

Insights into Lipid-Based Delivery Nanosystems of Protein-Tyrosine Kinase Inhibitors for Cancer Therapy


Abstract
:1. Introduction
2. Protein Kinase Inhibitors
2.1. Proteins with Kinase Activity
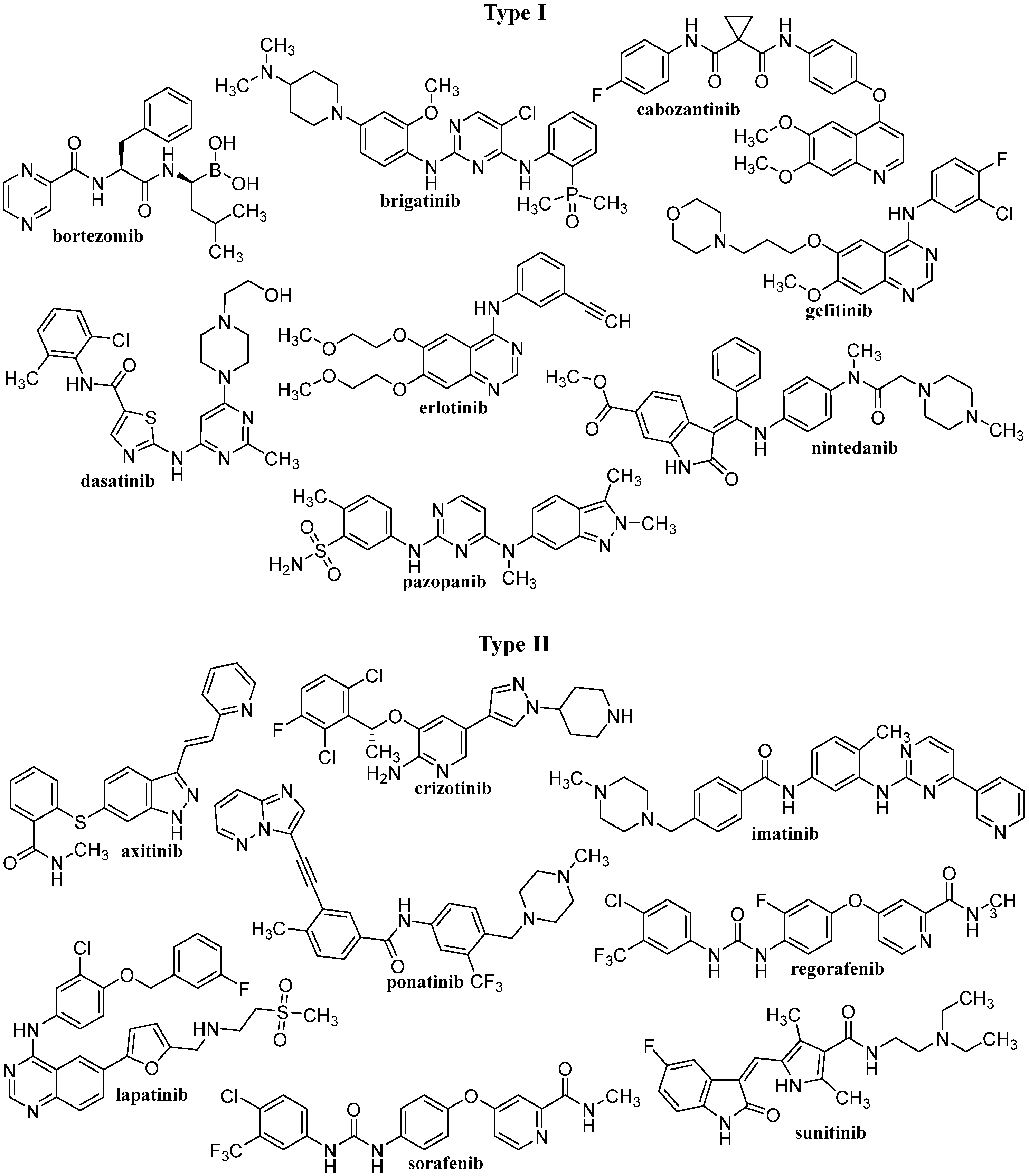
2.2. Tyrosine Kinase Inhibitors
3. Nanoformulations of TKIs
3.1. Self-Nanoemulsifying Drug Delivery System
3.1.1. In Vitro Tested SNEDDS-Based TKI Formulations
3.1.2. In Vivo Tested SNEDDS-Based TKI Formulations
3.2. Liposomes
3.2.1. In Vitro Tested Liposomal TKI Formulations
3.2.2. In Vivo Tested Liposomal TKI Formulations
3.3. Solid Lipid Nanoparticles
3.3.1. SLN-Based TKI Formulations Tested In Vitro
3.3.2. In Vivo Tested SLN-Based TKI Formulations
3.4. Nanostructured Lipid Carriers
3.4.1. NLC-Based TKI Formulations Tested In Vitro
3.4.2. In Vivo Tested NLC-Based TKI Formulations
4. Clinical Applicability of TKI Lipid-Based Delivery Nanosystems
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO–Cancer, 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 30 October 2022).
- WHO–Global Health Observatory. Cancer Mortality and Morbidity. Available online: http://www.who.int/gho/ncd/mortality_morbidity/cancer_text/en/index.html (accessed on 30 October 2022).
- Baguley, B.C. A brief history of cancer chemotherapy. In Anticancer Drug Development; Baguley, B.C., Kerr, D.J., Eds.; Academic Press: London, UK, 2002. [Google Scholar]
- Goodman, L.S.; Wintrobe, M.M.; Damesek, W.; Goodman, M.J.; Gilman, A.; McLennan, M.T. Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodginkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. JAMA 1984, 251, 2255–2261. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A. The initial clinical trial of nitrogen mustard. Am. J. Surg. 1963, 105, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Kumar, S.; Prasad, D.N.; Bhardwaj, T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018, 151, 401–433. [Google Scholar] [PubMed]
- DrugBank: Cyclophosphamide. Available online: http://www.drugbank.ca/drugs/DB00531 (accessed on 30 October 2022).
- DrugBank: Folic Acid. Available online: http://www.drugbank.ca/drugs/DB00158 (accessed on 30 October 2022).
- Wills, L.; Clutterbuck, P.W.; Evans, B.D.F. A new factor in the production and cure of certain macrocytic anaemias. Lancet 1937, 229, 311–314. [Google Scholar] [CrossRef]
- Wills, L.; Clutterbuck, P.W.; Evans, B.D.F. CCLX. A new factor in the production and cure of macrocytic anaemias and its relation to other haemopoetic principles curative in pernicious anaemia. Biochem. J. 1937, 31, 2136–2147. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.R. A tribute to Sydney Farber–the father of modern chemotherapy. Brit. J. Haematol. 2006, 134, 20–26. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F., Jr.; Wolff, J.A. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (aminopterin). N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- DrugBank: Methotrexate. Available online: http://www.drugbank.ca/drugs/DB00563 (accessed on 30 October 2022).
- Chabner, B.A.; Roberts, T.G. Chemotherapy and war on cancer. Nat. Rew. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- GlaxoSmithKline: History and Heritage. Available online: https://www.gsk.com/en-gb/company/history-and-heritage (accessed on 1 November 2022).
- Hitchings, G.H.; Elion, G.B.; Falco, E.A.; Russell, P.B.; Vanderwerff, H. Studies on analogs of purines and pyrimidines. Ann. N. Y. Acad. Sci. 1950, 52, 1318. [Google Scholar] [CrossRef]
- DrugBank: Mercaptopurine. Available online: http://www.drugbank.ca/drugs/DB01030 (accessed on 30 October 2022).
- Skipper, H.E.; Thomson, J.R.; Elion, G.B.; Hitchings, G.H. Observatory on the anticancer activity of 6-mercaptopurine. Cancer Res. 1954, 14, 294–298. [Google Scholar]
- DrugBank: Fluorouracil. Available online: http://www.drugbank.ca/drugs/DB00544 (accessed on 30 October 2022).
- National Cancer Institute. Developmental Therapeutics Program: DTP 50th Anniversary Timeline. Available online: https://dtp.cancer.gov/timeline/noflash/index.htm (accessed on 30 October 2022).
- Roche, V.F.; Zito, S.W.; Lemke, T.; Williams, D.A. Foye’s Principles of Medicinal Chemistry, 8th ed.; Wolters Kluwer: Baltimore, MD, USA, 2019. [Google Scholar]
- WHO Collaborating Centre for Drug Statistics Methodology: International Language for Drug Utilization Research ATC/DDD. Available online: http://www.whocc.no (accessed on 30 October 2022).
- Zahavi, D.; Weiner, L. Monoclonal antibodies in cancer therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Sig. Transduct. Target Ther. 2022, 7, 39. [Google Scholar]
- Bologna, M.; Vicentini, C.; Muzi, P.; Pace, G.; Angelucci, A. Cancer multitarget pharmacology in prostate tumors: Tyrosine kinase inhibitors and beyond. Curr. Med. Chem. 2011, 18, 2827. [Google Scholar] [CrossRef]
- Roskoski, R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [PubMed]
- LiverTox. Protein kinase inhibitors. In Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548591 (accessed on 30 October 2022).
- Fabbro, D.; Ruetz, S.; Buchdunger, E.; Cowan-Jacob, S.W.; Fendrich, G.; Liebetanz, J.; Mestan, J.; O’Reilly, T.; Traxler, P.; Chaudhuri, B.; et al. Protein kinases as targets for anticancer agents: From inhibitors to useful drugs. Pharmacol. Ther. 2002, 93, 79–98. [Google Scholar] [CrossRef]
- Sierra, J.R.; Cepero, V.; Giordano, S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol. Cancer 2010, 9, 75. [Google Scholar] [PubMed] [Green Version]
- Mongre, R.K.; Mishra, C.B.; Shukla, A.K.; Prakash, A.; Jung, S.; Ashraf-Uz-Zaman, M.; Lee, M.S. Emerging importance of tyrosine kinase inhibitors against cancer: Quo vadis to cure? Int. J. Mol. Sci. 2021, 22, 11659. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespee, R.; Sounni, N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol. Oncol. 2020, 13, 143. [Google Scholar]
- Kral, V.; Jampilek, J.; Havlicek, J.; Brusova, H.; Pekarek, T. Dosage Forms of Tyrosine Kinase Inhibitors. WO/2010/081443 A2, 22 July 2010.
- Kral, V.; Brusova, H.; Jampilek, J.; Havlicek, J.; Pekarek, T.; Tkadlecova, M. Imatinib Mesylate Polymorphs Generated by Crystallization in Aqueous Inorganic Salt Solutions. WO/2011/023146 A1, 3 March 2011.
- Yang, T.; Zhai, J.; Hu, D.; Yang, R.; Wang, G.; Li, Y.; Liang, G. “Targeting design” of nanoparticles in tumor therapy. Pharmaceutics 2022, 14, 1919. [Google Scholar] [CrossRef]
- Yin, Y.L.; Yuan, X.; Gao, H.; Yang, Q. Nanoformulations of small molecule protein tyrosine kinases inhibitors potentiate targeted cancer therapy. Int. J. Pharm. 2020, 573, 118785. [Google Scholar] [CrossRef] [PubMed]
- Foroughi-Nia, B.; Barar, J.; Memar, M.Y.; Aghanejad, A.; Davaran, S. Progresses in polymeric nanoparticles for delivery of tyrosine kinase inhibitors. Life Sci. 2021, 278, 119642. [Google Scholar] [CrossRef] [PubMed]
- Smidova, V.; Michalek, P.; Goliasova, Z.; Eckschlager, T.; Hodek, P.; Adam, V.; Heger, Z. Nanomedicine of tyrosine kinase inhibitors. Theranostics 2021, 11, 1546–1567. [Google Scholar] [CrossRef]
- Russo, E.; Spallarossa, A.; Tasso, B.; Villa, C.; Brullo, C. Nanotechnology of tyrosine kinase inhibitors in cancer therapy: A perspective. Int. J. Mol. Sci. 2021, 22, 6538. [Google Scholar]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Vasantha Rupasinghe, H.P. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [PubMed] [Green Version]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Krieger, M.; Bretscher, A.; Ploegh, H.; Amon, A.; Martin, K.C. Molecular Cell Biology, 8th ed.; W.H. Freeman and Macmillan: New York NY, USA, 2016. [Google Scholar]
- DrugBank: Imatinib. Available online: http://www.drugbank.ca/drugs/DB00619 (accessed on 30 October 2022).
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A History of chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.; Cervera, J.; Such, E.; Barragan, E.; Bolufer, P.; Fuster, O.; Collado, R.; Martinez, J.; Sanz, M.A. Complex variant t(9;22) chromosome translocations in five cases of chronic myeloid leukemia. Adv. Hematol. 2009, 2009, 187125. [Google Scholar] [CrossRef] [Green Version]
- Druker, B.J.; Lydon, N.B. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic myelogenous leukemia. J. Clin. Investig. 2000, 105, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.R.; Wu, Y.M.; Lin, S.F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [Green Version]
- Gocek, E.; Moulas, A.N.; Studzinski, G.P. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit. Rev. Clin. Lab. Sci. 2014, 51, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Murray, R.K.; Granner, D.K.; Mayes, P.A.; Rodwell, V.W. Harper’s Illustrated Biochemistry, 26th ed; Lange Medical Books/McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Secko, D. Proteinphosphorylation: A global regulator of cellular activity. Sci. Creat. Quart. 2011, 6, 7–12. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.C.; Qi, R.Z.; Paudel, H.; Zhu, H.J. Regulation and function of protein kinases and phosphatases. Enzym. Res. 2011, 2011, 794089. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor tyrosine kinase-targeted cancer therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Receptor Tyrosine Kinases (RTKs). IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=304 (accessed on 25 October 2022).
- McDonell, L.M.; Kernohan, K.D.; Boycott, K.M.; Sawyer, S.L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: Two sides of the same coin. Hum. Mol. Genet. 2015, 24, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K. Protein Kinases-Promising Targets for Anticancer Drug Research; IntechOpen: Rijeka, Croatia, 2021; Available online: https://www.intechopen.com/books/8977 (accessed on 30 October 2022).
- Ren, H. Tyrosine Kinases as Druggable Targets in Cancer; IntechOpen: Rijeka, Croatia, 2019; Available online: https://www.intechopen.com/books/8660 (accessed on 30 October 2022).
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cance with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Cicenas, J.; Zalyte, E.; Bairoch, A.; Gaudet, P. Kinases and cancer. Cancers 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojjat-Farsangi, M. Targeting non-receptor tyrosine kinases using small molecule inhibitors: An overview of recent advances. J. Drug Target. 2016, 24, 192–211. [Google Scholar] [CrossRef]
- Thomson, R.J.; Moshirfar, M.; Ronquillo, Y. Tyrosine Kinase Inhibitors; StatPearls Publishing: Tampa, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK563322 (accessed on 30 October 2022).
- Bhatia, R.; Singh, R.K. Introductory chapter: Protein kinases as promising targets for drug design against cancer. In Protein Kinases; Singh, R.K., Ed.; IntechOpen: Rijeka, Croatia, 2021; Available online: https://www.intechopen.com/chapters/78859 (accessed on 30 October 2022).
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Ann. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, D.; Cowan-Jacob, S.W.; Moebitz, H. Ten things you should know about protein kinases: IUPHAR Review 14. Br. J. Pharmacol. 2015, 172, 2675–2700. [Google Scholar] [PubMed] [Green Version]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door”: Exploiting the active kinase conformation. J. Med. Chem. 2009, 53, 2681–2694. [Google Scholar] [CrossRef]
- Rimassa, L.; Danesi, R.; Pressiani, T.; Merle, P. Management of adverse events associated with tyrosine kinase inhibitors: Improving outcomes for patients with hepatocellular carcinoma. Cancer Treat. Rev. 2019, 77, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Kantarjian, H.; Boddu, P.C.; Nogueras-Gonzalez, G.M.; Verstovsek, S.; Garcia-Manero, G.; Borthakur, G.; Sasaki, K.; Kadia, T.M.; Sam, P.; et al. Analysis of cardiovascular and arteriothrombotic adverse events in chronic-phase CML patients after frontline TKIs. Blood Adv. 2019, 26, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Basak, D.; Arrighi, S.; Darwiche, Y.; Deb, S. Comparison of anticancer drug toxicities: Paradigm shift in adverse effect profile. Life 2022, 12, 48. [Google Scholar] [CrossRef]
- Eisenmann, E.D.; Talebi, Z.; Sparreboom, A.; Baker, S.D. Boosting the oral bioavailability of anticancer drugs through intentional drug-drug interactions. Basic Clin. Pharmacol. Toxicol. 2022, 130, 23–35. [Google Scholar] [CrossRef]
- Stuurman, F.E.; Nuijen, B.; Beijnen, J.H.; Schellens, J.H. Oral anticancer drugs: Mechanisms of low bioavailability and strategies for improvement. Clin. Pharmacokinet. 2013, 52, 399–414. [Google Scholar] [PubMed]
- Podeszwa, B.; Niedbala, H.; Polanski, J.; Musiol, R.; Tabak, D.; Finster, J.; Serafin, K.; Milczarek, M.; Wietrzyk, J.; Boryczka, S.; et al. Investigating the antiproliferative activity of quinoline-5,8-diones and styrylquinolinecarboxylic acids on tumor cell lines. Bioorg. Med. Chem. Lett. 2007, 17, 6138–6141. [Google Scholar] [CrossRef]
- Mrozek-Wilczkiewicz, A.; Kalinowski, D.S.; Musiol, R.; Finster, J.; Szurko, A.; Serafin, K.; Knas, M.; Kamalapuram, S.K.; Kovacevic, Z.; Jampilek, J.; et al. Investigating the anti-proliferative activity of styrylazanaphthalenes and azanaphthalenediones. Bioorg. Med. Chem. 2010, 18, 2664–2671. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured lipid carriers for delivery of chemotherapeutics: A review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svenson, S. What nanomedicine in the clinic right now really forms nanoparticles? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Jampilek, J.; Kralova, K. Application of nanobioformulations for controlled release and targeted biodistribution of drugs. In Nanobiomaterials: Applications in Drug Delivery; Sharma, A.K., Keservani, R.K., Kesharwani, R.K., Eds.; CRC Press: Warentown, NJ, USA, 2018; pp. 131–208. [Google Scholar]
- Jampilek, J.; Kralova, K. Nanotechnology based formulations for drug targeting to central nervous system. In Nanoparticulate Drug Delivery Systems; Keservani, R.K., Sharma, A.K., Eds.; Apple Academic Press & CRC Press: Warentown, NJ, USA, 2019; pp. 151–220. [Google Scholar]
- Jampilek, J.; Kralova, K. Recent Advances in lipid nanocarriers applicable in the fight against cancer. In Nanoarchitectonics in Biomedicine; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 219–294. [Google Scholar]
- Jampilek, J.; Kralova, K.; Campos, E.V.R.; Fraceto, L.F. Bio-based nanoemulsion formulations applicable in agriculture, medicine and food industry. In Nanobiotechnology in Bioformulations; Prasad, R., Kumar, V., Kumar, M., Choudhary, D.K., Eds.; Springer: Cham, Germany, 2019; pp. 33–84. [Google Scholar]
- Jampilek, J.; Kralova, K. Nanoformulations–valuable tool in therapy of viral diseases attacking humans and animals. In Nanotheranostic–Applications and Limitations; Rai, M., Jamil, B., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 137–178. [Google Scholar]
- Jampilek, J.; Kralova, K.; Novak, P.; Novak, M. Nanobiotechnology in neurodegenerative diseases. In Nanobiotechnology in Neurodegenerative Diseases; Rai, M., Yadav, A., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2019; pp. 65–138. [Google Scholar]
- Jampilek, J.; Kralova, K. Natural biopolymeric nanoformulations for brain drug delivery. In Nanocarriers for Brain Targetting: Principles and Applications; Keservani, R.K., Sharma, A.K., Kesharwani, R.K., Eds.; Apple Academic Press & CRC Press: Warentown, NJ, USA, 2019; pp. 131–203. [Google Scholar]
- Jampilek, J.; Kralova, K. Nanotechnology: New frontiers in anti-HIV therapy. In Nanotechnological Applications in Virology; Rai, M., Yadav, A., Eds.; Academic Press & Elsevier: London, UK, 2022; pp. 129–171. [Google Scholar]
- Jampilek, J.; Kralova, K. Potential of nanonutraceuticals in increasing immunity. Nanomaterials 2020, 10, 2224. [Google Scholar] [CrossRef] [PubMed]
- Placha, D.; Jampilek, J. Chronic inflammatory diseases, anti-inflammatory agents and their delivery nanosystems. Pharmaceutics 2021, 13, 642019. [Google Scholar]
- Jampilek, J.; Kralova, K. Advances in nanostructures for antimicrobial therapy. Materials 2022, 15, 2388. [Google Scholar]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [PubMed]
- Wu, J. The enhanced permeability and retention (EPR) effect: The significance of the concept and methods to enhance its application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Onzi, G.; Guterres, S.S.; Pohlmann, A.R.; Frank, L.A. Passive Targeting and the enhanced permeability and retention (EPR) effect. In The ADME Encyclopedia; Springer: Cham, Switzerland, 2021; pp. 1–13. [Google Scholar]
- Niculescu, A.G.; Grumezescu, A.M. Novel tumor-targeting nanoparticles for cancer treatment—A review. Int. J. Mol. Sci. 2022, 23, 5253. [Google Scholar] [CrossRef] [PubMed]
- Basile, L.; Pignatello, R.; Passirani, C. Active targeting strategies for anticancer drug nanocarriers. Curr. Drug Deliv. 2012, 9, 255–268. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Dong, S.J.; Li, Z.M.; Feng, X.R.; Xu, W.G.; Tulinao, C.M.S.; Jiang, Y.; Ding, J.X. Biointerface engineering nanoplatforms for cancer-targeted drug delivery. Asian J. Pharm. Sci. 2020, 15, 397–415. [Google Scholar]
- Mishra, P.; Nayak, B.; Dey, R. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Anirudhan, T.S.; Nair, A.S. Temperature and ultrasound sensitive gatekeepers for the controlled release of chemotherapeutic drugs from mesoporous silica nanoparticles. J. Mater. Chem. B 2018, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Zhang, Y.; Ma, J.; Li, Q.; Li, Y.; Zhou, X.; Zhao, D.; Song, H.; Chen, Q.; Zhu, X. Light/magnetic hyperthermia triggered drug released from multifunctional thermo-sensitive magneto liposomes for precise cancer synergetic theranostics. J. Control. Release 2017, 272, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Kashkooli, F.M.; Soltani, M.; Souri, M. Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies. J. Control. Release 2020, 327, 316–349. [Google Scholar]
- Fahr, A. Voigt’s Pharmaceutical Technology, 12th ed.; John Wiley & Sons: New York, NY, USA, 2018. [Google Scholar]
- Muller, R.H.; Shegokar, R.; Keck, C.M. 20 Years of lipid nanoparticles (SLN & NLC): Present state of development & industrial applications. Curr. Drug Discov. Technol. 2011, 8, 207–227. [Google Scholar]
- Jampilek, J.; Kos, J.; Kralova, K. Potential of nanomaterial applications in dietary supplements and foods for special medical purposes. Nanomaterials 2019, 9, 296. [Google Scholar] [CrossRef] [Green Version]
- Mehanna, M.M.; Mneimneh, A.T. Formulation and applications of lipid-based nanovehicles: Spotlight on self-emulsifying systems. Adv. Pharm. Bull. 2021, 11, 56–67. [Google Scholar] [CrossRef]
- Kesharwani, R.; Jaiswal, P.; Patel, D.K.; Yadav, P.K. Lipid-based drug delivery system (LBDDS): An emerging paradigm to enhance oral bioavailability of poorly soluble drugs. Biomed. Mater. Devices 2022. [Google Scholar] [CrossRef]
- Korani, M.; Korani, S.; Zendehdel, E.; Jaafari, M.R.; Sathyapalan, T.; Sahebkar, A. Utilization of lipid-based nanoparticles to improve the therapeutic benefits of bortezomib. Anticancer Agents Med. Chem. 2020, 20, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, Z.; Barghi, L. Novel approaches for efficient delivery of tyrosine kinase inhibitors. J. Pharm. Pharm. Sci. 2019, 22, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvam, C.; Prabu, S.L.; Jordan, B.C.; Purushothaman, Y.; Umamaheswari, A.; Zare, M.S.H.; Thilagavathi, R. Molecular mechanisms of curcumin and its analogs in colon cancer prevention and treatment. Life Sci. 2019, 239, 117032. [Google Scholar]
- Lorincz, A.; Mihaly, J.; Wacha, A.; Nemeth, C.; Besztercei, B.; Gyulavari, P.; Varga, Z.; Petak, I.; Bota, A. Combination of multifunctional ursolic acid with kinase inhibitors for anti-cancer drug carrier vesicles. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 131, 112481. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Desai, N.; Dixit, R.; Nagarsenker, M. Self-nanoemulsifying drug delivery systems: Formulation insights, applications and advances. Nanomedicine 2010, 5, 1595–1616. [Google Scholar] [CrossRef] [PubMed]
- Sakthi, U.M.; Lobo, F.J.R.; Uppuluri, K.B. Self nano emulsifying drug delivery systems for oral delivery of hydrophobic drugs. Biomed. Pharmacol. J. 2013, 6, 355–362. [Google Scholar]
- Buya, A.B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Self-nano-emulsifying drug-delivery systems: From the development to the current applications and challenges in oral drug delivery. Pharmaceutics 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Ujhelyi, Z.; Vecsernyes, M.; Feher, P.; Kosa, D.; Arany, P.; Nemes, D.; Sinka, D.; Vasvari, G.; Fenyvesi, F.; Varadi, J.; et al. Physico-chemical characterization of self-emulsifying drug delivery systems. Drug Discov. Today Technol. 2018, 27, 81–86. [Google Scholar] [CrossRef]
- Teaima, M.; Hababeh, S.; Khanfar, M.; Alanazi, F.; Alshora, D.; El-Nabarawi, M. Design and optimization of pioglitazone hydrochloride self-nanoemulsifying drug delivery system (SNEDDS) incorporated into an orally disintegrating tablet. Pharmaceutics 2022, 14, 425. [Google Scholar] [CrossRef]
- Zafar, A.; Yasir, M.; Alruwaili, N.K.; Imam, S.S.; Alsaidan, O.A.; Alshehri, S.; Ghoneim, M.M.; Alquraini, A.; Rawaf, A.; Ansari, M.J.; et al. Formulation of self-nanoemulsifying drug delivery system of cephalexin: Physiochemical characterization and antibacterial evaluation. Polymers 2022, 14, 1055. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.; Simon, L.; Brabet, P.; Legrand, P.; Dorandeu, C.; Him, J.L.K.; Durand, T.; Crauste, C.; Begu, S. Formulation and evaluation of SNEDDS loaded with original lipophenol for the oral route to prevent dry AMD and Stragardt’s disease. Pharmaceutics 2022, 14, 1029. [Google Scholar] [CrossRef]
- Kim, K.S.; Yang, E.S.; Kim, D.S.; Kim, D.W.; Yoo, H.H.; Yong, C.S.; Youn, Y.S.; Oh, K.T.; Jee, J.P.; Kim, J.O.; et al. A novel solid self-nanoemulsifying drug delivery system (S-SNEDDS) for improved stability and oral bioavailability of an oily drug, 1-palmitoyl-2-linoleoyl-3-acetyl-rac-glycerol. Drug Deliv. 2017, 24, 1018–1025. [Google Scholar] [CrossRef]
- Baloch, J.; Sohail, M.F.; Sarwar, H.S.; Kiani, M.H.; Khan, G.M.; Jahan, S.; Rafay, M.; Chaudhry, M.T.; Yasinzai, M.; Shahnaz, G. Self-nanoemulsifying drug delivery system (SNEDDS) for improved oral bioavailability of chlorpromazine: In Vitro and in vivo evaluation. Medicina 2019, 55, 210. [Google Scholar] [CrossRef] [Green Version]
- Alhasani, K.F.; Kazi, M.; Ibrahim, M.A.; Shahba, A.A.; Alanazi, F.K. Self-nanoemulsifying ramipril tablets: A novel delivery system for the enhancement of drug dissolution and stability. Int. J. Nanomed. 2019, 14, 5435–5448. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.J.; Alnakhli, M.; Al-Otaibi, T.; Al Meanazel, O.; Anwer, M.K.; Ahmed, M.M.; Alshahrani, S.M.; Alshetaili, A.; Aldawsari, M.F.; Alalaiwe, A.S.; et al. Improvement of solubility, in vitro release, ex-vivo permeation and anticancer activity. J. Drug Deliv. Sci. Technol. 2021, 61, 102204. [Google Scholar] [CrossRef]
- Wang, C.Q.; Wang, M.T.; Chen, P.; Wang, J.X.; Le, Y. Dasatinib nanoemulsion and nanocrystal for enhanced oral drug delivery. Pharmaceutics 2022, 14, 197. [Google Scholar] [CrossRef]
- Bhattacharya, S. Double w/o/w self-nano emulsifying drug delivery system of imatinib mesylate for colon cancer treatment. J. Mol. Liq. 2021, 341, 117368. [Google Scholar] [CrossRef]
- Karimi, M.; Karimian, K.; Heli, H. A nanoemulsion-based delivery system for imatinib and in vitro anticancer efficacy. Braz. J. Pharm. Sci. 2020, 56, e18973. [Google Scholar] [CrossRef]
- Izadiyan, Z.; Basri, M.; Masoumi, H.R.F.; Karjiban, R.A.; Salim, N.; Kalantari, K. Improvement of physicochemical properties of nanocolloidal carrier loaded with low water solubility drug for parenteral cancer treatment by response surface methodology. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 94, 841–849. [Google Scholar] [CrossRef]
- Izadiyan, Z.; Basri, M.; Masoumi, H.R.F.; Karjiban, R.A.; Salim, N.; Shameli, K. Modeling and optimization of nanoemulsion containing sorafenib for cancer treatment by response surface methodology. Chem. Cent. J. 2017, 11, 21. [Google Scholar] [CrossRef] [Green Version]
- Alshahrani, S.M.; Alshetaili, A.S.; Alalaiwe, A.; Alsulays, B.B.; Anwer, M.K.; Al-Shdefat, R.; Imam, F.; Shakeel, F. Anticancer efficacy of self-nanoemulsifying drug delivery system of sunitinib malate. AAPS PharmSciTech 2018, 19, 123–133. [Google Scholar] [CrossRef]
- Alkhatib, M.H.; Nori, D.A.; Al-Ghamdi, M.A. Nanoemulsion formulated with flaxseed oil running title: Antitumor activity and hepatotoxicity effect of sorafenib. Int. J. Pharm. Sci. Rev. Res. 2017, 6, 175–188. [Google Scholar]
- Alkhatib, M.H.; Alnahdi, N.S.; Backer, W.S. Antitumor activity, hematoxicity and hepatotoxicity of sorafenib formulated in a nanoemulsion based on the carrot seed oil. Int. J. Life Sci. Biotechnol. Pharma Res. 2018, 8, 50–57. [Google Scholar]
- Nazari-Vanani, R.; Azarpira, N.; Heli, H.; Karimian, K.; Sattarahmady, N. A novel self-nanoemulsifying formulation for sunitinib: Evaluation of anticancer efficacy. Colloids Surf. B Biointerfaces 2017, 160, 65–72. [Google Scholar] [CrossRef]
- Mazur, F.; Bally, M.; Städler, B.; Chandrawati, R. Liposomes and lipid bilayers in biosensors. Adv. Colloid Interface Sci. 2017, 249, 88–99. [Google Scholar] [CrossRef]
- Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Silver Spring, MD, USA, 2018. Available online: https://www.fda.gov/media/70837/download (accessed on 30 October 2022).
- Xu, L.T.; Wang, X.; Liu, Y.; Yang, G.Z.; Falconer, R.J.; Zhao, C.X. Lipid nanoparticles for drug delivery. Adv. NanoBiomed Res. 2021, 2, 2100109. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A review of liposomes as a drug delivery system: Current status of approved products, regulatory. environments, and future perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Jain, A.; Tiwari, A.; Verma, A.; Saraf, S.; Jain, S.K. Combination cancer therapy using multifunctional liposomes. Crit. Rev. Ther. Drug Carr. Syst. 2020, 37, 105–134. [Google Scholar] [CrossRef]
- Li, F.Q.; Mei, H.; Xie, X.D.; Zhang, H.J.; Liu, J.; Lv, T.T.; Nie, H.F.; Gao, Y.; Jia, L. Aptamer-conjugated chitosan-anchored liposomal complexes for targeted delivery of erlotinib to EGFR-mutated lung cancer cells. AAPS J. 2017, 19, 814–826. [Google Scholar] [CrossRef]
- Negi, L.M.; Jaggi, M.; Joshi, V.; Ronodip, K.; Talegaonkar, S. Hyaluronan coated liposomes as the intravenous platform for delivery of imatinib mesylate in MDR colon cancer. Int. J. Biol. Macromol. 2015, 73, 222–235. [Google Scholar] [CrossRef]
- Peres-Filho, M.J.; dos Santos, A.P.; Nascimento, T.L.; de Avila, R.I.; Ferreira, F.S.; Valadares, M.C.; Lima, E.M. Antiproliferative activity and VEGF expression reduction in MCF7 and PC-3 cancer cells by paclitaxel and imatinib co-encapsulation in folate-targeted liposomes. AAPS PharmSciTech 2018, 19, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Jose, A.; Ninave, K.M.; Karnam, S.; Venuganti, V.V.K. Temperature-sensitive liposomes for co-delivery of tamoxifen and imatinib for synergistic breast cancer treatment. J. Liposome Res. 2019, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Varshosaz, J.; Raghami, F.; Rostami, M.; Jahanian, A. PEGylated trimethylchitosan emulsomes conjugated to octreotide for targeted delivery of sorafenib to hepatocellular carcinoma cells of HepG2. J. Liposome Res. 2019, 29, 383–398. [Google Scholar] [CrossRef] [PubMed]
- Abshire, C.; Murad, H.Y.; Arora, J.S.; Liu, J.; Mandava, S.H.; John, V.T.; Khismatullin, D.B.; Lee, B.R. Focused ultrasound-triggered release of tyrosine kinase inhibitor from thermosensitive liposomes for treatment of renal cell carcinoma. J. Pharm. Sci. 2017, 106, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Almurshedi, A.S.; Radwan, M.; Omar, S.; Alaiya, A.A.; Badran, M.M.; Elsaghire, H.; Saleem, I.Y.; Hutcheon, G.A. A novel pH-sensitive liposome to trigger delivery of afatinib to cancer cells: Impact on lung cancer therapy. J. Mol. Liq. 2018, 259, 154–166. [Google Scholar] [CrossRef]
- Ravar, F.; Saadat, E.; Kelishadi, P.D.; Dorkoosh, F.A. Liposomal formulation for co-delivery of paclitaxel and lapatinib, preparation, characterization and optimization. J. Liposome Res. 2016, 26, 175–187. [Google Scholar] [CrossRef]
- Patel, K.; Bothiraja, C.; Mali, A.; Kamble, R. Investigation of sorafenib tosylate loaded liposomal dry powder inhaler for the treatment of non-small cell lung cancer. Part. Sci. Technol. 2021, 39, 990–999. [Google Scholar] [CrossRef]
- Salmaso, S.; Mastrotto, F.; Roverso, M.; Gandin, V.; De Martin, S.; Gabbia, D.; De Franco, M.; Vaccarin, C.; Verona, M.; Chilin, A.; et al. Tyrosine kinase inhibitor prodrug-loaded liposomes for controlled release at tumor microenvironment. J. Control. Release 2021, 340, 318–330. [Google Scholar] [CrossRef]
- Kulkarni, A.A.; Vijaykumar, V.E.; Natarajan, S.K.; Sengupta, S.; Sabbisetti, V.S. Sustained inhibition of cMET-VEGFR2 signaling using liposome-mediated delivery increases efficacy and reduces toxicity in kidney cancer. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Spring, B.Q.; Sears, R.B.; Zheng, L.Z.; Mai, Z.M.; Watanabe, R.; Sherwood, M.E.; Schoenfeld, D.A.; Pogue, B.W.; Pereira, S.P.; Villa, E.; et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat. Nanotechnol. 2016, 11, 378–387. [Google Scholar] [CrossRef]
- Pardeshi, S.; Tiwari, A.; Titame, U.; Singh, P.K.; Yadav, P.K.; Chourasia, M.K. Development of asolectin-based liposomal formulation for controlled and targeted delivery of erlotinib as a model drug for EGFR monotherapy. J. Liposome Res. 2022. [Google Scholar] [CrossRef]
- Li, F.Q.; Mei, H.; Gao, Y.; Xie, X.D.; Nie, H.F.; Li, T.; Zhang, H.J.; Jia, L. Co-delivery of oxygen and erlotinib by aptamer-modified liposomal complexes to reverse hypoxia-induced drug resistance in lung cancer. Biomaterials 2017, 145, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Du, B.; Cheng, G.Y. Reshaping tumor blood vessels to enhance drug penetration with a multistrategy synergistic nanosystem. Mol. Pharm. 2020, 17, 3151–3164. [Google Scholar] [CrossRef]
- Sanlier, S.H.; Ak, G.; Yilmaz, H.; Unal, A.; Bozkaya, U.F.; Taniyan, G.; Yildirim, Y.; Turkyilmaz, G.Y. Development of ultrasound-triggered and magnetic-targeted nanobubble system for dual-drug delivery. J. Pharm. Sci. 2019, 108, 1272–1283. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Ramasamy, T.; Tran, H.; Ku, S.K.; Shin, B.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Systemic delivery of axitinib with nanohybrid liposomal nanoparticles inhibits hypoxic tumor growth. J. Mater. Chem. B 2015, 3, 408–416. [Google Scholar] [CrossRef]
- Ye, P.; Zhang, W.D.; Yang, T.; Lu, Y.; Lu, M.; Gai, Y.K.; Ma, X.; Xiang, G.Y. Folate receptor-targeted liposomes enhanced the antitumor potency of imatinib through the combination of active targeting and molecular targeting. Int. J. Nanomed. 2014, 9, 2167–2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amiri, M.; Gholami, T.; Amiri, O.; Pardakhti, A.; Ahmadi, M.; Akbari, A.; Amanatfard, A.; Salavati-Niasari, M. The magnetic inorganic-organic nanocomposite based on ZnFe2O4-Imatinib-liposome for biomedical applications, in vivo and in vitro study. J. Alloys Compd. 2020, 849, 156604. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, Y.; Zhao, P.X.; Zhang, S.S.; Li, M.S.; He, C.C.; Zhang, X.J.; Yang, T.; Yan, R.C.; Ye, P.; et al. Co-delivery of doxorubicin and imatinib by pH sensitive cleavable PEGylated nanoliposomes with folate-mediated targeting to overcome multidrug resistance. Int. J. Pharm. 2018, 542, 266–279. [Google Scholar] [CrossRef]
- Zhao, M.J.; Lee, S.H.; Song, J.G.; Kim, H.Y.; Han, H.K. Enhanced oral absorption of sorafenib via the layer-by-layer deposition of a pH-sensitive polymer and glycol chitosan on the liposome. Int. J. Pharm. 2018, 544, 14–20. [Google Scholar] [CrossRef]
- He, Q.Y.; He, X.X.; Deng, B.; Shi, C.; Lin, L.P.; Liu, P.; Yang, Z.; Yang, S.L.; Xu, Z.S. Sorafenib and indocyanine green co-loaded in photothermally sensitive liposomes for diagnosis and treatment of advanced hepatocellular carcinoma. J. Mater. Chem. 2018, 6, 5823–5834. [Google Scholar] [CrossRef]
- Bianchini, F.; De Santis, A.; Portioli, E.; Krauss, I.R.; Battistini, L.; Curti, C.; Peppicelli, S.; Calorini, L.; D’Errico, G.; Zanardi, F.; et al. Integrin-targeted AmpRGD sunitinib liposomes as integrated antiangiogenic tools. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.J.; Su, Y.; Liu, W.; Chang, J.B.A.; Zhang, Z.Z. A nanoliposome-based photoactivable drug delivery system for enhanced cancer therapy and overcoming treatment resistance. Int. J. Nanomed. 2017, 12, 8257–8275. [Google Scholar] [CrossRef]
- Yang, X.; Li, H.P.; Qian, C.G.; Guo, Y.X.; Li, C.Z.; Gao, F.; Yang, Y.; Wang, K.K.; Oupicky, D.; Sun, M.J. Near-infrared light-activated IR780-loaded liposomes for anti-tumor angiogenesis and photothermal therapy. Nanomedicine 2018, 14, 2283–2294. [Google Scholar] [CrossRef] [PubMed]
- Kallus, S.; Englinger, B.; Senkiv, J.; Laemmerer, A.; Heffeter, P.; Berger, W.; Kowol, C.R.; Keppler, B.K. Nanoformulations of anticancer FGFR inhibitors with improved therapeutic index. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2632–2643. [Google Scholar] [CrossRef]
- Mehnert, W.; Mader, K. Solid lipid nanoparticles: Production, characterization and applications. Adv. Drug Deliv. Rev. 2012, 64, 83–101. [Google Scholar] [CrossRef]
- Kammari, R.; Das, N.G.; Das, S.K. Nanoparticulate systems for therapeutic and diagnostic applications. Emerg. Nanotechnol. Diagn. Drug Deliv. Med. Dev. 2017, 2017, 105–144. [Google Scholar]
- Mishra, V.; Bansal, K.K.; Verma, A.; Yadav, N.; Thakur, S.; Sudhakar, K.; Rosenholm, J.M. Solid lipid nanoparticles: Emerging colloidal nano drug delivery system. Pharmaceutics 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, N.; Awasthi, R.; Sharma, B.; Kharkwal, H.; Kulkarni, G.T. Lipid nanoparticles as carriers for bioactive delivery. Front. Chem. 2021, 9, 580118. [Google Scholar] [CrossRef] [PubMed]
- Muller, R.H.; Mader, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery-a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, H.A.; Javadzadeh, Y.; Hamidi, M.; Jalali, M.B. Repaglinide-loaded solid lipid nanoparticles: Effect of using different surfactants/stabilizers on physicochemical properties of nanoparticles. Daru 2015, 23, 46. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.; Patro, C.S. Solid lipid nanoparticles for efficient oral delivery of tyrosine kinase inhibitors: A nano targeted cancer drug delivery. Adv. Pharm. Bull. 2022, 12, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Mandal, B.; Bhattacharjee, H.; Mittal, N.; Sah, H.; Balabathula, P.; Thoma, L.A.; Wood, G.C. Core–shell-type lipid–polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine 2013, 9, 474–491. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Lu, S.T.; Deng, K.; Yu, H.; Cui, C.; Zhang, Y.; Wu, M.; Zhuo, R.X.; Xu, H.B.; Huang, S.W. MRI-guided targeting delivery of doxorubicin with reduction-responsive lipid-polymer hybrid nanoparticles. Int. J. Nanomed. 2017, 12, 6871–6882. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Waters, A.K.; Kalyan, P.; Achrol, A.S.; Kesari, S.; Yenugonda, V.M. Lipid–polymer hybrid nanoparticles as a next-generation drug delivery platform: State of the art, emerging technologies, and perspectives. Int. J. Nanomed. 2019, 14, 1937–1952. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.M.; Fatima, F.; Anwer, M.K.; Aldawsari, M.F.; Alsaidan, Y.S.M.; Alfaiz, S.A.; Haque, A.; Alanazi, A.Z.; Alhazzani, K. Development and characterization of brigatinib loaded solid lipid nanoparticles: In-vitro cytotoxicity against human carcinoma A549 lung cell lines. Chem. Phys. Lipids 2020, 233, 105003. [Google Scholar] [CrossRef]
- Satari, N.; Taymouri, S.; Varshosaz, J.; Rostami, M.; Mirian, M. Preparation and evaluation of inhalable dry powder containing glucosamine-conjugated gefitinib SLNs for lung cancer therapy. Drug Dev. Ind. Pharm. 2020, 46, 1265–1277. [Google Scholar] [CrossRef]
- Naseri, N.; Zakeri-Milani, P.; Hamishehkar, H.; Pilehvar-Soltanahmadi, Y.; Valizadeh, H. Development, in vitro characterization, antitumor and aerosol performance evaluation of respirable prepared by self-nano-emulsification method. Drug Res. 2017, 67, 343–348. [Google Scholar]
- Bakhtiary, Z.; Barar, J.; Aghanejad, A.; Saei, A.A.; Nemati, E.; Dolatabadi, J.E.N.; Omidi, Y. Microparticles containing erlotinib-loaded solid lipid nanoparticles for treatment of non-small cell lung cancer. Drug Dev. Ind. Pharm. 2017, 43, 1244–1253. [Google Scholar] [CrossRef]
- Mandal, B.; Mittal, N.K.; Balabathula, P.; Thoma, L.A.; Wood, G.C. Development and in vitro evaluation of core-shell type lipid-polymer hybrid nanoparticles for the delivery of erlotinib in non-small cell lung cancer. Eur. J. Pharm. Sci. 2016, 81, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Poudel, B.K.; Regmi, S.; Pathak, S.; Ruttala, H.B.; Gautam, M.; An, G.J.; Jeong, J.H.; Choi, H.G.; Yong, C.S.; et al. Paclitaxel and erlotinib-co-loaded solid lipid core nanocapsules: Assessment of physicochemical characteristics and cytotoxicity in non-small cell lung cancer. Pharm. Res. 2018, 35, 96. [Google Scholar] [CrossRef] [PubMed]
- Siram, K.; Karuppaiah, A.; Gautam, M.; Sankar, V. Fabrication of hyaluronic acid surface modified solid lipid nanoparticles loaded with imatinib mesylate for targeting human breast cancer MCF-7 cells. J. Clust. Sci. 2022. [Google Scholar] [CrossRef]
- Tang, S.J.; Li, Y.J. Sorafenib-loaded ligand-functionalized polymer-lipid hybrid nanoparticles for enhanced therapeutic effect against liver cancer. J. Nanosci. Nanotechnol. 2019, 19, 6866–6871. [Google Scholar] [CrossRef] [PubMed]
- Benizri, S.; Ferey, L.; Alies, B.; Mebarek, N.; Vacher, G.; Appavoo, A.; Staedel, C.; Gaudin, K.; Barthelemy, P. Nucleoside-lipid-based nanocarriers for sorafenib delivery. Nanoscale Res. Lett. 2018, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Tahir, N.; Madni, A.; Li, W.; Correia, A.; Khan, M.M.; Rahim, M.A.; Santos, H.A. Microfluidic fabrication and characterization of Sorafenib-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery. Int. J. Pharm. 2020, 581, 119275. [Google Scholar] [CrossRef]
- Younis, M.A.; Khalil, I.A.; Abd Elwakil, M.M.; Harashima, H. A multifunctional lipid-based nanodevice for the highly specific codelivery of sorafenib and midkine siRNA to hepatic cancer cells. Mol. Pharm. 2019, 16, 4031–4044. [Google Scholar] [CrossRef]
- Farinha, D.; Migawa, M.; Sarmento-Ribeiro, A.; Faneca, H. A combined antitumor strategy mediated by a new targeted nanosystem to hepatocellular carcinoma. Int. J. Nanomed. 2021, 16, 3385–3405. [Google Scholar] [CrossRef]
- Arduino, I.; Liu, Z.H.; Rahikkala, A.; Figueiredo, P.; Correia, A.; Cutrignelli, A.; Denora, N.; Santos, H.A. Preparation of cetyl palmitate-based PEGylated solid lipid nanoparticles by microfluidic technique. Acta Biomater. 2021, 121, 566–578. [Google Scholar] [CrossRef]
- Grillone, A.; Riva, E.R.; Mondini, A.; Forte, C.; Calucci, L.; Innocenti, C.; de Julian Fernandez, C.; Cappello, V.; Gemmi, M.; Moscato, S.; et al. Active targeting of sorafenib: Preparation, characterization, and in vitro testing of drug-loaded magnetic solid lipid nanoparticles. Adv. Healthc. Mater. 2015, 4, 1681–1690. [Google Scholar] [CrossRef]
- Khaledian, S.; Kahrizi, D.; Moradi, S.; Martinez, F. An experimental and computational study to evaluation of chitosan/gum tragacanth coated-natural lipid-based nanocarriers for sunitinib delivery. J. Mol. Liq. 2021, 334, 116075. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Anwer, M.K.; Fatima, F.; Aldawsari, M.F.; Alalaiwe, A.; Alali, A.S.; Alharthi, A.I.; Abul Kalam, M. Boosting the anticancer activity of sunitinib malate in breast cancer through lipid polymer hybrid nanoparticles approach. Polymers 2022, 14, 2459. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.T.; Lin, H.; Wang, C.S.; Chang, C.H.; Lin, A.M.Y.; Yang, J.C.H.; Lo, Y.L. Improving the anticancer effect of afatinib and microRNA by using lipid polymeric nanoparticles conjugated with dual pH-responsive and targeting peptides. J. Nanobiotechnol. 2019, 17, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.L.; Lin, H.C.; Hong, S.T.; Chang, C.H.; Wang, C.S.; Lin, A.M.Y. Lipid polymeric nanoparticles modified with tight junction-modulating peptides promote afatinib delivery across a blood-brain barrier model. Cancer Nanobiotechnol. 2021, 12, 13. [Google Scholar] [CrossRef]
- Fu, D.H.; Li, C.; Huang, Y.W. Lipid-polymer hybrid nanoparticle-based combination treatment with cisplatin and EGFR/HER2 receptor-targeting afatinib to enhance the treatment of nasopharyngeal carcinoma. OncoTargets Ther. 2021, 14, 2449–2461. [Google Scholar] [CrossRef]
- Vivek, R.; Jose, S. Development, evaluation and targeting of imatinib mesylate loaded solid lipid nanoparticles to the lymphatic system. Int. J. Pharm. Sci. Res. 2018, 9, 2359–2368. [Google Scholar]
- Molaahmadi, M.R.; Varshosaz, J.; Taymouri, S.; Akbari, V. Lipid nanocapsules for imatinib delivery: Design, optimization and evaluation of anticancer activity against melanoma cell line. Iran. J. Pharm. Sci. 2019, 18, 1676–1693. [Google Scholar]
- Zinger, A.; Baudo, G.; Naoi, T.; Giordano, F.; Lenna, S.; Massaro, M.; Ewing, A.; Kim, H.R.; Tasciotti, E.; Yustein, J.T.; et al. Reproducible and characterized method for ponatinib encapsulation into biomimetic lipid nanoparticles as a Platform for multi-tyrosine kinase-targeted therapy. ACS Appl. Bio Mater. 2020, 3, 6737–6745. [Google Scholar] [CrossRef]
- Nadaf, S.J.; Killedar, S.G.; Kumbar, V.M.; Bhagwat, D.A.; Gurav, S.S. Pazopanib-laden lipid based nanovesicular delivery with augmented oral bioavailability and therapeutic efficacy against non-small cell lung cancer. Int. J. Pharm. 2022, 628, 122287. [Google Scholar] [CrossRef]
- Yang, K.M.; Shin, I.C.; Park, J.W.; Kim, K.S.; Kim, D.K.; Park, K.; Kim, K. Nanoparticulation improves bioavailability of Erlotinib. Drug Dev. Ind. Pharm. 2017, 43, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Rampaka, R.; Ommi, K.; Chella, N. Role of solid lipid nanoparticles as drug delivery vehicles on the pharmacokinetic variability of Erlotinib HCl. J. Drug Deliv. Sci. Technol. 2021, 66, 102886. [Google Scholar] [CrossRef]
- Kim, J.; Ramasamy, T.; Choi, J.Y.; Kim, S.T.; Youn, Y.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. PEGylated polypeptide lipid nanocapsules to enhance the anticancer efficacy of erlotinib in non-small cell lung cancer. Colloids Surf. B Biointerfaces 2017, 150, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Wang, G.X. Redox-responsive and pH-sensitive nanoparticles enhanced stability and anticancer ability of erlotinib to treat lung cancer in vivo. Drug Des. Devel. Ther. 2017, 11, 3519–3529. [Google Scholar] [CrossRef] [Green Version]
- Ganthala, P.D.; Alavala, S.; Chella, N.; Andugulapati, S.B.; Bathini, N.B.; Sistla, R. Co-encapsulated nanoparticles of Erlotinib and Quercetin for targeting lung cancer through nuclear EGFR and PI3K/AKT inhibition. Colloids Surf. B Biointerfaces 2022, 211, 112305. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Su, Z.G.; Xue, L.J.; Xu, H.; Zhang, C. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J. Control. Release 2016, 229, 80–92. [Google Scholar] [CrossRef]
- Pang, J.T.; Xing, H.X.; Sun, Y.G.; Feng, S.; Wang, S.Z. Non-small cell lung cancer combination therapy: Hyaluronic acid modified, epidermal growth factor receptor targeted, pH sensitive lipid-polymer hybrid nanoparticles for the delivery of erlotinib plus bevacizumab. Biomed. Pharmacother. 2020, 125, 109861. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Ramasamy, T.; Kim, S.Y.; Kim, J.; Ku, S.K.; Youn, Y.S.; Kim, J.R.; Jeong, J.H.; Choi, H.G.; Yong, C.S.; et al. PEGylated lipid bilayer-supported mesoporous silica nanoparticle composite for synergistic co-delivery of axitinib and celastrol in multi-targeted cancer therapy. Acta Biomater. 2016, 39, 94–105. [Google Scholar] [CrossRef]
- Gao, H.L.; Chen, C.; Xi, Z.J.; Chen, J.; Zhang, Q.Z.; Cao, S.L.; Jiang, X.G. In vivo behavior and safety of lapatinib-incorporated lipid nanoparticles. Curr. Pharm. Biotechnol. 2013, 14, 1062–1071. [Google Scholar] [CrossRef]
- Gao, H.L.; Cao, S.L.; Chen, C.; Cao, S.J.; Yang, Z.; Pang, Z.Q.; Xi, Z.J.; Pan, S.Q.; Zhang, Q.Z.; Jiang, X.G. Incorporation of lapatinib into lipoprotein-like nanoparticles with enhanced water solubility and anti-tumor effect in breast cancer. Nanomedicine 2013, 8, 1429–1442. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Ruan, S.B.; Zhang, Q.Y.; He, Q.; Ga, H.L. Lapatinib-incorporated lipoprotein-like nanoparticles: Preparation and a proposed breast cancer-targeting mechanism. Acta Pharmacol. Sin. 2014, 35, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Huo, Z.J.; Wang, S.J.; Wang, Z.Q.; Zuo, W.S.; Liu, P.; Pang, B.; Liu, K. Novel nanosystem to enhance the antitumor activity of lapatinib in breast cancer treatment: Therapeutic efficacy evaluation. Cancer Sci. 2015, 106, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsakiris, N.; Papavasileiou, M.; Bozzato, E.; Lopes, A.; Vigneron, A.M.; Preat, V. Combinational drug-loaded lipid nanocapsules for the treatment of cancer. Int. J. Pharm. 2019, 569, 118588. [Google Scholar] [CrossRef] [PubMed]
- Clavreul, A.; Roger, E.; Pourbaghi-Masouleh, M.; Lemaire, L.; Tetaud, C.; Menei, P. Development and characterization of sorafenib-loaded lipid nanocapsules for the treatment of glioblastoma. Drug Deliv. 2018, 25, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.W.; Niu, B.H.; Chen, J.; He, X.Y.; Bao, X.Y.; Zhu, J.H.; Yu, H.J.; Li, Y.P. The use of lipid-coated nanodiamond to improve bioavailability and efficacy of sorafenib in resisting metastasis of gastric cancer. Biomaterials 2014, 35, 4565–4572. [Google Scholar] [CrossRef]
- Yang, S.M.; Zhang, B.; Gong, X.W.; Wang, T.Q.; Liu, Y.J.; Zhang, N. In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer. Int. J. Nanomed. 2016, 11, 2329–2343. [Google Scholar]
- Zhang, J.; Wang, T.Q.; Mu, S.J.; Olerile, L.D.; Yu, X.Y.; Zhang, N. Biomacromolecule/lipid hybrid nanoparticles for controlled delivery of sorafenib in targeting hepatocellular carcinoma therapy. Nanomedicine 2017, 12, 911–925. [Google Scholar] [CrossRef]
- Yang, F.; Li, A.M.; Liu, H.; Zhang, H.R. Gastric cancer combination therapy: Synthesis of a hyaluronic acid and cisplatin containing lipid prodrug coloaded with sorafenib in a nanoparticulate system to exhibit enhanced anticancer efficacy and reduced toxicity. Drug Des. Devel. Ther. 2018, 12, 3321–3333. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.D.; Liu, Y. Targeted and synergistic therapy for hepatocellular carcinoma: Monosaccharide modified lipid nanoparticles for the co-delivery of doxorubicin and sorafenib. Drug Des. Devel. Ther. 2018, 12, 2149–2161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.M.; Hu, J.; Chan, H.F.; Skibba, M.; Liang, G.; Chen, M.W. iRGD decorated lipid-polymer hybrid nanoparticles for targeted co-delivery of doxorubicin and sorafenib to enhance anti-hepatocellular carcinoma efficacy. Nanotechnol. Biol. Med. 2016, 12, 1303–1311. [Google Scholar] [CrossRef]
- Wang, Z.F.; Duan, X.X.; Lv, Y.H.; Zhao, Y.F. Low density lipoprotein receptor (LDLR)-targeted lipid nanoparticles for the delivery of sorafenib and dihydroartemisinin in liver cancers. Life Sci. 2019, 239, 117013. [Google Scholar] [CrossRef]
- Wang, C.; Su, L.; Wu, C.S.; Wu, J.L.; Zhu, C.B.; Yuan, G.Y. RGD peptide targeted lipid-coated nanoparticles for combinatorial delivery of sorafenib and quercetin against hepatocellular carcinoma. Drug Dev. Ind. Pharm. 2016, 42, 1938–1944. [Google Scholar] [CrossRef]
- Wang, Z.F.; Zhao, K.; Zhang, Y.X.; Duan, X.X.; Zhao, Y.F. Anti-GPC3 antibody tagged cationic switchable lipid-based nanoparticles for the co-delivery of anti-miRNA27a and sorafenib in liver cancers. Pharm. Res. 2019, 36, 145. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, R.M.; Vischio, F.; Arduino, I.; Canepa, F.; Laquintana, V.; Notarnicola, M.; Scavo, M.P.; Bianco, G.; Fanizza, E.; Lopedota, A.A.; et al. Magnetic implants in vivo guiding sorafenib liver delivery by superparamagnetic solid lipid nanoparticles. J. Colloid Interface Sci. 2022, 608, 239–254. [Google Scholar] [CrossRef]
- Park, W.; Chen, J.; Cho, S.; Park, S.J.; Larson, A.C.; Na, K.; Kim, D.H. Acidic pH-triggered drug-eluting nanocomposites for magnetic resonance imaging-monitored intra-arterial drug delivery to hepatocellular carcinoma. ACS Appl. Mater. Interfaces 2016, 8, 12711–12719. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.L.; Su, G.X.; Yin, X.Q.; Luo, J.; Gu, R.R.; Wang, S.; Feng, J.; Chen, B.H. Non-small cell lung cancer-targeted, redox-sensitive lipid-polymer hybrid nanoparticles for the delivery of a second-generation irreversible epidermal growth factor inhibitor-Afatinib: In Vitro and in vivo evaluation. Biomed. Pharmacother. 2019, 120, 109493. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Z.W.; Li, J.Y.; Mo, Z.R.; Huang, Y.; Ma, C.; Wang, W.H.; Pan, X.; Wu, C.B. PLGA porous microspheres dry powders for codelivery of afatinib-loaded solid lipid nanoparticles and paclitaxel: Novel therapy for EGFR tyrosine kinase inhibitors resistant nonsmall cell lung cancer. Adv. Healthc. Mater. 2019, 8, 1900965. [Google Scholar] [CrossRef]
- Akbari, J.; Saeedi, M.; Ahmadi, F.; Hashemi, S.M.H.; Babaei, A.; Yaddollahi, S.; Seyyed Rostamkalaei, S.S.; Asare-Addom, K.; Nokhodchi, A. Solid lipid nanoparticles and nanostructured lipid carriers: A review of the methods of manufacture and routes of administration. Pharm. Dev. Technol. 2022, 27, 525–544. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinís, M.A.; Rodriguez-Gascon, A.; Almeida, A.J.; Preat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine 2016, 12, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Selvamuthukumar, S.; Velmurugan, R. Nanostructured lipid carriers: A potential drug carrier for cancer chemotherapy. Lipids Health Dis. 2012, 11, 159. [Google Scholar]
- Rizwanullah, M.; Ahmad, J.; Amin, S. Nanostructured lipid carriers: A novel platform for chemotherapeutics. Curr. Drug Deliv. 2016, 13, 4–26. [Google Scholar] [CrossRef]
- Gaballu, F.A.; Abbaspour-Ravasjani, S.; Mansoori, B.; Yekta, R.; Hamishehkar, H.; Mohammadi, A.; Dehghan, G.; Shokouhi, B.; Dehbokri, S.G.; Baradaran, B. Comparative of in-vitro evaluation between erlotinib loaded nanostructured lipid carriers and liposomes against A549 lung cancer cell line. Iran. J. Pharm. Sci. 2019, 18, 1168–1179. [Google Scholar]
- Majumder, J.; Minko, T. Multifunctional lipid-based nanoparticles for codelivery of anticancer drugs and siRNA for treatment of non-small cell lung cancer with different level of resistance and EGFR mutations. Pharmaceutics 2021, 13, 1063. [Google Scholar] [CrossRef] [PubMed]
- Gundogdu, E.; Demir, E.S.; Ekinci, M.; Ozgenc, E.; Ilem-Ozdemir, D.; Senyigit, Z.; Gonzalez-Alvarez, I.; Bermejo, M. An innovative formulation based on nanostructured lipid carriers for imatinib delivery: Pre-formulation, cellular uptake and cytotoxicity studies. Nanomaterials 2022, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Makeen, H.A.; Mohan, S.; Al-Kasim, M.A.; Sultan, M.H.; Albarraq, A.; Ahmed, R.A.; Alhazmi, H.A.; Alam, M.I. Preparation, characterization, and anti-cancer activity of nanostructured lipid carriers containing imatinib. Pharmaceutics 2021, 13, 1086. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.L.; Botto, C.; Amore, E.; Emma, M.R.; Augello, G.; Craparo, E.F.; Cervello, M. Lipid nanocarriers containing sorafenib inhibit colonies formation in human hepatocarcinoma cells. Int. J. Pharm. 2015, 493, 75–85. [Google Scholar] [CrossRef]
- Taymouri, S.; Alem, M.; Varshosaz, J.; Rostami, M.; Akbari, V.; Firoozpour, L. Biotin decorated sunitinib loaded nanostructured lipid carriers for tumor targeted chemotherapy of lung cancer. J. Drug Deliv. Sci. Technol. 2019, 50, 237–247. [Google Scholar] [CrossRef]
- Gilani, S.J.; Bin-Jumah, M.N.; Imam, S.S.; Zafar, A.; Yasir, M.; Alshehri, S.; Ghuneim, M.M. Formulation of osimertinib nano lipid carriers: Optimization, characterization and cytotoxicity assessment. J. Clust. Sci. 2022. [Google Scholar] [CrossRef]
- Gorle, A. Design, Development and Characterization of Nanostructure Lipidcarriers (Nlcs) by Hph Method Loaded with Anticancer Drug. SSRN 2022. Available online: https://ssrn.com/abstract=4054657 (accessed on 30 October 2022). [CrossRef]
- Patel, P.; Patel, M. Enhanced oral bioavailability of nintedanib esylate with nanostructured lipid carriers by lymphatic targeting: In Vitro, cell line and in vivo evaluation. Eur. J. Phar. Sci. 2021, 159, 105715. [Google Scholar] [CrossRef]
- Gupta, B.; Poudel, B.K.; Tran, T.H.; Pradhan, R.; Cho, H.J.; Jeong, J.H.; Shin, B.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Modulation of pharmacokinetic and cytotoxicity profile of imatinib base by employing optimized nanostructured lipid carriers. Pharm. Res. 2015, 32, 2912–2927. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/search?distance=50&cond=cancer&term=Nanoparticle&viewType=Table&limit=100&page=1 (accessed on 22 November 2022).
- ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/search?distance=50&cond=cancer&term=Nanoparticle&viewType=Table&limit=100&page=2 (accessed on 22 November 2022).
- ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/search?distance=50&cond=cancer&term=Nanoparticle&viewType=Table&limit=100&page=3 (accessed on 22 November 2022).
- ClinicalTrials.gov. Available online: https://beta.clinicaltrials.gov/search?distance=50&cond=cancer&term=Nanoparticle&viewType=Table&limit=100&page=4 (accessed on 22 November 2022).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INN | Brand Name | Approval for the 1st Indication | Type | Kinase Target | Major Therapeutical Uses |
|---|---|---|---|---|---|
| afatinib | Gilotrif | 2013 | V | EGFR, HER2 | NSCLC |
| axitinib | Inlyta | 2012 | II | VEGFR1-3, PDGFR | RCC |
| brigatinib | Alunbrig | 2017 | I | ALK, ROS1, IGF-1R, Flt3, EGFR | NSCLC |
| bortezomib | Velcade | 2003 | I | proteasome | multiple myeloma, MCL |
| cabozantinib | Cometriq, Cabometyx | 2012 | I | RET, HGFR, VEGFR1-3, Kit, TrkB, Flt3, Axl, Tie2, ROS1 | MTC, RCC, HCC |
| crizotinib | Xalkori | 2011 | II | ALK, HGFR, ROS1, MST1R | NSCLC |
| dasatinib | Sprycel | 2006 | I | BCR-ABL, EGFR, PDGFR Src, Lck, Yes, Fyn, Kit, EphA2 | CML, ALL |
| erlotinib | Tarceva | 2004 | I | EGFR, HER1 | NSCLC, SCLC, PaC |
| gefitinib | Iressa | 2009 | I | EGFR | NSCLC |
| imatinib | Gleevec | 2001 | II | BCR-ABL, c-Kit, PDGFR | CML, ALL, DFSP, HES, GIST, MDS/MDP |
| lapatinib | Tykerb | 2007 | II | EGFR, HER2 | breast cancer |
| nintedanib | Ofev | 2014 | I | VEGFR, FGFR, PDGFR | pulmonary fibrosis |
| osimertinib | Tagrisso | 2015 | V | EGFR | NSCLC |
| pazopanib | Votrient | 2009 | I | VEGFR1-3, PDGFR, FGFR1/3, Kit, Lck, Fms, Itk | RCC, STS |
| ponatinib | Iclusig | 2013 | II | BCR-ABL, VEGFR, PDGFR, FGFR, EphR, Src, Kit, RET, Tie2, Flt3 | CML, ALL |
| regorafenib | Stivarga | 2012 | II | BCR-ABL, VEGFR, BRAF, c-Kit, PDGFR, RET, FGFR, Tie2, Eph | CRC, GIST |
| selumetinib | Koselugo | 2020 | V | MEK1/2 | NF1 |
| sorafenib | Nexavar | 2005 | II | B/C-Raf, BRAF, c-Kit, Flt3, RET, VEGFR1-3, PDGFR | RCC, DTC, HCC, ThC |
| sunitinib | Sutent | 2006 | II | PDGF, VEGFR1-3, c-Kit, Flt3, CSF-1R, RET | CML, RCC, GIST, PNET |
| trametinib | Mekinist | 2013 | III | MEK1/2 | melanoma, NSCLC |
| TKI | Ingredients | Particle Size | Tested Human Cancer Cell Lines | Benefits | Refs. |
|---|---|---|---|---|---|
| brigatinib | oleic acid, Tween® 20, diethylene glycol monoethylether | ca. 50 nm | lung adenocarcinoma A549 cells | ↑ solubility (205×) ↑ intestinal permeability ↑ anticancer effect | [118] |
| dasatinib | oleic acid, Cremophor® RH-40, 1,2-propanediol | ca. 16 nm | MDA-MB-231 breast cancer cells | ↑ intestinal permeability ↑ anticancer effect | [119] |
| imatinib | Cremophor® EL, Labrasol® ALF, Lauroglycol™ 90 | ca. 47 nm | MDST-8 colon carcinoma cells | ↑ anticancer effect | [120] |
| imatinib | ethyl oleate, Tween® 80, polyethylene glycol 600 | ca. 81 nm | MCF-7 breast cancer cells | ↑ anticancer effect | [121] |
| sorafenib | medium-chain triglycerides, lecithin, Tween® 80 | ca. 43 nm | HT-29 colorectal adenocarcinoma cells | ↑ anticancer effect | [122] |
| sorafenib | glycerol, Lipoid S75, Tween® 80 | 75–107 nm | HepG2 liver cancer cells | ↑ anticancer effect | [123] |
| sunitinib | Lauroglycol™ 90, Triton-X100, Transcutol®-P | ca. 42 nm | HT-29 colorectal adenocarcinoma cells | ↑ anticancer effect | [124] |
| TKI | Ingredients | Particle Size | Tested Human Cancer Cell Lines | Benefits | Refs. |
|---|---|---|---|---|---|
| erlotinib | lecithin, cholesterol, chitosan, anti-EGFR aptamer | 70–200 nm | lung adenocarcinoma PC-9, H-1975 cells | long-term stability effective targeting ↑ drug accumulation ↑ anticancer effect | [135] |
| imatinib | sodium-deoxycholate, hyaluronic acid, lecithin | ca. 159 nm | HT-29 colorectal adenocarcinoma cellsColo-320-DMF colon carcinoma | ↑ cellular uptake ↑ anticancer effect | [136] |
| imatinib + paclitaxel | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000], cholesterol, folic acid, phosphatidylcholine | ca. 122 nm | MCF-7 breast cancer cellsPC-3 prostate cancer cells | ↑ internalization and accumulation in cancer cells ↑ anticancer effect | [137] |
| imatinib + tamoxifen | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, monopalmitoyl-2-hydroxy- sn-glycero-3-phosphocholine, sorbitan monooleate | ca. 168 nm | MCF-7, MDA-MB-231 breast cancer cells | synergistic inhibition ↑ anticancer effect | [138] |
| sorafenib | maleimide-polyethylen glycol-N-hydroxysuccinimide, soya lecithin, trimethyl chitosan, octreotide | ca. 127 nm | HepG2 hepatocellular carcinoma cells | sustained release ↑ accumulation in cancer cells ↑ anticancer effect | [139] |
| sorafenib | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine, hydrogenated soya phosphatidylcholine, cholesterol | ca. 107 nm | renal carcinoma cells | sustained release ↑ cellular uptake ↑ anticancer effect | [140] |
| afatinib | 1,2-distearoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-sn-glycero- 3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phosphocholine, 1,2-dioleoyl-3-trimethylammo- nium-propane chloride, cholesteryl hemisuccinate | 46–57 nm | lung adenocarcinoma H-1975, H-1650, HCC-827 cells | ↑ tumor-targetability induction apoptosis in H-1975 cells ↑ anticancer effect | [141] |
| TKI | Ingredients | Particle Size | Tested Human Cancer Cell Lines | Benefits | Refs. |
|---|---|---|---|---|---|
| brigatinib | stearic acid, soya lecithin | 176–787 nm | lung adenocarcinoma A549 cells | sustained release ↑ anticancer effect | [171] |
| gefitinib | Pluronic® F127, lecithin, polyethylene glycol 2000, stearic acid, cholesterol, glucosamine | ca. 187 nm | lung adenocarcinoma A549 cells | ↑ anticancer effect | [172] |
| erlotinib | Pluronic® F68, Transcutol®-P, glycerol monostearate | 300–800 nm | lung adenocarcinoma A549 cells | sustained release ↑ anticancer effect | [173] |
| erlotinib | Compritol® ATO 888, Tween® 80, Pluronic® 407 | <100 nm | lung adenocarcinoma A549 cells | ↑ anticancer effect | [174] |
| erlotinib | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(poly- ethylene glycol)-2000], hydrogenated soya phosphatidylcholine, polycaprolactone | ca. 170 nm | lung adenocarcinoma A549 cells | sustained release ↑ anticancer effect | [175] |
| erlotinib+ paclitaxel | Pluronic® 188, methoxy- poly(ethylene glycol)-b- poly(L-aspartic acid sodium, soya lecithin, glyceryl monostearate, didodecyldimethylammonium bromide | ca. 195 nm | lung adenocarcinoma NCI-H23 cells | pH-dependent and sustained release induction of apoptosis ↑ anticancer effect | [176] |
| imatinib | glyceryl palmitostearate, quillaja saponin, hyaluronic acid | ca. 92 nm | MCF-7 breast cancer cells | sustained release CD44 targeting ↑ cellular uptake ↑ anticancer effect | [177] |
| sorafenib | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(poly- ethylene glycol)-2000], folic acid, chitosan, chondroitin sulfate | ca. 178 nm | hepatocarcinoma SMMC-7721 cells | sustained release induction of apoptosis ↑ anticancer effect | [178] |
| sorafenib | thymidine 3′-(1,2-dipalmitoyl-sn- glycero-3-phosphate), 2′,3′-dioleyl- 5′-deoxy-5′-trimethyl-ammonium-uridine | ca. 200 nm | hepatocarcinoma HuH7, HepG2 cells breast cancer MDA-MB-134, T-47D cells | ↑ anticancer effect | [179] |
| sorafenib | poly(D,L-lactic-co-glycolic acid), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(poly- ethylene glycol)-2000], lecithin | ca. 190 nm | breast cancer MDA-MB-231 cells prostate cancer PC3-MM2 cells | sustained release long-term stability ↑ anticancer effect | [180] |
| sorafenib + MK-siRNA | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide- (polyethylene glycol)-2000], cholesterol, polyethylenimine, 1-methyl-4,4-bis[(9Z,12Z)-9,12-octadecadien-1-yloxy] piperidine, egg phosphatidylcoline, SP94 targeting peptide | 140–160 nm | HepG2 hepatocellular carcinoma cells | specific delivery targeting ↑ drug accumulation ↑ anticancer effect | [181] |
| sorafenib + selumetinib | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1,2-distearoyl-sn- glycero-3-phosphoethanolamine- N-[maleimide(polyethylene glycol)-2000], poly(D,L-lactic-co- glycolic acid), polyvinyl alcohol, cholesterol, N-acetylgalactos- amine | ca. 170 nm | hepatocellular carcinoma HepG2, Hep3B cells glioblastoma DBTRG-05MG cells | induction of apoptosis ↑ anticancer effect | [182] |
| sorafenib + paclitaxel | distearoyl phosphoethanolamine-polyethylene glycol, (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (poly(ethylene glycol)-2000], cetyl palmitate, Pluronic® F68, polyvinyl alcohol | ca. 200 nm | human glioblastoma U87-MG cells lung adenocarcinoma A549 cells | ↑ drug accumulation ↑ cellular uptake avoided drug efflux pumps ↑ anticancer effect | [183] |
| sorafenib + iron oxide | cetyl palmitate, Tween® 80, EMG1300 (iron oxide with surfactant) | ca. 420 nm | HepG2 hepatocellular carcinoma cells | magnetically driven accumulation ↑ drug accumulation ↑ cellular uptake ↑ anticancer effect | [184] |
| sunitinib | Roghan Kermanshahi Ghee oil, fat tail sheep, dioctyl sulfosuccinate sodium salt, chitosan, gum tragacanth | 110–156 nm | acute myeloid leukemia THP-1 cells | sustained release ↑ anticancer effect | [185] |
| sunitinib | Phospholipon® 90H, soya lecithin, polyvinyl alcohol, chitosan | ca. 439 nm | MCF-7 breast cancer cells | induction of apoptosis ↑ anticancer effect | [186] |
| afatinib | (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (poly(ethylene glycol)-2000], poly(D,L-lactic-co-glycolic acid), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy- (polyethylene glycol)-2000], lecithin | 147–183 nm | colorectal adenocarcinoma Caco-2 cells | ↑ targeting pH-sensitive penetration ↑ cellular uptake ↑ anticancer effect | [187] |
| afatinib | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(poly- ethylene glycol)-2000], 1,2-di- stearoyl-sn-glycero-3-phospho- ethanolamine-N-[methoxy(poly- ethylene glycol)-2000], lecithin, tight junction-modulating short peptides FD7/CCD | ca. 66 nm | lung adenocarcinoma PC9 cells | tight junctions perturbation blood-brain barrier permeation sustained release ↑ targeting ↑ anticancer effect | [188] |
| afatinib + cisplatin | 1,2-dilauroyl-sn-glycero-3-phosphocholine, 1,2-distearoyl-sn-gly- cero-3-phosphoethanolamine- N-[amino(polyethylene glycol)- 2000], poly(DL-lactide- co-glycolide) | ca. 138 nm | nasopharyngeal carcinoma HONE1cells | reduced cell viability induction of apoptosis synergistic efficacy ↑ anticancer effect | [189] |
| TKI | Ingredients | Particle Size | Tested Human Cancer Cell Lines | Benefits | Refs. |
|---|---|---|---|---|---|
| erlotinib | Precirol®, Miglyol®, poloxamer 407 | ca. 109 nm | lung adenocarcinoma A549 cells | ↑ cellular uptake apoptosis induction ↑ anticancer effect | [225] |
| gefitinib + paclitaxel | trilaurin, α-tocopherol, 1,2-distearoyl-sn-glycero-3-phosphocholine, (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000], luteinizing hormone release hormone | 100–300 nm | lung adenocarcinoma A549, H-1975, PC-9, PC-9GR cells | ↑ anticancer effect | [226] |
| imatinib | Compritol® ATO 888, oleic acid, Gelucire® 48/16, Gelucire® 43/01, Span 80, Tween® 80 | ca. 96 nm | gastric adenocarcinoma CRL-1739 cells | sustained release ↑ anticancer effect | [227] |
| imatinib | stearic acid, sesame oil, sodium lauryl sulphate, Tween® 80 | ca. 104 nm | MCF-7 breast cancer cells | ↑ anticancer effect | [228] |
| sorafenib | tripalmitin, Captex® 355 EP/NF | <300 nm | hepatocellular carcinoma HepG2, Hep3B, Huh7, PLC/PRF/5 cells | sustained release ↑ cellular uptake ↑ anticancer effect | [229] |
| sunitinib | Pluronic® F127, cholesterol, Labrafac™, biotin, stearylamine | ca. 125 nm | lung adenocarcinoma A549 cells | sustained release (>8 h) ↑ cellular uptake ↑ anticancer effect | [230] |
| osimertinib | stearic acid, Labrafil® M 1944CS, poloxamer 407 | ca. 162 nm | lung adenocarcinoma A549 cells | sustained release ↑ permeation ↑ anticancer effect | [231] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jampilek, J.; Kralova, K. Insights into Lipid-Based Delivery Nanosystems of Protein-Tyrosine Kinase Inhibitors for Cancer Therapy. Pharmaceutics 2022, 14, 2706. https://doi.org/10.3390/pharmaceutics14122706
Jampilek J, Kralova K. Insights into Lipid-Based Delivery Nanosystems of Protein-Tyrosine Kinase Inhibitors for Cancer Therapy. Pharmaceutics. 2022; 14(12):2706. https://doi.org/10.3390/pharmaceutics14122706
Chicago/Turabian StyleJampilek, Josef, and Katarina Kralova. 2022. "Insights into Lipid-Based Delivery Nanosystems of Protein-Tyrosine Kinase Inhibitors for Cancer Therapy" Pharmaceutics 14, no. 12: 2706. https://doi.org/10.3390/pharmaceutics14122706
APA StyleJampilek, J., & Kralova, K. (2022). Insights into Lipid-Based Delivery Nanosystems of Protein-Tyrosine Kinase Inhibitors for Cancer Therapy. Pharmaceutics, 14(12), 2706. https://doi.org/10.3390/pharmaceutics14122706