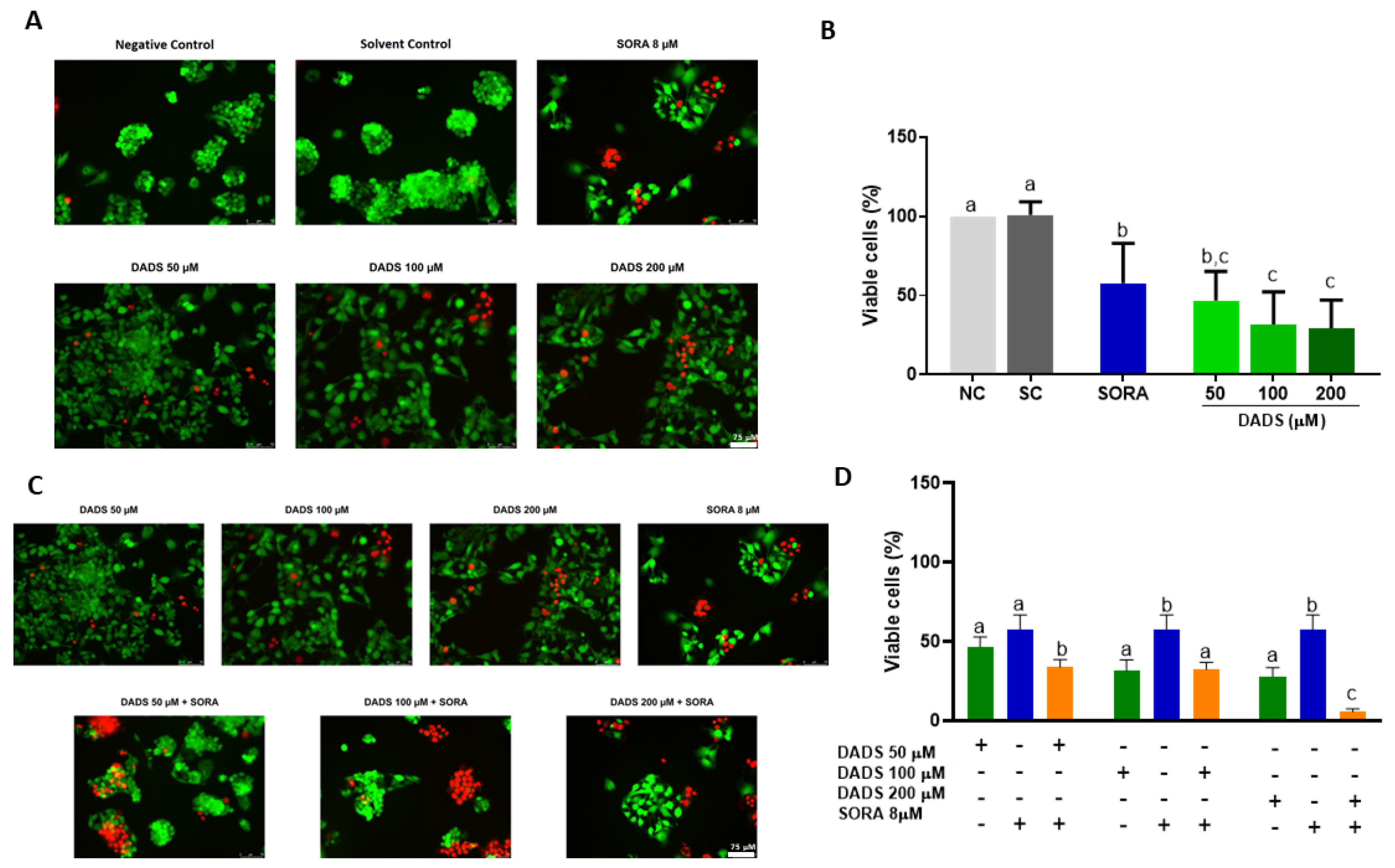
3.1. DADS and SORA Combination Effectively Induces Cytotoxicity in HepG2 and HUVEC Cells
3.2. DADS and SORA Combination Decreases Survival Fraction in HepG2 and HUVEC Cells
3.3. DADS and SORA Combination Modulates Autophagy-Mediated Cell Death
3.4. DADS and SORA Combination Modulates Cell Cycle
3.5. DADS and SORA Combination Effectively Suppresses Cell Migration and Invasion
3.6. DADS and SORA Combination Induces Expression of Genes and Proteins Related to Cell Death Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chacko, S.; Samanta, S. Hepatocellular carcinoma: A life-threatening disease. Biomed. Pharmacother. 2016, 84, 1679–1688. [Google Scholar] [CrossRef]
- Hsieh, T.C.; Wu, Y.C.; Sun, S.S.; Yen, K.Y.; Kao, C.H. Treating hepatocellular carcinoma with (90)Y-bearing microspheres: A review. Biomedicine 2016, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, O.; Gnoni, A.; Licchetta, A.; Longo, V.; Calabrese, A.; Argentiero, A.; Delcuratolo, S.; Solimando, A.G.; Casadei-Gardini, A.; Silvestris, N. Predictive and Prognostic Factors in HCC Patients Treated with Sorafenib. Medicina 2019, 55, 707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Hamid, N.M.; Abass, S.A.; Mohamed, A.A.; Muneam Hamid, D. Herbal management of hepatocellular carcinoma through cutting the pathways of the common risk factors. Biomed. Pharmacother. 2018, 107, 1246–1258. [Google Scholar] [CrossRef]
- Pezzuto, J.M. Plant-derived anticancer agents. Biochem. Pharmacol. 1997, 53, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Holbeck, S.; Sausville, E.A. Natural products and derivatives as leads to cell cycle pathway targets in cancer chemotherapy. Curr. Cancer Drug Targets 2002, 2, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef] [Green Version]
- Sak, K. Chemotherapy and dietary phytochemical agents. Chemother. Res. Pract. 2012, 2012, 282570. [Google Scholar] [CrossRef] [Green Version]
- Bahman, A.A.; Abaza, M.S.I.; Khoushiash, S.I.; Al-Attiyah, R.J. Sequencedependent effect of sorafenib in combination with natural phenolic compounds on hepatic cancer cells and the possible mechanism of action. Int. J. Mol. Med. 2018, 42, 1695–1715. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, K.; Gu, C.; Yu, G.; Zhao, D.; Mai, W.; Zhong, Y.; Liu, S.; Nie, Y.; Yang, H. Berberine, a natural plant alkaloid, synergistically sensitizes human liver cancer cells to sorafenib. Oncol. Rep. 2018, 40, 1525–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Su, Q. Molecular mechanisms for the anti-cancer effects of diallyl disulfide. Food Chem. Toxicol. 2013, 57, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Lin, J.; Su, J.; Oyang, L.; Wang, H.; Tan, S.; Tang, Y.; Chen, X.; Liu, W.; Luo, X.; et al. Diallyl disulfide inhibits colon cancer metastasis by suppressing Rac1-mediated epithelial-mesenchymal transition. OncoTargets Ther. 2019, 12, 5713–5728. [Google Scholar] [CrossRef] [Green Version]
- Shang, A.; Cao, S.Y.; Xu, X.Y.; Gan, R.Y.; Tang, G.Y.; Corke, H.; Mavumengwana, V.; Li, H.B. Bioactive Compounds and Biological Functions of Garlic (Allium sativum L.). Foods 2019, 8, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.A. Preclinical perspectives on garlic and cancer. J. Nutr. 2006, 136, 827S–831S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miroddi, M.; Calapai, F.; Calapai, G. Potential beneficial effects of garlic in oncohematology. Mini Rev. Med. Chem. 2011, 11, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.Y.; Yao, S.Q.; Zu, X.Y.; Huang, Z.X.; Liu, L.J.; Zhong, M.; Zhu, B.Y.; Tang, S.S.; Liao, D.F. Apoptosis induced by diallyl disulfide in human breast cancer cell line MCF-7. Acta Pharmacol. Sin. 2008, 29, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Song, J.D.; Lee, S.K.; Kim, K.M.; Park, S.E.; Park, S.J.; Kim, K.H.; Ahn, S.C.; Park, Y.C. Molecular mechanism of diallyl disulfide in cell cycle arrest and apoptosis in HCT-116 colon cancer cells. J. Biochem. Mol. Toxicol. 2009, 23, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Gunadharini, D.N.; Arunkumar, A.; Krishnamoorthy, G.; Muthuvel, R.; Vijayababu, M.R.; Kanagaraj, P.; Srinivasan, N.; Aruldhas, M.M.; Arunakaran, J. Antiproliferative effect of diallyl disulfide (DADS) on prostate cancer cell line LNCaP. Cell Biochem. Funct. 2006, 24, 407–412. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, Y.; Chen, X.; Shen, L.; Li, G.C.; Xu, M. Enhancement of diallyl disulfide-induced apoptosis by inhibitors of MAPKs in human HepG2 hepatoma cells. Biochem. Pharmacol. 2004, 68, 323–331. [Google Scholar] [CrossRef]
- Coriat, R.; Nicco, C.; Chereau, C.; Mir, O.; Alexandre, J.; Ropert, S.; Weill, B.; Chaussade, S.; Goldwasser, F.; Batteux, F. Sorafenib-induced hepatocellular carcinoma cell death depends on reactive oxygen species production in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 2284–2293. [Google Scholar] [CrossRef] [Green Version]
- Han, S.X.; Ma, J.L.; Lv, Y.; Huang, C.; Liang, H.H.; Duan, K.M. Secretory Transactivating Transcription-apoptin fusion protein induces apoptosis in hepatocellular carcinoma HepG2 cells. World J. Gastroenterol. 2008, 14, 3642–3649. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, K.; Mohamed, H.; Shah, A.M.; Yu, S.; Akhlaq, M.; Xiao, H.; Li, S.; Naz, T.; Nosheen, S.; Bai, X.; et al. In Vitro Anticancer Potential of Berberis lycium Royle Extracts against Human Hepatocarcinoma (HepG2) Cells. Biomed. Res. Int. 2020, 2020, 8256809. [Google Scholar] [CrossRef]
- Vakili Zahir, N.; Nakhjavani, M.; Hajian, P.; Shirazi, F.H.; Mirzaei, H. Evaluation of Silibinin Effects on the Viability of HepG2 (Human hepatocellular liver carcinoma) and HUVEC (Human Umbilical Vein Endothelial) Cell Lines. Iran J. Pharm. Res. 2018, 17, 261–267. [Google Scholar] [PubMed]
- Xiang, L.; Chi, T.; Tang, Q.; Yang, X.; Ou, M.; Chen, X.; Yu, X.; Chen, J.; Ho, R.J.; Shao, J.; et al. A pentacyclic triterpene natural product, ursolic acid and its prodrug US597 inhibit targets within cell adhesion pathway and prevent cancer metastasis. Oncotarget 2015, 6, 9295–9312. [Google Scholar] [CrossRef] [Green Version]
- Yurdacan, B.; Egeli, U.; Eskiler, G.G.; Eryilmaz, I.E.; Cecener, G.; Tunca, B. The role of usnic acid-induced apoptosis and autophagy in hepatocellular carcinoma. Hum. Exp. Toxicol. 2019, 38, 201–215. [Google Scholar] [CrossRef]
- Bal-Price, A.; Coecke, S. Guidance on Good Cell Culture Practice (GCCP). Neuromethods 2011, 56, 1–25. [Google Scholar] [CrossRef]
- Edginton, A.N.; Zimmerman, E.I.; Vasilyeva, A.; Baker, S.D.; Panetta, J.C. Sorafenib metabolism, transport, and enterohepatic recycling: Physiologically based modeling and simulation in mice. Cancer Chemother. Pharmacol. 2016, 77, 1039–1052. [Google Scholar] [CrossRef] [Green Version]
- Tandia, M.; Mhiri, A.; Paule, B.; Saffroy, R.; Cailliez, V.; Noe, G.; Farinotti, R.; Bonhomme-Faivre, L. Correlation between clinical response to sorafenib in hepatocellular carcinoma treatment and polymorphisms of P-glycoprotein (ABCB1) and of breast cancer resistance protein (ABCG2): Monocentric study. Cancer Chemother. Pharmacol. 2017, 79, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Auger, J.; Ginies, C.; Siess, M.H.; Teyssier, C. In vivo metabolism of diallyl disulphide in the rat: Identification of two new metabolites. Xenobiotica 2002, 32, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Ren, F.; Ma, H.; Xu, M. The roles of p38MAPK and caspase-3 in DADS-induced apoptosis in human HepG2 cells. J. Exp. Clin. Cancer Res. 2010, 29, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iciek, M.; Kwiecien, I.; Chwatko, G.; Sokolowska-Jezewicz, M.; Kowalczyk-Pachel, D.; Rokita, H. The effects of garlic-derived sulfur compounds on cell proliferation, caspase 3 activity, thiol levels and anaerobic sulfur metabolism in human hepatoblastoma HepG2 cells. Cell Biochem. Funct. 2012, 30, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of cell cycle by flow cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutasim, K.A.; Nystrom, M.L.; Thomas, G.J. Cell migration and invasion assays. Methods Mol. Biol. 2011, 731, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Antoni, L.; Sodha, N.; Collins, I.; Garrett, M.D. CHK2 kinase: Cancer susceptibility and cancer therapy-two sides of the same coin? Nat. Rev. Cancer 2007, 7, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Jiang, G.; Tang, J.; Wang, H.; Feng, G.; Chen, F.; Tu, Z.; Liu, G.; Zhao, Y.; Peng, M.J.; et al. c-Fos over-expression promotes radioresistance and predicts poor prognosis in malignant glioma. Oncotarget 2016, 7, 65946–65956. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef]
- Littlepage, L.E.; Sternlicht, M.D.; Rougier, N.; Phillips, J.; Gallo, E.; Yu, Y.; Williams, K.; Brenot, A.; Gordon, J.I.; Werb, Z. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010, 70, 2224–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, A.H.; Gao, B.T.; Goldsmith, Z.K.; Irvine, A.S.; Saleh, N.; Lee, R.P.; Lendermon, J.B.; Bheemreddy, R.; Zhang, Q.; Brennan, R.C.; et al. Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in in vitro models of retinoblastoma. BMC Cancer 2017, 17, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.C.; Kim, S.H.; Baek, H.S.; Moon, C.; Kang, S.S.; Kim, S.H.; Kim, Y.B.; Shin, I.S.; Kim, J.C. The involvement of Nrf2 in the protective effects of diallyl disulfide on carbon tetrachloride-induced hepatic oxidative damage and inflammatory response in rats. Food Chem. Toxicol. 2014, 63, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death Dis. 2019, 10, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.X.; Zhu, Q.G.; Zhang, S.M.; Guan, L.; Li, T.; Zhang, L.; Wang, S.Y.; Ren, W.L.; Chen, X.M.; Zhao, J.; et al. Precision medicine for hepatocellular carcinoma: Driver mutations and targeted therapy. Oncotarget 2017, 8, 55715–55730. [Google Scholar] [CrossRef]
- Wu, W.D.; Chen, P.S.; Omar, H.A.; Arafa, E.A.; Pan, H.W.; Jeng, J.; Hung, J.H. Antrodia cinnamomea boosts the anti-tumor activity of sorafenib in xenograft models of human hepatocellular carcinoma. Sci. Rep. 2018, 8, 12914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmageed, M.M.; El-Naga, R.N.; El-Demerdash, E.; Elmazar, M.M. Indole-3- carbinol enhances sorafenib cytotoxicity in hepatocellular carcinoma cells: A mechanistic study. Sci. Rep. 2016, 6, 32733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celano, M.; Maggisano, V.; Bulotta, S.; Allegri, L.; Pecce, V.; Abballe, L.; Damante, G.; Russo, D. Quercetin improves the effects of sorafenib on growth and migration of thyroid cancer cells. Endocrine 2020, 67, 496–498. [Google Scholar] [CrossRef]
- Chen, W.L.; Hsieh, C.L.; Chen, J.H.; Huang, C.S.; Chen, W.T.; Kuo, Y.C.; Chen, C.Y.; Hsu, F.T. Amentoflavone enhances sorafenib-induced apoptosis through extrinsic and intrinsic pathways in sorafenib-resistant hepatocellular carcinoma SK-Hep1 cells in vitro. Oncol. Lett. 2017, 14, 3229–3234. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Deng, C.; Dang, F. Synergistic antitumor effect of resveratrol and sorafenib on hepatocellular carcinoma through PKA/AMPK/eEF2K pathway. Food Nutr. Res. 2021, 65. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.K.; Chen, T.Y.; Lin, J.H.; Wang, C.Y.; Wang, B.S. Protective effects of five allium derived organosulfur compounds against mutation and oxidation. Food Chem. 2016, 197, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, A.; Jafari, D.; Kamarul, T.; Bagheri, A.; Sharifi, A.M. Evaluating the Protective Effects and Mechanisms of Diallyl Disulfide on Interlukin-1beta-Induced Oxidative Stress and Mitochondrial Apoptotic Signaling Pathways in Cultured Chondrocytes. J. Cell. Biochem. 2017, 118, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Day, R.M.; Suzuki, Y.J. Cell proliferation, reactive oxygen and cellular glutathione. Dose Response 2006, 3, 425–442. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Chiang, I.T.; Hsu, F.T. Protein Kinase B Inactivation Is Associated with Magnolol-Enhanced Therapeutic Efficacy of Sorafenib in Hepatocellular Carcinoma In Vitro and In Vivo. Cancers 2019, 12, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Feng, C.; Han, L.; Ma, Y.; Jiao, Y.; Wang, J.; Jia, L.; Jing, F.; Gao, X.; Zhang, Y.; et al. Diallyl disulfide inhibits the metastasis of type esophagealgastric junction adenocarcinoma cells via NF-kappaB and PI3K/AKT signaling pathways in vitro. Oncol. Rep. 2018, 39, 784–794. [Google Scholar] [CrossRef]
- Maher, J.M.; Dieter, M.Z.; Aleksunes, L.M.; Slitt, A.L.; Guo, G.; Tanaka, Y.; Scheffer, G.L.; Chan, J.Y.; Manautou, J.E.; Chen, Y.; et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology 2007, 46, 1597–1610. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Santisteban, I.; Llopis, A.; Krenning, L.; Vallejo-Rodriguez, J.; van den Broek, B.; Zubiaga, A.M.; Medema, R.H. Sustained CHK2 activity, but not ATM activity, is critical to maintain a G1 arrest after DNA damage in untransformed cells. BMC Biol. 2021, 19, 35. [Google Scholar] [CrossRef] [PubMed]
- Aliouat-Denis, C.M.; Dendouga, N.; Van den Wyngaert, I.; Goehlmann, H.; Steller, U.; van de Weyer, I.; Van Slycken, N.; Andries, L.; Kass, S.; Luyten, W.; et al. p53-independent regulation of p21Waf1/Cip1 expression and senescence by Chk2. Mol. Cancer Res. 2005, 3, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, J.; Hu, X.; Liu, S.; He, B. Prognostic and Therapeutic Values of Tumor Necrosis Factor-Alpha in Hepatocellular Carcinoma. Med. Sci. Monit. 2016, 22, 3694–3704. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.C.; Ning, F.; Wang, H.F.; Chen, D.Y.; Cai, Y.N.; Sheng, H.Y.; Lash, G.E.; Liu, L.; Du, J. Epidermal growth factor and tumor necrosis factor alpha cooperatively promote the motility of hepatocellular carcinoma cell lines via synergistic induction of fibronectin by NF-kappaB/p65. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2568–2582. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Luo, X.; Li, W.; Zhong, J.; Cao, J.; Zhu, S.; Chen, X.; Zhou, R.; Shang, C.; Chen, Y. TNF-alpha is a potential therapeutic target to overcome sorafenib resistance in hepatocellular carcinoma. EBioMedicine 2019, 40, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besancon, F.; Bauvy, C.; Souquere, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, M.J.; Wang, D.; McDonnell, M.A.; Lehmann, U.; Kelekar, A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007, 14, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Dourmashkin, R.R.; Allen, P.D.; Gray, A.B.; Newland, A.C.; Kelsey, S.M. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997, 98, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copple, I.M.; Lister, A.; Obeng, A.D.; Kitteringham, N.R.; Jenkins, R.E.; Layfield, R.; Foster, B.J.; Goldring, C.E.; Park, B.K. Physical and functional interaction of sequestosome 1 with Keap1 regulates the Keap1-Nrf2 cell defense pathway. J. Biol. Chem. 2010, 285, 16782–16788. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, J.M.; Hallahan, A.R. p38 MAP kinase: A convergence point in cancer therapy. Trends Mol. Med. 2004, 10, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Kim, N.D.; Kim, G.Y.; Hwang, H.J.; Kim, B.W.; Kim, W.J.; Choi, Y.H. Inhibitory effects of diallyl disulfide on the production of inflammatory mediators and cytokines in lipopolysaccharide-activated BV2 microglia. Toxicol. Appl. Pharmacol. 2012, 262, 177–184. [Google Scholar] [CrossRef]
- Nakagawa, H.; Tsuta, K.; Kiuchi, K.; Senzaki, H.; Tanaka, K.; Hioki, K.; Tsubura, A. Growth inhibitory effects of diallyl disulfide on human breast cancer cell lines. Carcinogenesis 2001, 22, 891–897. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, A.R.T.; Tuttis, K.; Santos, P.W.d.S.; Aissa, A.F.; Antunes, L.M.G. Diallyl Disulfide Induces Chemosensitization to Sorafenib, Autophagy, and Cell Cycle Arrest and Inhibits Invasion in Hepatocellular Carcinoma. Pharmaceutics 2022, 14, 2582. https://doi.org/10.3390/pharmaceutics14122582
Machado ART, Tuttis K, Santos PWdS, Aissa AF, Antunes LMG. Diallyl Disulfide Induces Chemosensitization to Sorafenib, Autophagy, and Cell Cycle Arrest and Inhibits Invasion in Hepatocellular Carcinoma. Pharmaceutics. 2022; 14(12):2582. https://doi.org/10.3390/pharmaceutics14122582
Chicago/Turabian StyleMachado, Ana Rita Thomazela, Katiuska Tuttis, Patrick Wellington da Silva Santos, Alexandre Ferro Aissa, and Lusânia Maria Greggi Antunes. 2022. "Diallyl Disulfide Induces Chemosensitization to Sorafenib, Autophagy, and Cell Cycle Arrest and Inhibits Invasion in Hepatocellular Carcinoma" Pharmaceutics 14, no. 12: 2582. https://doi.org/10.3390/pharmaceutics14122582
APA StyleMachado, A. R. T., Tuttis, K., Santos, P. W. d. S., Aissa, A. F., & Antunes, L. M. G. (2022). Diallyl Disulfide Induces Chemosensitization to Sorafenib, Autophagy, and Cell Cycle Arrest and Inhibits Invasion in Hepatocellular Carcinoma. Pharmaceutics, 14(12), 2582. https://doi.org/10.3390/pharmaceutics14122582