
Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line

 , ,
, , 
Abstract
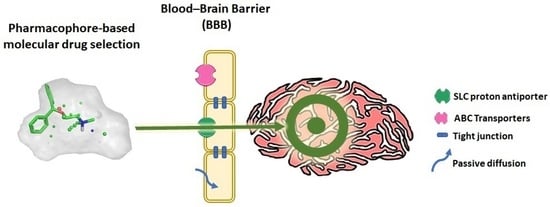
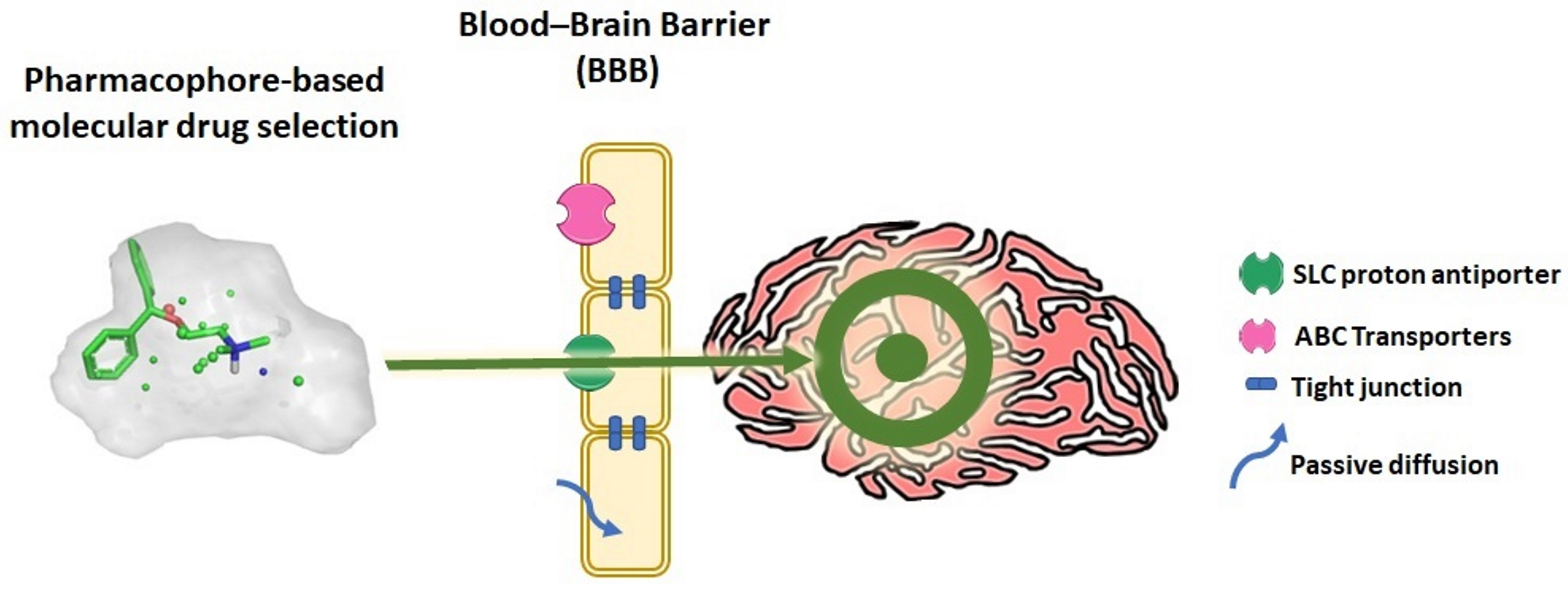
:
1. Introduction
2. Materials and Methods
2.1. Drugs and Chemicals
2.2. Cell Culture
2.3. In Vitro [3H]-Clonidine Trans-Stimulation Experiments in hCMEC/D3 Cells for Substrate Screening
2.4. Quantification of the Trans-Stimulation Effect for Selected Substrates
2.5. Determination and Classification of Substrate Candidates by [3H]-Clonidine Trans-Stimulation Experiments in hCMEC/D3 Cells
2.6. Data Transport Analysis
2.7. Pharmacophore Generation
2.8. Virtual Screening
3. Results
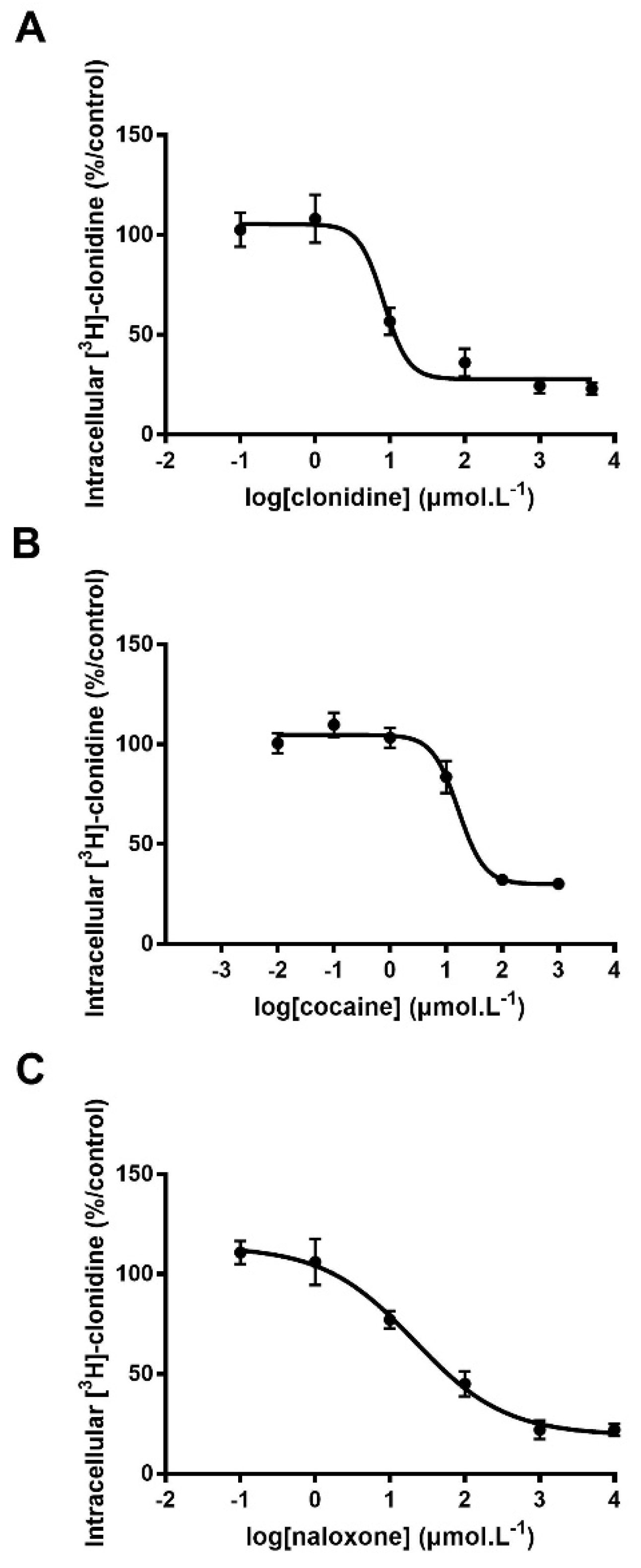
3.1. Characterization of Substrates by Trans-Stimulation Experiments
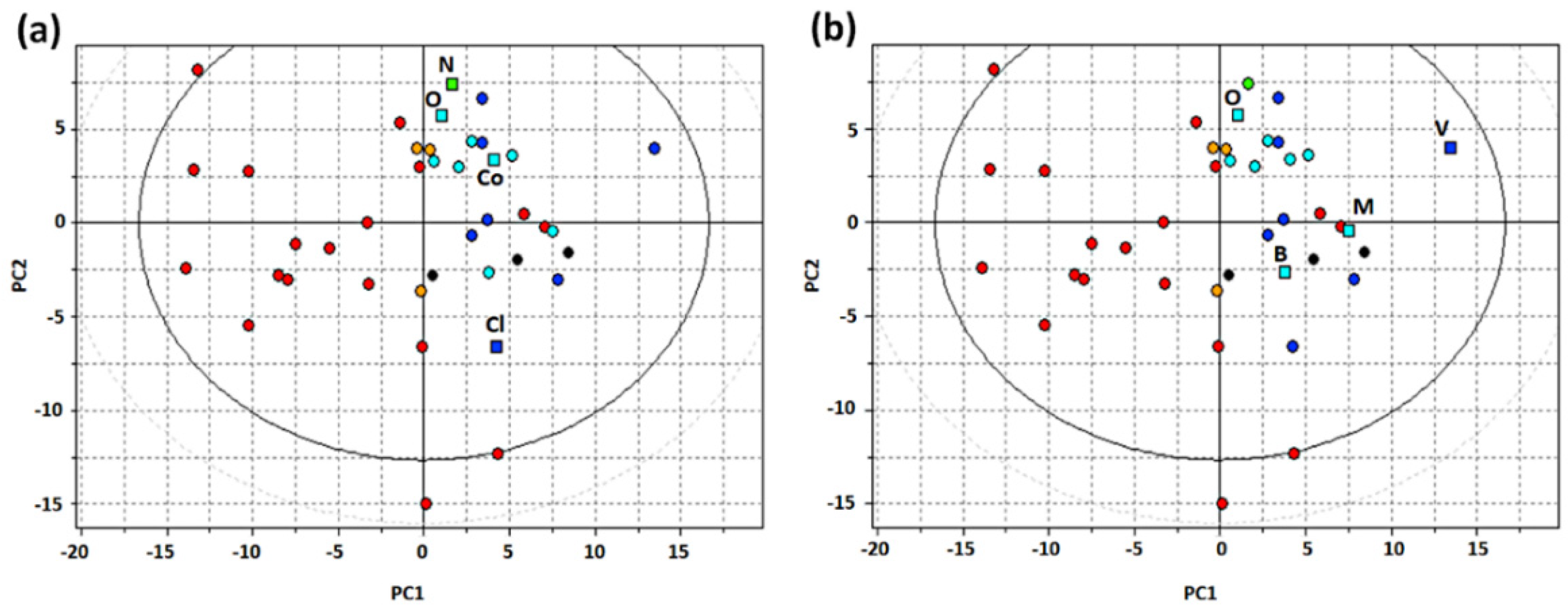
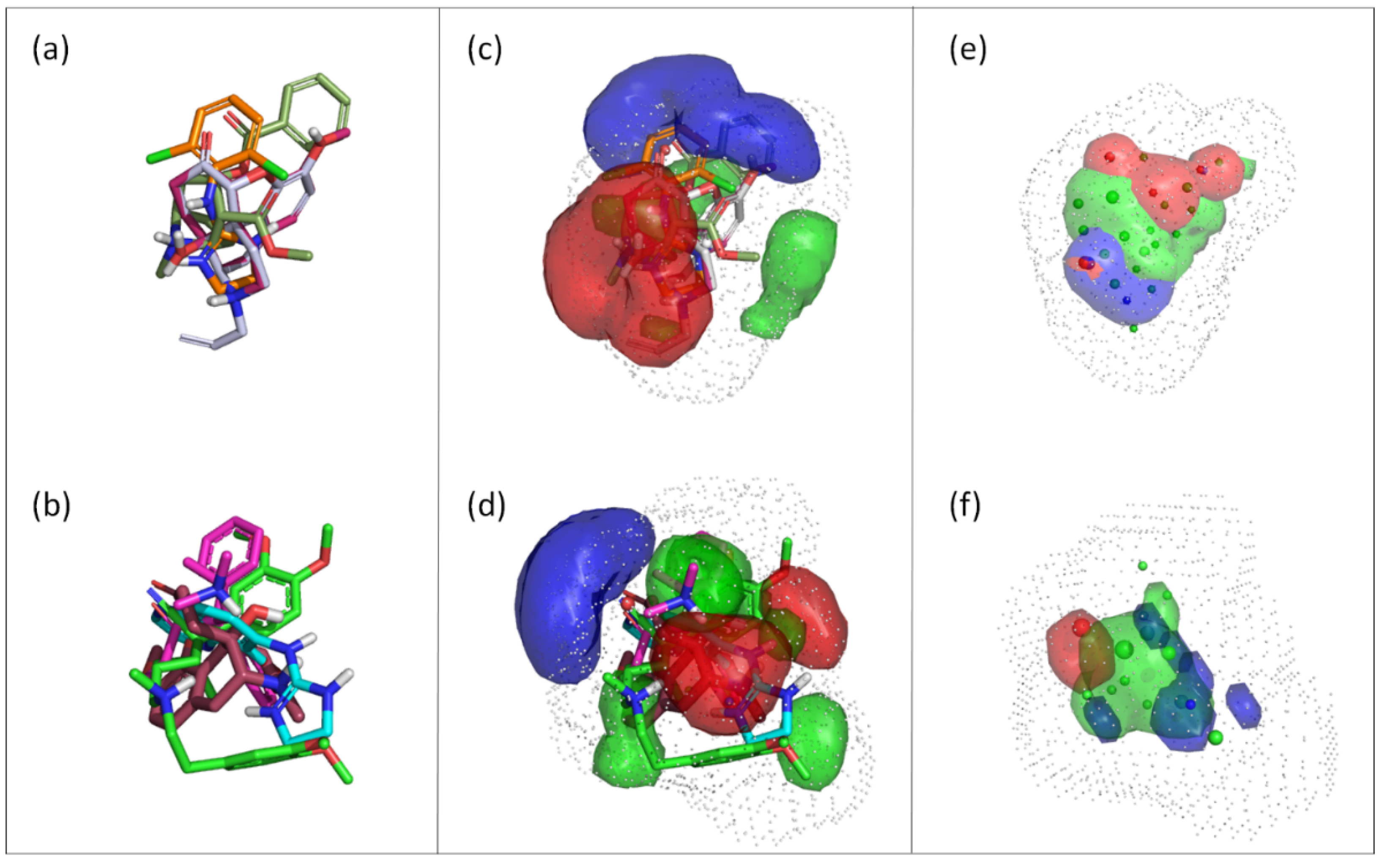
3.2. Compound Selection for Pharmacophore Generation
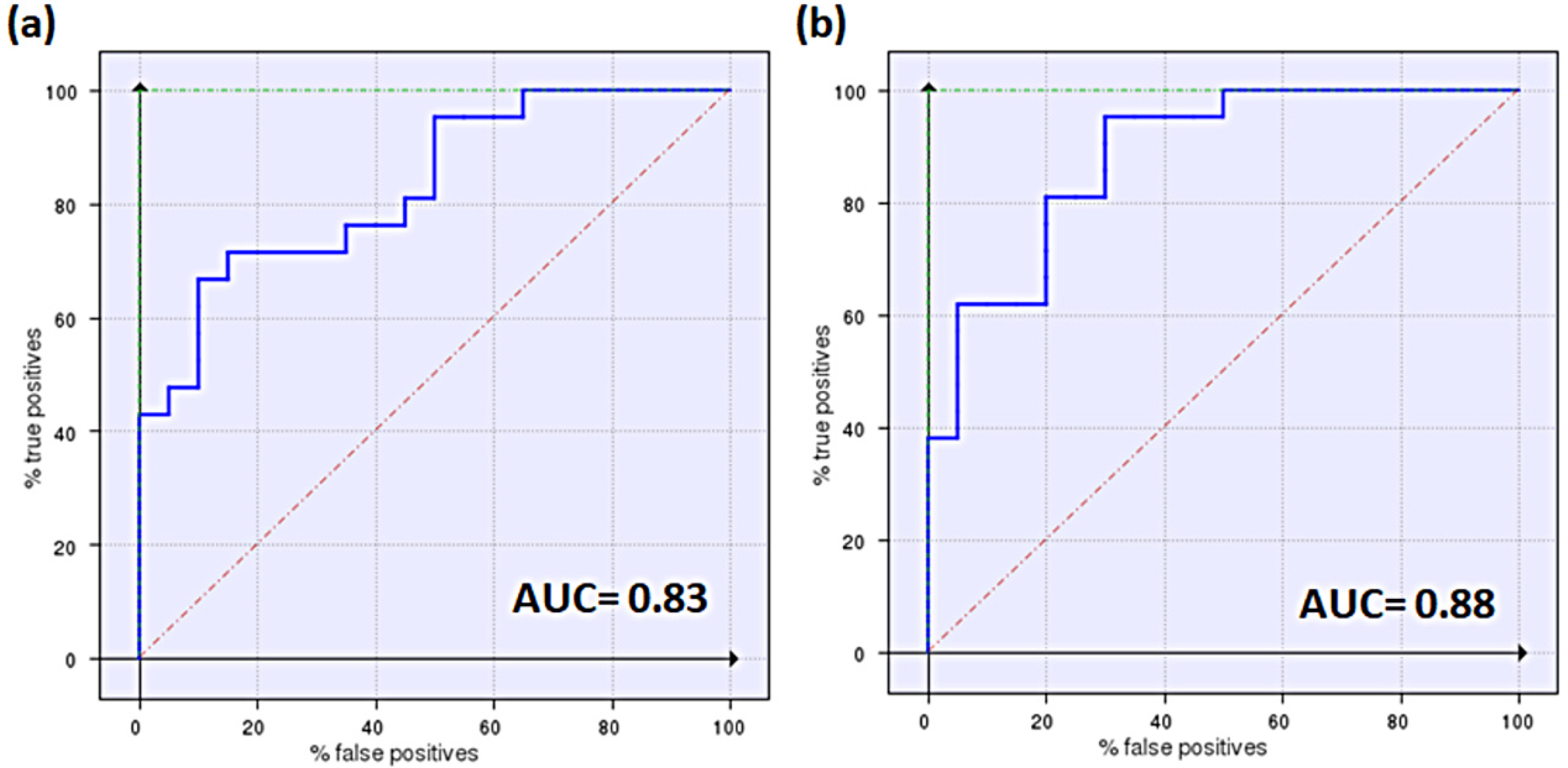
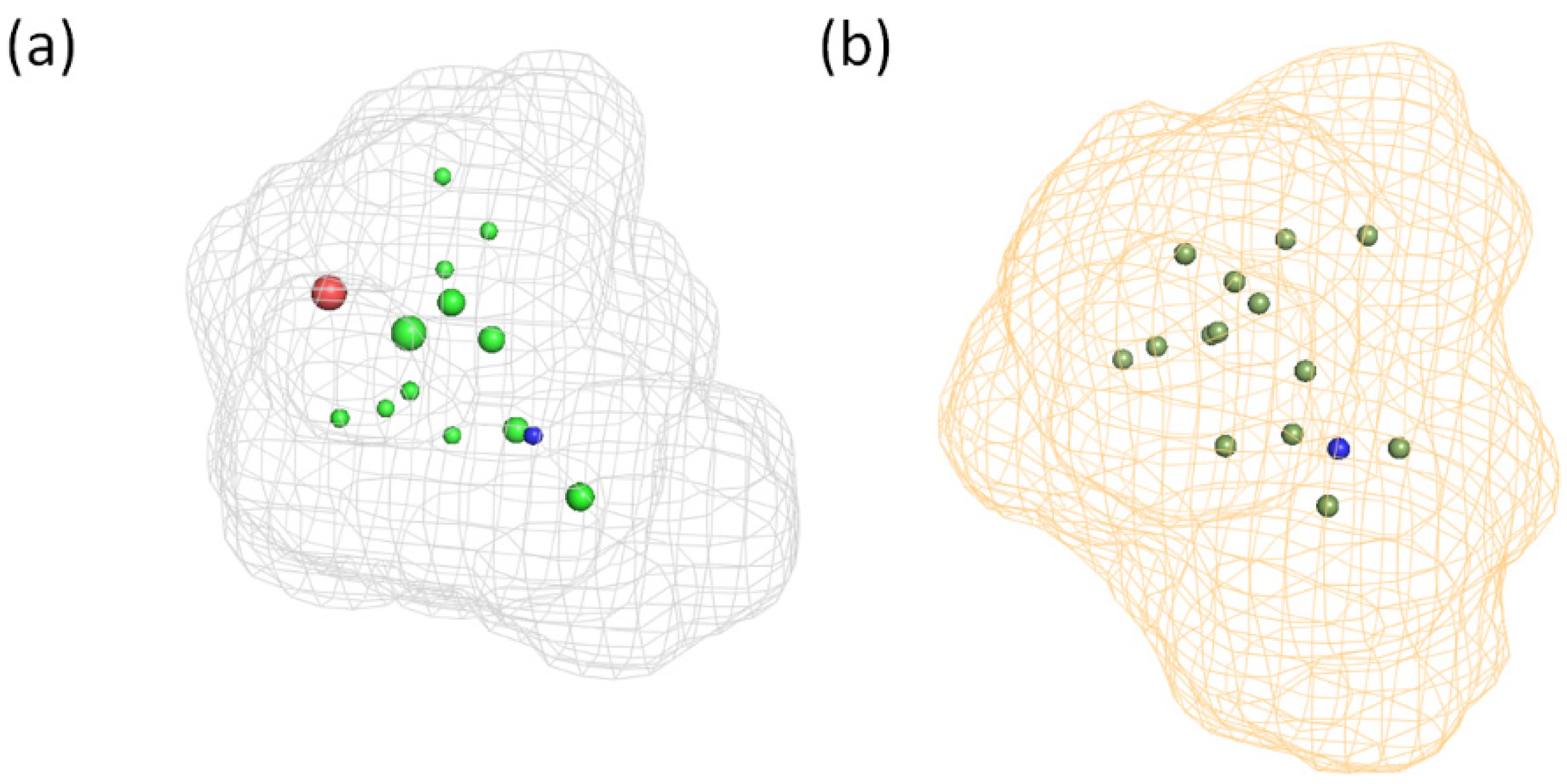
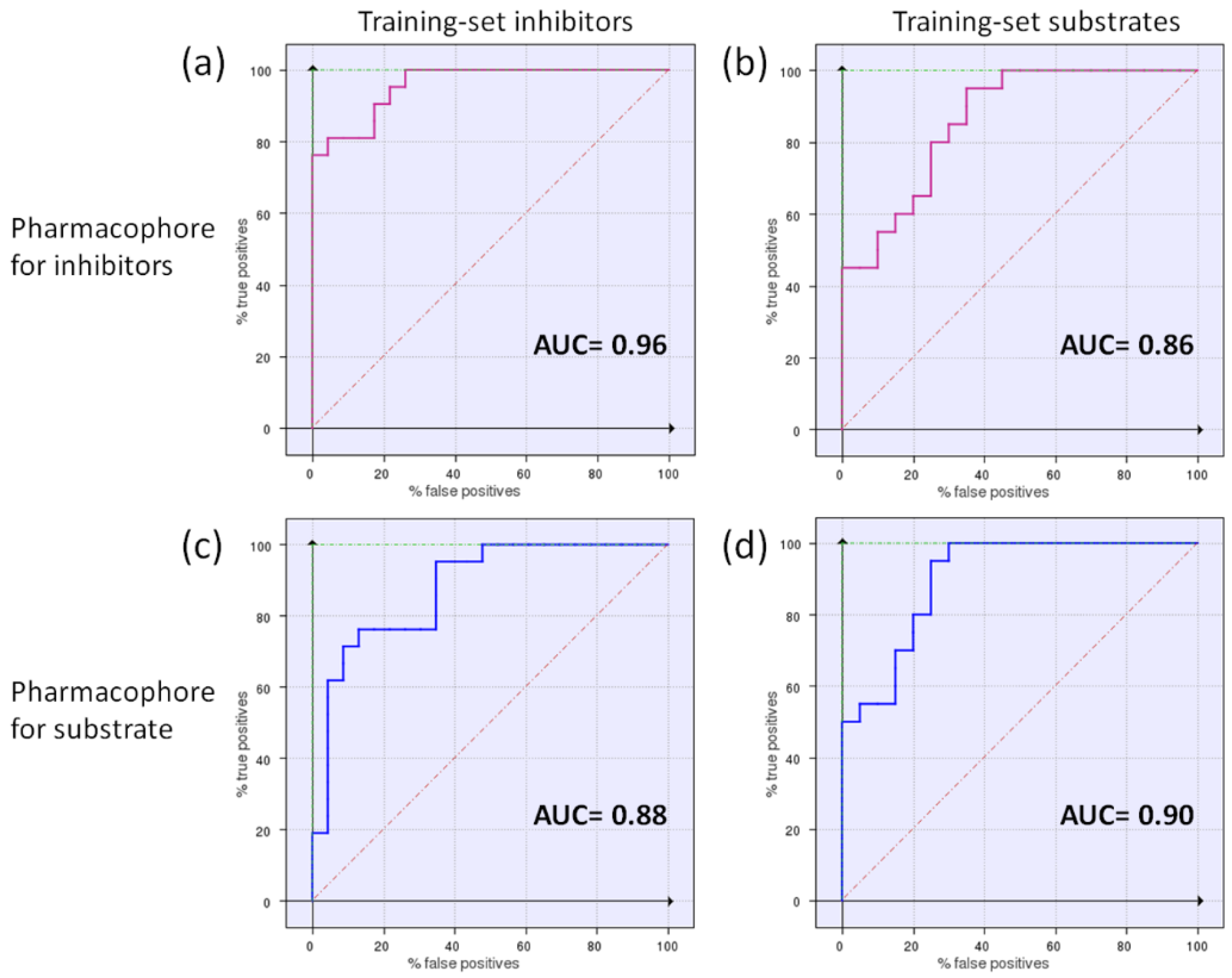
3.3. Generation and Validation of a Pharmacophore Model for Substrates of the Proton-Antiporter
3.4. Pharmacophore-Based Selection of Possible Novel Substrates by Virtual Screening
3.5. Additional Screenings on Databases Containing Drugs, Natural and Endogenous Compounds, and Their Evaluation by Trans-Stimulation Assay
3.6. Comparison of the Pharmacophores for Inhibitors and Substrates
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Remião, F.; Cisternino, S.; Decleves, X. Opioids and the Blood-Brain Barrier: A Dynamic Interaction with Consequences on Drug Disposition in Brain. Curr. Neuropharmacol. 2017, 15, 1156–1173. [Google Scholar] [CrossRef] [PubMed]
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. Int. J. Mol. Sci. 2020, 21, 7890. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Campanelli, F.; Chapy, H.; Gomez-Zepeda, D.; Glacial, F.; Smirnova, M.; Taghi, M.; Pallud, J.; Perrière, N.; Declèves, X.; et al. An Interspecies Molecular and Functional Study of Organic Cation Transporters at the Blood-Brain Barrier: From Rodents to Humans. Pharmaceutics 2020, 12, 308. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Saubamea, B.; Cochois-Guégan, V.; Marie-Claire, C.; Cattelotte, J.; Smirnova, M.; Schinkel, A.H.; Scherrmann, J.-M.; Cisternino, S. Transport of Biogenic Amine Neurotransmitters at the Mouse Blood–Retina and Blood–Brain Barriers by Uptake1 and Uptake2. J. Cereb. Blood Flow Metab. 2012, 32, 1989–2001. [Google Scholar] [CrossRef] [Green Version]
- César-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Superti-Furga, G.; Lackner, D.; Wiedmer, T.; Ingles-Prieto, A.; Barbosa, B.; Girardi, E.; Goldmann, U.; Gürtl, B.; Klavins, K.; Klimek, C.; et al. The RESOLUTE consortium: Unlocking SLC transporters for drug discovery. Nat. Rev. Drug Discov. 2020, 19, 429–430. [Google Scholar] [CrossRef]
- André, P.; Debray, M.; Scherrmann, J.-M.; Cisternino, S. Clonidine Transport at the Mouse Blood—Brain Barrier by a New H+ Antiporter that Interacts with Addictive Drugs. J. Cereb. Blood Flow Metab. 2009, 29, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Chapy, H.; Goracci, L.; Vayer, P.; Parmentier, Y.; Carrupt, P.-A.; Decleves, X.; Scherrmann, J.-M.; Cisternino, S.; Cruciani, G. Pharmacophore-based discovery of inhibitors of a novel drug/proton antiporter in human brain endothelial hCMEC/D3 cell line. Br. J. Pharmacol. 2015, 172, 4888–4904. [Google Scholar] [CrossRef]
- Auvity, S.; Chapy, H.; Goutal, S.; Caillé, F.; Hosten, B.; Smirnova, M.; Decleves, X.; Tournier, N.; Cisternino, S. Diphenhydramine as a selective probe to study H+-antiporter function at the blood–brain barrier: Application to [11C]diphenhydramine positron emission tomography imaging. J. Cereb. Blood Flow Metab. 2016, 37, 2185–2195. [Google Scholar] [CrossRef] [Green Version]
- Chapy, H.; Smirnova, M.; André, P.; Schlatter, J.; Chiadmi, F.; Couraud, P.-O.; Scherrmann, J.-M.; Decleves, X.; Cisternino, S. Carrier-Mediated Cocaine Transport at the Blood-Brain Barrier as a Putative Mechanism in Addiction Liability. Int. J. Neuropsychopharmacol. 2014, 18, pyu001. [Google Scholar] [CrossRef] [Green Version]
- Cisternino, S.; Chapy, H.; André, P.; Smirnova, M.; Debray, M.; Scherrmann, J.-M. Coexistence of Passive and Proton Antiporter-Mediated Processes in Nicotine Transport at the Mouse Blood–Brain Barrier. AAPS J. 2012, 15, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Okura, T.; Hattori, A.; Takano, Y.; Sato, T.; Hammarlund-Udenaes, M.; Terasaki, T.; Deguchi, Y. Involvement of the Pyrilamine Transporter, a Putative Organic Cation Transporter, in Blood-Brain Barrier Transport of Oxycodone. Drug Metab. Dispos. 2008, 36, 2005–2013. [Google Scholar] [CrossRef]
- Goldberg, M.J.; Spector, R.; Chiang, C.K. Transport of diphenhydramine in the central nervous system. J. Pharmacol. Exp. Ther. 1987, 240, 717–722. [Google Scholar]
- Wang, X.; Qi, B.; Su, H.; Li, J.; Sun, X.; He, Q.; Fu, Y.; Zhang, Z. Pyrilamine-sensitive proton-coupled organic cation (H+/OC) antiporter for brain-specific drug delivery. J. Control. Release 2017, 254, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Au-Yeung, S.C.S.; Rurak, D.W.; Gruber, N.; Riggs, K.W. A pharmacokinetic study of diphenhydramine transport across the blood-brain barrier in adult sheep: Potential involvement of a carrier-mediated mechanism. Drug Metab. Dispos. 2006, 34, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Sachkova, A.; Doetsch, D.A.; Jensen, O.; Brockmöller, J.; Ansari, S. How do psychostimulants enter the human brain? Analysis of the role of the proton-organic cation antiporter. Biochem. Pharmacol. 2021, 192, 114751. [Google Scholar] [CrossRef]
- Chapy, H.; Saubamea, B.; Tournier, N.; Bourasset, F.; Behar-Cohen, F.; Decleves, X.; Scherrmann, J.-M.; Cisternino, S. Blood-brain and retinal barriers show dissimilar ABC transporter impacts and concealed effect of P-glycoprotein on a novel verapamil influx carrier. Br. J. Pharmacol. 2016, 173, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Fischer, W.; Metzner, L.; Hoffmann, K.; Neubert, R.H.H.; Brandsch, M. Substrate Specificity and Mechanism of the Intestinal Clonidine Uptake by Caco-2 Cells. Pharm. Res. 2006, 23, 131–137. [Google Scholar] [CrossRef]
- Chapy, H.; André, P.; Decleves, X.; Scherrmann, J.-M.; Cisternino, S. A polyspecific drug/proton antiporter mediates diphenhydramine and clonidine transport at the mouse blood-retinal barrier. Br. J. Pharmacol. 2015, 172, 4714–4725. [Google Scholar] [CrossRef]
- Kuwayama, K.; Inoue, H.; Kanamori, T.; Tsujikawa, K.; Miyaguchi, H.; Iwata, Y.; Miyauchi, S.; Kamo, N.; Kishi, T. Uptake of 3,4-methylenedioxymethamphetamine and its related compounds by a proton-coupled transport system in Caco-2 cells. Biochim. Biophys. Acta 2008, 1778, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Mizuuchi, H.; Katsura, T.; Saito, H.; Hashimoto, Y.; Inui, K.I. Transport characteristics of diphenhydramine in human intestinal epithelial Caco-2 cells: Contribution of pH-dependent transport system. J. Pharmacol. Exp. Ther. 1999, 290, 388–392. [Google Scholar]
- Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. GRID-Based Three-Dimensional Pharmacophores I: FLAPpharm, a Novel Approach for Pharmacophore Elucidation. J. Chem. Inf. Model. 2012, 52, 2587–2598. [Google Scholar] [CrossRef]
- Goracci, L.; Buratta, S.; Urbanelli, L.; Ferrara, G.; Di Guida, R.; Emiliani, C.; Cross, S. Evaluating the risk of phospholipidosis using a new multidisciplinary pipeline approach. Eur. J. Med. Chem. 2015, 92, 49–63. [Google Scholar] [CrossRef]
- Sirci, F.; Goracci, L.; Rodríguez, D.; Van Muijlwijk-Koezen, J.; de Terán, H.G.; Mannhold, R. Ligand-, structure- and pharmacophore-based molecular fingerprints: A case study on adenosine A1, A2A, A2B, and A3 receptor antagonists. J. Comput. Aided Mol. Des. 2012, 26, 1247–1266. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.P.; Rohrbach, P.; McLean, J.E.; Fidock, D.A.; Stein, W.D.; Lanzer, M. Differences in trans-stimulated chloroquine efflux kinetics are linked to PfCRT in Plasmodium falciparum. Mol. Microbiol. 2007, 64, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Dresser, M.J.; Gray, A.T.; Giacomini, K.M. Kinetic and selectivity differences between rodent, rabbit, and human organic cation transporters (OCT1). J. Pharmacol. Exp. Ther. 2000, 292, 1146–1152. [Google Scholar]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the Large-neutral Amino Acid Transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [Green Version]
- Shafik, S.H.; Cobbold, S.A.; Barkat, K.; Richards, S.N.; Lancaster, N.S.; Llinás, M.; Hogg, S.J.; Summers, R.L.; McConville, M.J.; Martin, R.E. The natural function of the malaria parasite’s chloroquine resistance transporter. Nat. Commun. 2020, 11, 3922. [Google Scholar] [CrossRef]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Milletti, F.; Storchi, L.; Goracci, L.; Bendels, S.; Wagner, B.; Kansy, M.; Cruciani, G. Extending pKa prediction accuracy: High-throughput pKa measurements to understand pKa modulation of new chemical series. Eur. J. Med. Chem. 2010, 45, 4270–4279. [Google Scholar] [CrossRef]
- Cruciani, G.; Crivori, P.; Carrupt, P.-A.; Testa, B. Molecular fields in quantitative structure–permeation relationships: The VolSurf approach. J. Mol. Struct. Theochem. 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Sedykh, A.; Fourches, D.; Duan, J.; Hucke, O.; Garneau, M.; Zhu, H.; Bonneau, P.; Tropsha, A. Human intestinal transporter database: QSAR modeling and virtual profiling of drug uptake, efflux and interactions. Pharm. Res. 2012, 30, 996–1007. [Google Scholar] [CrossRef] [Green Version]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, Ó.; Stobbe, M.D.; et al. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419–425. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef]
- Goracci, L.; Deschamps, N.; Randazzo, G.M.; Petit, C.; Passos, C.D.S.; Carrupt, P.-A.; Simões-Pires, C.; Nurisso, A. A Rational Approach for the Identification of Non-Hydroxamate HDAC6-Selective Inhibitors. Sci. Rep. 2016, 6, 29086. [Google Scholar] [CrossRef] [Green Version]
- Tosco, P.; Mackey, M. Lessons and Successes in the Use of Molecular Fields. In Comprehensive Medicinal Chemistry III; Elsevier: Amsterdam, The Netherlands, 2017; pp. 253–296. [Google Scholar] [CrossRef]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Hammarlund-Udenaes, M.; Fridén, M.; Syvänen, S.; Gupta, A. On The Rate and Extent of Drug Delivery to the Brain. Pharm. Res. 2007, 25, 1737–1750. [Google Scholar] [CrossRef] [Green Version]
- Summerfield, S.G.; Zhang, Y.; Liu, H. Examining the Uptake of Central Nervous System Drugs and Candidates across the Blood-Brain Barrier. J. Pharmacol. Exp. Ther. 2016, 358, 294–305. [Google Scholar] [CrossRef]
- Doran, A.; Obach, R.S.; Smith, B.J.; Hosea, N.A.; Becker, S.; Callegari, E.; Chen, C.; Chen, X.; Choo, E.; Cianfrogna, J.; et al. The impact of P-Glycoprotein on the disposition of drugs targeted for indications of the central nervous system: Evaluation using the mdr1a/1B knockout mouse model. Drug Metab. Dispos. 2004, 33, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, C.L.; Osgood, S.M.; Mancuso, J.Y.; Doran, A.C. Diphenhydramine has Similar Interspecies Net Active Influx at the Blood–Brain Barrier. J. Pharm. Sci. 2014, 103, 1557–1562. [Google Scholar] [CrossRef]
- Sadiq, M.W.; Boström, E.; Keizer, R.; Bjorkman, S.; Hammarlund-Udenaes, M. Oxymorphone Active Uptake at the Blood–Brain Barrier and Population Modeling of its Pharmacokinetic–Pharmacodynamic Relationship. J. Pharm. Sci. 2013, 102, 3320–3331. [Google Scholar] [CrossRef]
- Verbeek, J.; Syvänen, S.; Schuit, R.C.; Eriksson, J.; de Lange, E.C.; Windhorst, A.D.; Luurtsema, G.; Lammertsma, A.A. Synthesis and preclinical evaluation of [11C]D617, a metabolite of (R)-[11C]verapamil. Nucl. Med. Biol. 2012, 39, 530–539. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Classification (a) | Compound | Classification (a) |
|---|---|---|---|
| Clonidine | G | Morphine | W |
| Desomorphine | G | Nicotine | W |
| Diphenhydramine | G | Agmatine | N-S |
| Heroine | G | Choline | N-S |
| Norbuprenorphine | G | Cimetidine | N-S |
| Tramadol | G | Dihydromorphine | N-S |
| Verapamil | G | Dopamine | N-S |
| 6-monoacetylmorphine | M-H | Ergothioneine | N-S |
| Brimonidine | M-H | Guanidine | N-S |
| Cocaethylene | M-H | Histamine | N-S |
| Cocaine | M-H | L-dopa | N-S |
| Codeine | M-H | L-carnitine | N-S |
| Methadone | M-H | Melatonin | N-S |
| Norcocaine | M-H | Milnacipran | N-S |
| Oxycodone | M-H | MPP | N-S |
| Pyrilamine | M-H | N-methylnaloxone | N-S |
| Apomorphine | M-H | Paraquat | N-S |
| MDMA | M-H | Serotonin | N-S |
| Naloxone | M-L | Tetraethylammonium | N-S |
| Hydromorphone | W | Tyramine | N-S |
| ID | Specs Compound Code | Structure | Similarity-Score (a) | Classification (b) |
|---|---|---|---|---|
| 1 | AI-204/34841050 |  | 0.41 | N-S |
| 2 | AO-476/43407062 |  | 0.39 | N-S |
| 3 | AE-641/00584045 |  | 0.38 | N-S |
| 4 | AE-641/00605007 |  | 0.37 | N-S |
| 5 | AE-641/11703012 |  | 0.37 | M-H |
| 6 | AN-329/43448732 |  | 0.36 | G |
| 7 | AE-562/12222547 |  | 0.35 | M-H |
| 8 | AE-641/00191008 |  | 0.34 | G |
| 9 | AK-968/10817037 |  | 0.33 | N-S |
| 10 | AE-641/30156016 |  | 0.29 | N-S |
| ID | Specs Compound Code | Structure | Similarity-Score (a) | Classification (b) |
|---|---|---|---|---|
| 11 | AE-641/00335028 |  | 0.21 | G |
| 12 | AE-907/30533025 |  | 0.21 | M-H |
| 13 | AO-476/43023620 |  | 0.21 | M-H |
| 14 | AO-365/41690614 |  | 0.19 | M-H |
| 15 | AN-465/41588344 |  | 0.18 | G |
| 16 | AG-205/14365199 |  | 0.17 | N-S |
| 17 | AO-365/43401585 |  | 0.17 | N-S |
| 18 | AP-906/42717100 |  | 0.17 | W |
| 19 | AF-399/37418022 |  | 0.14 | W |
| 20 | AI-942/13331402 |  | 0.14 | M-H |
| Substrate Candidate | Database | Pharmacophore | Classification |
|---|---|---|---|
| Amitriptyline | A | P2 | G |
| Buflomedil | A | P2 | G |
| Chlorpheniramine | A | P2 | G |
| Chlorpromazine | A | P2 | G |
| Cocaine | B | P1, P2 | M-H |
| Desipramine | A | P2 | G |
| Dextromethorphan | A | P1 | G |
| Diphenhydramine | C | P2 | G |
| Doxapram | A | P2 | M-L |
| Doxepin | A, C | P2 | G |
| Hydrocodone | C | P1, P2 | M-H |
| Hydromorphone | C | P1 | W |
| Hydroxyl-melatonin | B | P2 | N-S |
| Imipramine | A | P2 | G |
| Mecamylamine | C | P2 | G |
| Methadone | A, C | P2 | M-H |
| Methixene | A | P1, P2 | G |
| Nalbuphine | C | P1 | W |
| Naloxone | C | P1 | M-L |
| Oxycodone | C | P1, P2 | M-H |
| Oxymorphone | C | P1 | M-L |
| Pheniramine | A | P2 | M-H |
| Promazine | A | P2 | G |
| Promethazine | A | P2 | G |
| Sibutramine | C | P2 | G |
| Trihexyphenidyl | A | P2 | G |
| Triprolidine | C | P2 | G |
| Venlafaxine | A | P1, P2 | M-L |
| D617-verapamil metabolite | A | P2 | M-L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnova, M.; Goracci, L.; Cruciani, G.; Federici, L.; Declèves, X.; Chapy, H.; Cisternino, S. Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line. Pharmaceutics 2022, 14, 255. https://doi.org/10.3390/pharmaceutics14020255
Smirnova M, Goracci L, Cruciani G, Federici L, Declèves X, Chapy H, Cisternino S. Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line. Pharmaceutics. 2022; 14(2):255. https://doi.org/10.3390/pharmaceutics14020255
Chicago/Turabian StyleSmirnova, Maria, Laura Goracci, Gabriele Cruciani, Laetitia Federici, Xavier Declèves, Hélène Chapy, and Salvatore Cisternino. 2022. "Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line" Pharmaceutics 14, no. 2: 255. https://doi.org/10.3390/pharmaceutics14020255
APA StyleSmirnova, M., Goracci, L., Cruciani, G., Federici, L., Declèves, X., Chapy, H., & Cisternino, S. (2022). Pharmacophore-Based Discovery of Substrates of a Novel Drug/Proton-Antiporter in the Human Brain Endothelial hCMEC/D3 Cell Line. Pharmaceutics, 14(2), 255. https://doi.org/10.3390/pharmaceutics14020255