
Recombinant Alpha-1 Antitrypsin as Dry Powder for Pulmonary Administration: A Formulative Proof of Concept

 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Expression Vector
2.3. Culture Media
- –
- LB medium: 10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl.
- –
- Trace elements solution: 6 g/L CaCl2 2 H2O, 6 g/L FeSO4 7H2O, 1.15 g/L MnCl2 4 H2O, 0.8 g/L CoCl2 6H2O, 0.7 g/L ZnSO4 7H2O, 0.3 g/L CuCl2 2H2O, 0.02 g/L H3BO3, 0.25 g/L (NH4)6Mo7O24 4H2O, 5 g/L ethylenediaminetetraacetic acid (EDTA). The components were dissolved in the minimum amount of HCl and added to water containing EDTA. The solution was autoclaved and stored in the dark at 4 °C.
- –
- MgCl2 solution: 1 M MgCl2, filter-sterilized.
- –
- HCDC medium [33]: 10 mL/L of the trace element solution, 10 mL/L of the MgCl2 solution, 2 g/L yeast extract, 10 mL/L glycerol, 10 g/L K2HPO4, 10 g/L KH2PO4, 91 g/L Na2HPO4 2H2O, 1.1 g/L NH4Cl, 3.7 g/L KCl, 6.6 g (NH4)2SO4. The medium (7 L) was heat-sterilized inside the bioreactor.
2.4. Shake-Flask Experiments
2.5. Fed-Batch Culture
2.6. Protein Purification
2.7. Endotoxin Removal and Evaluation of the Proinflammatory Activity of rAAT Solutions
2.8. Activity Assays
2.9. SEC Chromatography
2.10. Circular Dichroism
2.11. Evaluation of Oxidation by Mass Spectrometry
2.12. Set Up of Spray-Drying Conditions
2.13. Protein Formulation
2.14. Thermal Analysis of Spray-Dried Powder
2.15. Flowability of Powders
2.16. Analysis of Particle Size Distribution
2.17. In Vitro Assessment of Aerodynamic Performance
2.18. Scanning Electron Microscopy (SEM)
2.19. Assessment of AAT Content in the Powder
2.20. Dynamic Light Scattering (DLS)
3. Results
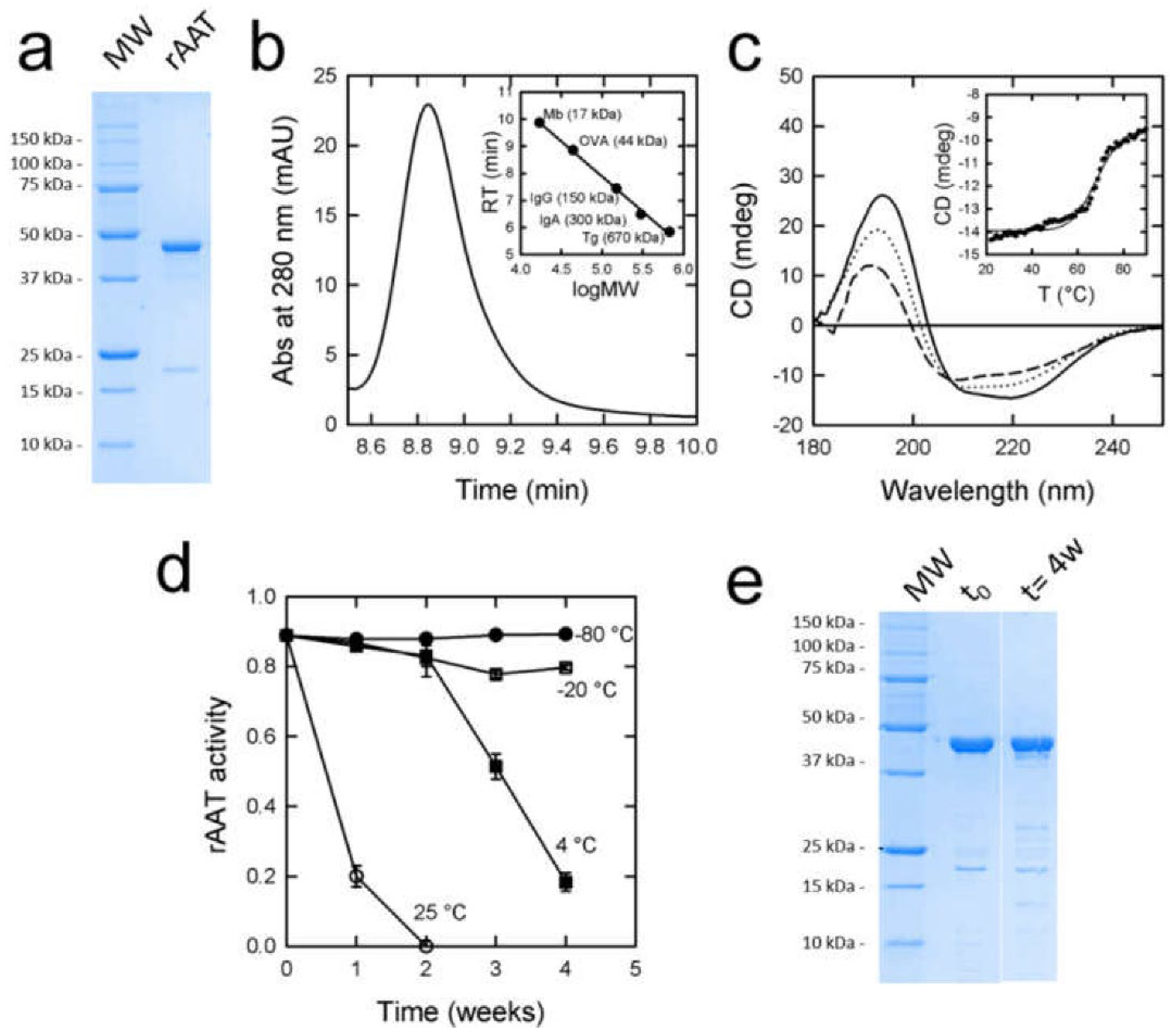
3.1. Expression and Purification of rAAT
3.2. Endotoxin Removal
3.3. rATT Characterization
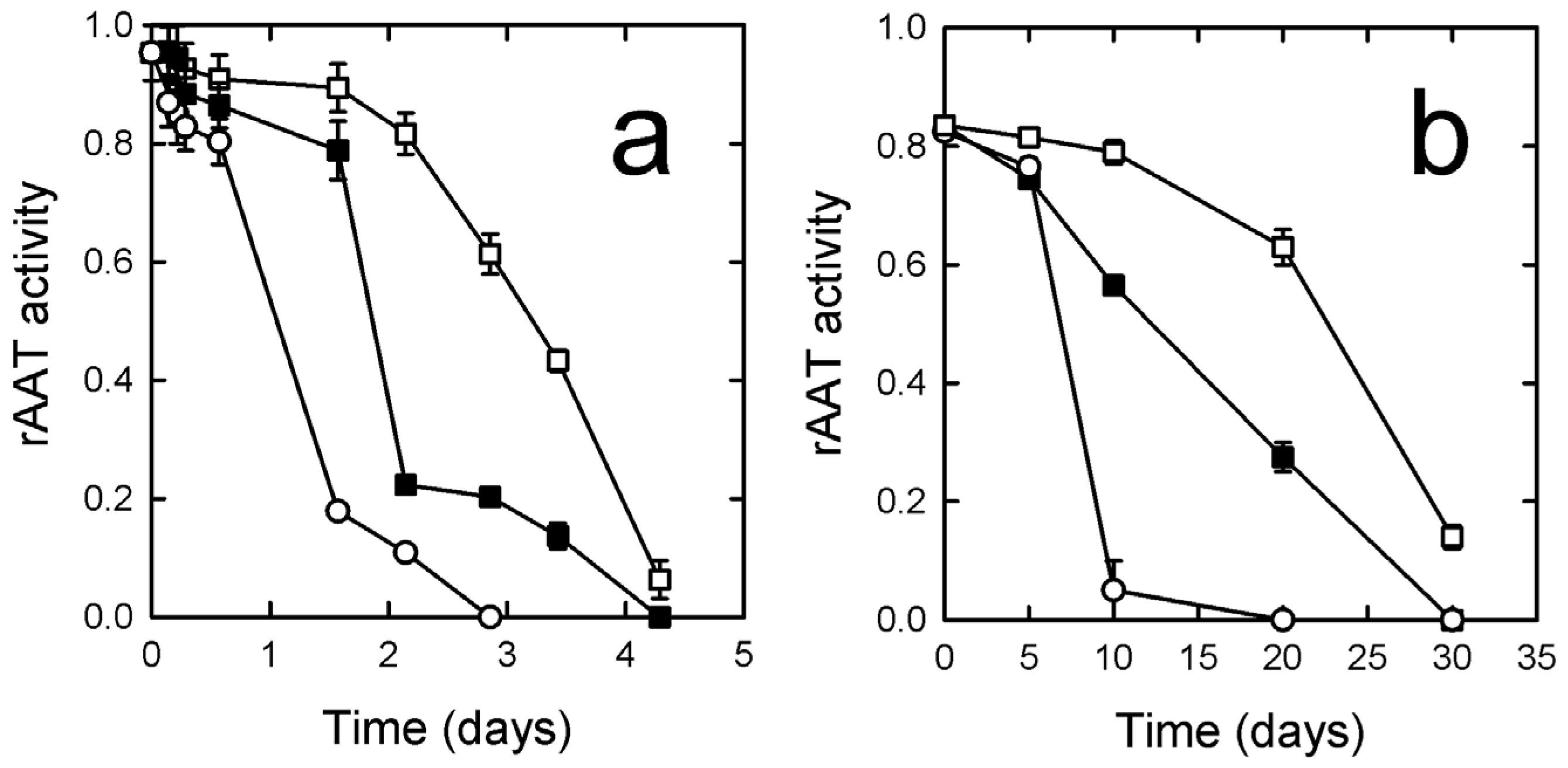
3.4. rATT Stability in Solution
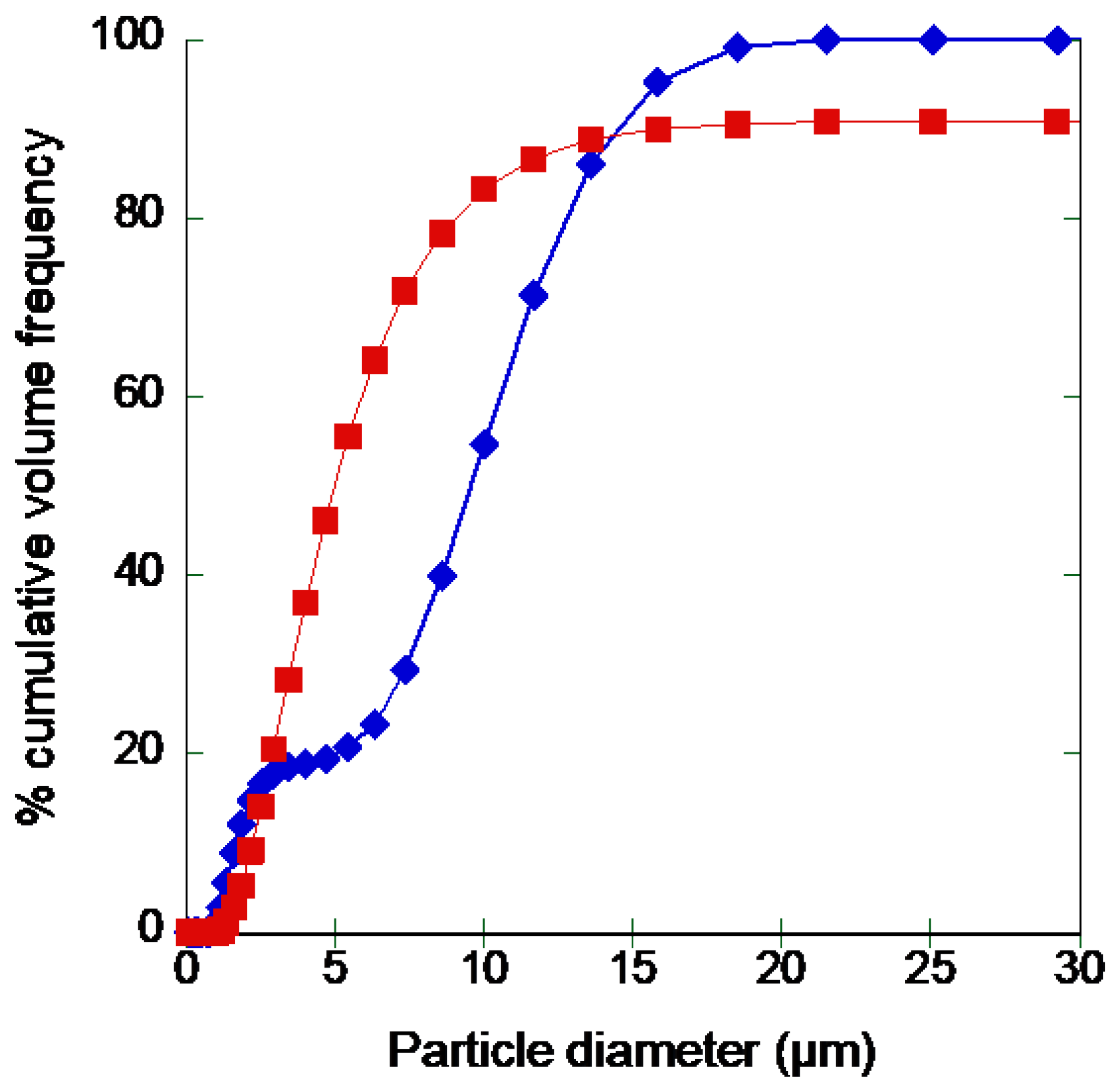
3.5. Formulation of Recombinant rAAT as Dry Powder
3.5.1. Effect of Buffering Agents and Excipients
3.5.2. Formulation of rAAT
3.5.3. Protein Stability and Activity upon Formulation
3.5.4. Formulation of hAAT
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Köhnlein, T.; Welte, T. The Discovery of A1-Antitrypsin and Its Role in Health and Disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, I.G.; Hendy, J.; Coughlin, P.; Horvath, A.; Lévesque, J.-P. Serine Protease Inhibitors Serpina1 and Serpina3 Are Down-Regulated in Bone Marrow during Hematopoietic Progenitor Mobilization. J. Exp. Med. 2005, 201, 1077–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.-S.; Kim, J.-S.; Lee, S.M.; Park, S.J. Additional N-Glycosylation in the N-Terminal Region of Recombinant Human Alpha-1 Antitrypsin Enhances the Circulatory Half-Life in Sprague-Dawley Rats. Glycoconj. J. 2016, 33, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Parfrey, H. A1-Antitrypsin Deficiency • 4: Molecular Pathophysiology. Thorax 2004, 59, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Wintrode, P.L. Effects of Glycosylation on the Stability and Flexibility of a Metastable Protein: The Human Serpin A1-Antitrypsin. Int. J. Mass Spectrom. 2011, 302, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, C.T.N. Neutrophil Serine Proteases: Specific Regulators of Inflammation. Nat. Rev. Immunol. 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Stoller, J.K.; Aboussouan, L.S. A Review of A1-Antitrypsin Deficiency. Am. J. Respir. Crit. Care Med. 2012, 185, 246–259. [Google Scholar] [CrossRef]
- Hazari, Y.M.; Bashir, A.; Habib, M.; Bashir, S.; Habib, H.; Qasim, M.A.; Shah, N.N.; Haq, E.; Teckman, J.; Fazili, K.M. Alpha-1-Antitrypsin Deficiency: Genetic Variations, Clinical Manifestations and Therapeutic Interventions. Mutat. Res. Rev. Mutat. Res. 2017, 773, 14–25. [Google Scholar] [CrossRef]
- Dunlea, D.M.; Fee, L.T.; McEnery, T.; McElvaney, N.G.; Reeves, E.P. The Impact of Alpha-1 Antitrypsin Augmentation Therapy on Neutrophil-Driven Respiratory Disease in Deficient Individuals. J. Inflamm. Res. 2018, 11, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Hoenderdos, K.; Condliffe, A. The Neutrophil in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- Turner, A.M. Alpha-1 Antitrypsin Deficiency: New Developments in Augmentation and Other Therapies. BioDrugs 2013, 27, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Mohanka, M.; Khemasuwan, D.; Stoller, J.K. A Review of Augmentation Therapy for Alpha-1 Antitrypsin Deficiency. Expert Opin. Biol. Ther. 2012, 12, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, K.D.; Tavill, A.S. Liver Disease in Alpha 1-Antitrypsin Deficiency: A Review. Am. J. Gastroenterol. 2008, 103, 2136–2141; quiz 2142. [Google Scholar] [CrossRef] [PubMed]
- Bianchera, A.; Alomari, E.; Bruno, S. Augmentation Therapy with Alpha-1 Antitrypsin: Present and Future of Production, Formulation, and Delivery. Curr. Med. Chem. 2022, 29, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Herth, F.J.F.; Sandhaus, R.A.; Turner, A.M.; Sucena, M.; Welte, T.; Greulich, T. Alpha 1 Antitrypsin Therapy in Patients with Alpha 1 Antitrypsin Deficiency: Perspectives from a Registry Study and Practical Considerations for Self-Administration During the COVID-19 Pandemic. Int. J. Chron. Obstruct. Pulmon. Dis. 2021, 16, 2983–2996. [Google Scholar] [CrossRef]
- Griese, M.; Scheuch, G. Delivery of Alpha-1 Antitrypsin to Airways. Ann. ATS 2016, 13, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Hertel, S.P.; Winter, G.; Friess, W. Protein Stability in Pulmonary Drug Delivery via Nebulization. Adv. Drug Deliv. Rev. 2015, 93, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Monk, R.; Graves, M.; Williams, P.; Strange, C. Inhaled Alpha 1-Antitrypsin: Gauging Patient Interest in a New Treatment. COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 411–415. [Google Scholar] [CrossRef]
- Durrani, M.J.; Kumar, H.; Kreiger, T.; Kabingue, K.; Mosher, V.; Barr, P.J.; Bathurst, I.C. Alpha 1-Antitrypsin Compositions and Treatment Methods Using Such Compositions. European Patent Application No. EP1684719, 2 August 2006. [Google Scholar]
- Nayar, R.; Manning, M.; Barr, P.; Pemberton, P.; Bathurst, I.; Gibson, H. Dry Recombinant Human Alpha 1-Antitrypsin Formulation. European Patent Application No. EP1685160, 26 May 2005. [Google Scholar]
- El Jamal, M.; Patton, J.S. Pulmonary Administration of Dry Powder Alpha 1-Antitrypsin. European Patent EP 0866726, 9 January 2008. [Google Scholar]
- Bar, L. Dry Powder Formulations of Alpha-1 Antitrypsin. European Patent Application No. EP3565584, 28 June 2018. [Google Scholar]
- Marante, T.; Viegas, C.; Duarte, I.; Macedo, A.S.; Fonte, P. An Overview on Spray-Drying of Protein-Loaded Polymeric Nanoparticles for Dry Powder Inhalation. Pharmaceutics 2020, 12, 1032. [Google Scholar] [CrossRef]
- Lockett, A.D.; Van Demark, M.; Gu, Y.; Schweitzer, K.S.; Sigua, N.; Kamocki, K.; Fijalkowska, I.; Garrison, J.; Fisher, A.J.; Serban, K.; et al. Effect of Cigarette Smoke Exposure and Structural Modifications on the α-1 Antitrypsin Interaction with Caspases. Mol. Med. 2012, 18, 445–454. [Google Scholar] [CrossRef]
- Cantin, A.M.; Woods, D.E.; Cloutier, D.; Héroux, J.; Dufour, E.K.; Leduc, R. Leukocyte Elastase Inhibition Therapy in Cystic Fibrosis: Role of Glycosylation on the Distribution of Alpha-1–Proteinase Inhibitor in Blood versus Lung. J. Aerosol. Med. 2002, 15, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Kee, S.; Weber, D.; Popp, B.; Nowak, T.; Schäfer, W.; Gröner, A.; Roth, N.J. Pathogen Safety and Characterisation of a Highly Purified Human Alpha1-Proteinase Inhibitor Preparation. Biologicals 2017, 47, 25–32. [Google Scholar] [CrossRef]
- Karnaukhova, E.; Ophir, Y.; Golding, B. Recombinant Human Alpha-1 Proteinase Inhibitor: Towards Therapeutic Use. Amino Acids 2006, 30, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Jha, S.; Sanyal, I.; Amla, D.V. Expression and Purification of Recombinant Human A1-Proteinase Inhibitor and Its Single Amino Acid Substituted Variants in Escherichia coli for Enhanced Stability and Biological Activity. J. Biotechnol. 2010, 147, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Johansen, H.; Sutiphong, J.; Sathe, G.; Jacobs, P.; Cravador, A.; Bollen, A.; Rosenberg, M.; Shatzman, A. High-Level Production of Fully Active Human Alpha 1-Antitrypsin in Escherichia coli. Mol. Biol. Med. 1987, 4, 291–305. [Google Scholar] [PubMed]
- Straus, S.D.; Fells, G.A.; Wewers, M.D.; Courtney, M.; Tessier, L.; Tolstoshev, P.; Lecocq, J.-P.; Crystal, R.G. Evaluation of Recombinant DNA-Directed E. Coli Produced A1-Antitrypsin as an Anti-Neutrophil Elastase for Potential Use as Replacement Therapy of A1-Antitrypsin Deficiency. Biochem. Biophys. Res. Commun. 1985, 130, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Courtney, M.; Buchwalder, A.; Tessier, L.H.; Jaye, M.; Benavente, A.; Balland, A.; Kohli, V.; Lathe, R.; Tolstoshev, P.; Lecocq, J.P. High-Level Production of Biologically Active Human Alpha 1-Antitrypsin in Escherichia coli. Proc. Natl. Acad. Sci. USA 1984, 81, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, B.; Hedstrom, L.; Hebert, D.N.; Gierasch, L.M.; Gershenson, A. Expression and Purification of Active Recombinant Human Alpha-1 Antitrypsin (AAT) from Escherichia coli. In Alpha-1 Antitrypsin Deficiency: Methods and Protocols; Borel, F., Mueller, C., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 195–209. ISBN 978-1-4939-7163-3. [Google Scholar]
- Khalilzadeh, R.; Shojaosadati, S.A.; Bahrami, A.; Maghsoudi, N. Over-Expression of Recombinant Human Interferon-Gamma in High Cell Density Fermentation of Escherichia coli. Biotechnol. Lett. 2003, 25, 1989–1992. [Google Scholar] [CrossRef]
- Lee, S.Y. High Cell-Density Culture of Escherichia Coli. Trends Biotechnol. 1996, 14, 98–105. [Google Scholar] [CrossRef]
- Porath, J. Immobilized Metal Ion Affinity Chromatography. Protein Expr. Purif. 1992, 3, 263–281. [Google Scholar] [CrossRef]
- Bieth, J.; Spiess, B.; Wermuth, C.G. The Synthesis and Analytical Use of a Highly Sensitive and Convenient Substrate of Elastase. Biochem. Med. 1974, 11, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, S.; Parumasivam, T.; Denman, J.A.; Gengenbach, T.; Tang, P.; Mao, S.; Chan, H.-K. L-Leucine as an Excipient against Moisture on in Vitro Aerosolization Performances of Highly Hygroscopic Spray-Dried Powders. Eur. J. Pharm. Biopharm. 2016, 102, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Seville, P.C.; Learoyd, T.P.; Li, H.-Y.; Birchall, J.C.; Williamson, I.J. Amino Acid-Modified Spray-Dried Powders with Enhanced Aerosolisation Properties for Pulmonary Drug Delivery. Powder Technol. 2007, 178, 40–50. [Google Scholar] [CrossRef]
- Mensink, M.A.; Frijlink, H.W.; van der Voort Maarschalk, K.; Hinrichs, W.L.J. How Sugars Protect Proteins in the Solid State and during Drying (Review): Mechanisms of Stabilization in Relation to Stress Conditions. Eur. J. Pharm. Biopharm. 2017, 114, 288–295. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an Online Server for Protein Secondary Structure Analyses from Circular Dichroism Spectroscopic Data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of Either Methionine 351 or Methionine 358 in Alpha 1-Antitrypsin Causes Loss of Anti-Neutrophil Elastase Activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Moraga, F.; Janciauskiene, S. Activation of Primary Human Monocytes by the Oxidized Form of A1-Antitrypsin *. J. Biol. Chem. 2000, 275, 7693–7700. [Google Scholar] [CrossRef] [Green Version]
- Faghihi, H.; Vatanara, A.; Najafabadi, A.R.; Ramezani, V.; Gilani, K. The Use of Amino Acids to Prepare Physically and Conformationally Stable Spray-Dried IgG with Enhanced Aerosol Performance. Int. J. Pharm. 2014, 466, 163–171. [Google Scholar] [CrossRef]
- Carpenter, J.F.; Pikal, M.J.; Chang, B.S.; Randolph, T.W. Rational Design of Stable Lyophilized Protein Formulations: Some Practical Advice. Pharm. Res. 1997, 14, 969–975. [Google Scholar] [CrossRef]
- Hydrodynamic Radius Converter. Available online: https://www.fluidic.com/toolkit/hydrodynamic-radius-converter/ (accessed on 30 November 2022).
- Kwon, K.S.; Yu, M.H. Effect of Glycosylation on the Stability of Alpha1-Antitrypsin toward Urea Denaturation and Thermal Deactivation. Biochim. Biophys. Acta 1997, 1335, 265–272. [Google Scholar] [CrossRef]
- Powell, L.M.; Pain, R.H. Effects of Glycosylation on the Folding and Stability of Human, Recombinant and Cleaved Alpha 1-Antitrypsin. J. Mol. Biol. 1992, 224, 241–252. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.; Saldova, R.; Wormald, M.R.; Rudd, P.M.; McElvaney, N.G.; Reeves, E.P. The Role and Importance of Glycosylation of Acute Phase Proteins with Focus on Alpha-1 Antitrypsin in Acute and Chronic Inflammatory Conditions. J. Proteome Res. 2014, 13, 3131–3143. [Google Scholar] [CrossRef] [PubMed]
- Cowden, D.I.; Fisher, G.E.; Weeks, R.L. A Pilot Study Comparing the Purity, Functionality and Isoform Composition of Alpha-1-Proteinase Inhibitor (Human) Products. Curr. Med. Res. Opin. 2005, 21, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Kolarich, D.; Turecek, P.L.; Weber, A.; Mitterer, A.; Graninger, M.; Matthiessen, P.; Nicolaes, G.A.F.; Altmann, F.; Schwarz, H.P. Biochemical, Molecular Characterization, and Glycoproteomic Analyses of Alpha(1)-Proteinase Inhibitor Products Used for Replacement Therapy. Transfusion 2006, 46, 1959–1977. [Google Scholar] [CrossRef] [PubMed]
- Cundell, A.M. Bacterial Endotoxin Requirements for Dry Powder Inhalants and Their Excipients: Are They Critical Quality Attributes? PDA J. Pharm. Sci. Technol. 2014, 68, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Loh, L.C.; Vyas, B.; Kanabar, V.; Kemeny, D.M.; O’Connor, B.J. Inhaled Endotoxin in Healthy Human Subjects: A Dose-Related Study on Systemic Effects and Peripheral CD4+ and CD8+ T Cells. Respir. Med. 2006, 100, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Steckel, H.; Furkert, F.H. Endotoxin Testing in Inhalation Grade Lactose—A Useful Quality Parameter? Int. J. Pharm. 2004, 275, 211–215. [Google Scholar] [CrossRef]
- Sou, T.; Bergström, C.A.S. Contemporary Formulation Development for Inhaled Pharmaceuticals. J. Pharm. Sci. 2021, 110, 66–86. [Google Scholar] [CrossRef]
- Andya, J.D.; Maa, Y.-F.; Costantino, H.R.; Nguyen, P.-A.; Dasovich, N.; Sweeney, T.D.; Hsu, C.C.; Shire, S.J. The Effect of Formulation Excipients on Protein Stability and Aerosol Performance of Spray-Dried Powders of a Recombinant Humanized Anti-IgE Monoclonal Antibody1. Pharm. Res. 1999, 16, 350–358. [Google Scholar] [CrossRef]
- Maa, Y.F.; Costantino, H.R.; Nguyen, P.A.; Hsu, C.C. The Effect of Operating and Formulation Variables on the Morphology of Spray-Dried Protein Particles. Pharm. Dev. Technol. 1997, 2, 213–223. [Google Scholar] [CrossRef]
- Maury, M.; Murphy, K.; Kumar, S.; Shi, L.; Lee, G. Effects of Process Variables on the Powder Yield of Spray-Dried Trehalose on a Laboratory Spray-Dryer. Eur. J. Pharm. Biopharm. 2005, 59, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Roe, K.D.; Labuza, T.P. Glass Transition and Crystallization of Amorphous Trehalose-Sucrose Mixtures. Int. J. Food Prop. 2005, 8, 559–574. [Google Scholar] [CrossRef]
- Weng, L.; Elliott, G.D. Distinctly Different Glass Transition Behaviors of Trehalose Mixed with Na2HPO4 or NaH2PO4: Evidence for Its Molecular Origin. Pharm. Res. 2015, 32, 2217–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüle, S.; Schulz-Fademrecht, T.; Garidel, P.; Bechtold-Peters, K.; Frieb, W. Stabilization of IgG1 in Spray-Dried Powders for Inhalation. Eur. J. Pharm. Biopharm. 2008, 69, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Costantino, H.R.; Andya, J.D.; Nguyen, P.A.; Dasovich, N.; Sweeney, T.D.; Shire, S.J.; Hsu, C.C.; Maa, Y.F. Effect of Mannitol Crystallization on the Stability and Aerosol Performance of a Spray-Dried Pharmaceutical Protein, Recombinant Humanized Anti-IgE Monoclonal Antibody. J. Pharm. Sci. 1998, 87, 1406–1411. [Google Scholar] [CrossRef]
- Chen, Y.; Ling, J.; Li, M.; Su, Y.; Arte, K.S.; Mutukuri, T.T.; Taylor, L.S.; Munson, E.J.; Topp, E.M.; Zhou, Q.T. Understanding the Impact of Protein–Excipient Interactions on Physical Stability of Spray-Dried Protein Solids. Mol. Pharm. 2021, 18, 2657–2668. [Google Scholar] [CrossRef]
- Sou, T.; Kaminskas, L.M.; Nguyen, T.-H.; Carlberg, R.; McIntosh, M.P.; Morton, D.A.V. The Effect of Amino Acid Excipients on Morphology and Solid-State Properties of Multi-Component Spray-Dried Formulations for Pulmonary Delivery of Biomacromolecules. Eur. J. Pharm. Biopharm. 2013, 83, 234–243. [Google Scholar] [CrossRef]
- Crystal, R.G. Alpha 1-Antitrypsin Deficiency, Emphysema, and Liver Disease. Genetic Basis and Strategies for Therapy. J. Clin. Investig. 1990, 85, 1343–1352. [Google Scholar] [CrossRef]
- Patton, J.S. Mechanisms of Macromolecule Absorption by the Lungs. Adv. Drug Deliv. Rev. 1996, 19, 3–36. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | −1 | +1 |
|---|---|---|
| Bulking agent | Sucrose | Mannitol |
| Leucine concentration (mg/mL) | 0.5 | 1 |
| Nozzle diameter (mm) | 0.5 | 0.7 |
| Sample ID | Input Parameters | Output | |||||
|---|---|---|---|---|---|---|---|
| x1 = Type of Bulking Agent | x2 = Leucine Concentration (mg/mL) | x3 = Nozzle Diameter (mm) | Yield (%) | Emitted Fraction (%) | Fine Particle Fraction (%) | DAR (°) | |
| S-0.5-0.5 | Sucrose | 0.5 | 0.5 | 46.66 | 84.4 ± 1.47 | 84.2 ± 17.2 | 42° ± 1° |
| S-0.5-0.7 | 0.7 | 72.19 | 85.07 ± 2.63 | 64.8 ± 9.9 | 37° ± 3° | ||
| S-1-0.5 | 1 | 0.5 | 54.4 | 81.25 ± 3.30 | 90.7 ± 7.6 | 45 ° ± 1° | |
| S-1-0.7 | 0.7 | 76.32 | 81.84 ± 2.61 | 72.9 ± 6.4 | 36° ± 3° | ||
| M-0.5-0.5 | Mannitol | 0.5 | 0.5 | 62.88 | 84.89 ± 1.07 | 86.3 ± 16.3 | 37° ± 2° |
| M-0.5-0.7 | 0.7 | 73.41 | 91.24 ± 9.58 | 78.8 ± 7.2 | 38° ± 1° | ||
| M-1-0.5 | 1 | 0.5 | 69.89 | 78.43 ± 3.17 | 92.8 ± 10.8 | 47° ± 1° | |
| M-1-0.7 | 0.7 | 72.27 | 98.56 ± 0.65 | 67.2 ± 0.2 | 37° ± 11° | ||
| Formulation | Dv10 μm | Dv50 μm | Dv90 μm | Span |
|---|---|---|---|---|
| S-0.5-0.5 | 1.15 | 2.34 | 5.49 | 1.85 |
| S-0.5-0.7 | 2.05 | 4.49 | 8.69 | 1.48 |
| S-1-0.5 | 1.04 | 1.98 | 8.94 | 3.99 |
| S-1-0.7 | 2.63 | 4.34 | 6.77 | 0.94 |
| M-0.5-0.5 | 1.06 | 2.12 | 5.44 | 2.07 |
| M-0.5-0.7 | 1.46 | 2.74 | 5.18 | 1.36 |
| M-1-0.5 | 1.06 | 2.12 | 5.44 | 2.07 |
| M-1-0.7 | 1.64 | 3.47 | 7.54 | 1.70 |
| Bulking Agent | Yield (%) | Emitted Fraction (%) | Fine Particle Fraction (%) | AAT Soluble Fraction | AAT Activity |
|---|---|---|---|---|---|
| Sucrose | 40.65 | 61.8 ± 3.4 | 39.2 ± 6.3 | 100% | 18% |
| Mannitol | 35.18 | 87.3 ± 0.4 | 41.54 ± 14.1 | 100% | 54% |
| Formulation | Yield (%) | Emitted Fraction (%) | Fine Particle Fraction (%) | AAT Soluble Fraction | AAT Activity on Elastase |
|---|---|---|---|---|---|
| SD-Prol | 43.44 | 60.9 ± 16.7 | 32.34 ± 25.39 | 100% | 67% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianchera, A.; Alomari, E.; Michielon, A.; Bazzoli, G.; Ronda, N.; Pighini, G.; Zanotti, I.; Giorgio, C.; Mozzarelli, A.; Bettini, R.; et al. Recombinant Alpha-1 Antitrypsin as Dry Powder for Pulmonary Administration: A Formulative Proof of Concept. Pharmaceutics 2022, 14, 2754. https://doi.org/10.3390/pharmaceutics14122754
Bianchera A, Alomari E, Michielon A, Bazzoli G, Ronda N, Pighini G, Zanotti I, Giorgio C, Mozzarelli A, Bettini R, et al. Recombinant Alpha-1 Antitrypsin as Dry Powder for Pulmonary Administration: A Formulative Proof of Concept. Pharmaceutics. 2022; 14(12):2754. https://doi.org/10.3390/pharmaceutics14122754
Chicago/Turabian StyleBianchera, Annalisa, Esraa’a Alomari, Annalisa Michielon, Gianluca Bazzoli, Nicoletta Ronda, Giovanni Pighini, Ilaria Zanotti, Carmine Giorgio, Andrea Mozzarelli, Ruggero Bettini, and et al. 2022. "Recombinant Alpha-1 Antitrypsin as Dry Powder for Pulmonary Administration: A Formulative Proof of Concept" Pharmaceutics 14, no. 12: 2754. https://doi.org/10.3390/pharmaceutics14122754
APA StyleBianchera, A., Alomari, E., Michielon, A., Bazzoli, G., Ronda, N., Pighini, G., Zanotti, I., Giorgio, C., Mozzarelli, A., Bettini, R., & Bruno, S. (2022). Recombinant Alpha-1 Antitrypsin as Dry Powder for Pulmonary Administration: A Formulative Proof of Concept. Pharmaceutics, 14(12), 2754. https://doi.org/10.3390/pharmaceutics14122754