
Targeted Self-Emulsifying Drug Delivery Systems to Restore Docetaxel Sensitivity in Resistant Tumors

 ,
,  ,
, 
 , ,
, ,  and
and 
Abstract
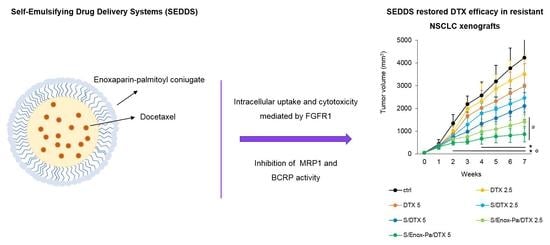
:
1. Introduction
2. Materials and Methods
2.1. Materials and Cell Lines
2.2. Methods
2.2.1. Solubility Studies and Determination of Distribution Coefficient of Docetaxel (Log DSEDDS/release medium)
2.2.2. Synthesis of Enoxaparin-Palmitoyl Conjugate (Enox-Pa)
2.2.3. Preparation and Characterization of SEDDS Coated with Enox-Pa and Loaded with DTX
2.2.4. Stability Studies in Biological Media
Stability in Bovine Serum Albumin (BSA) Solution and Human Plasma
Hemolysis Assay
2.2.5. Sterilization of SEDDS
2.2.6. In Vitro Studies
Cell Viability
Cell Silencing and Knock-Out
Immunoblotting
SEDDS Uptake
ATPases Activity
2.2.7. In Vivo Experiments
2.2.8. Statistical Analysis
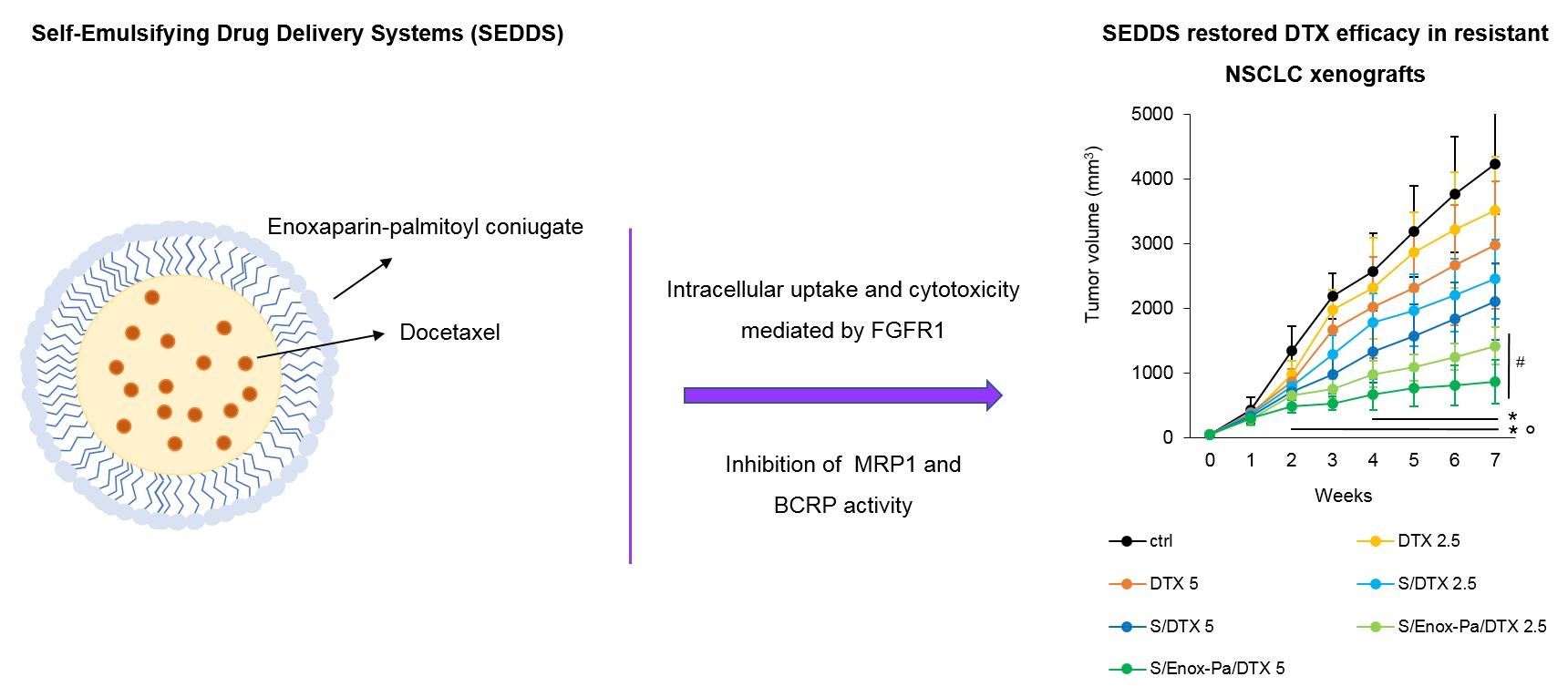
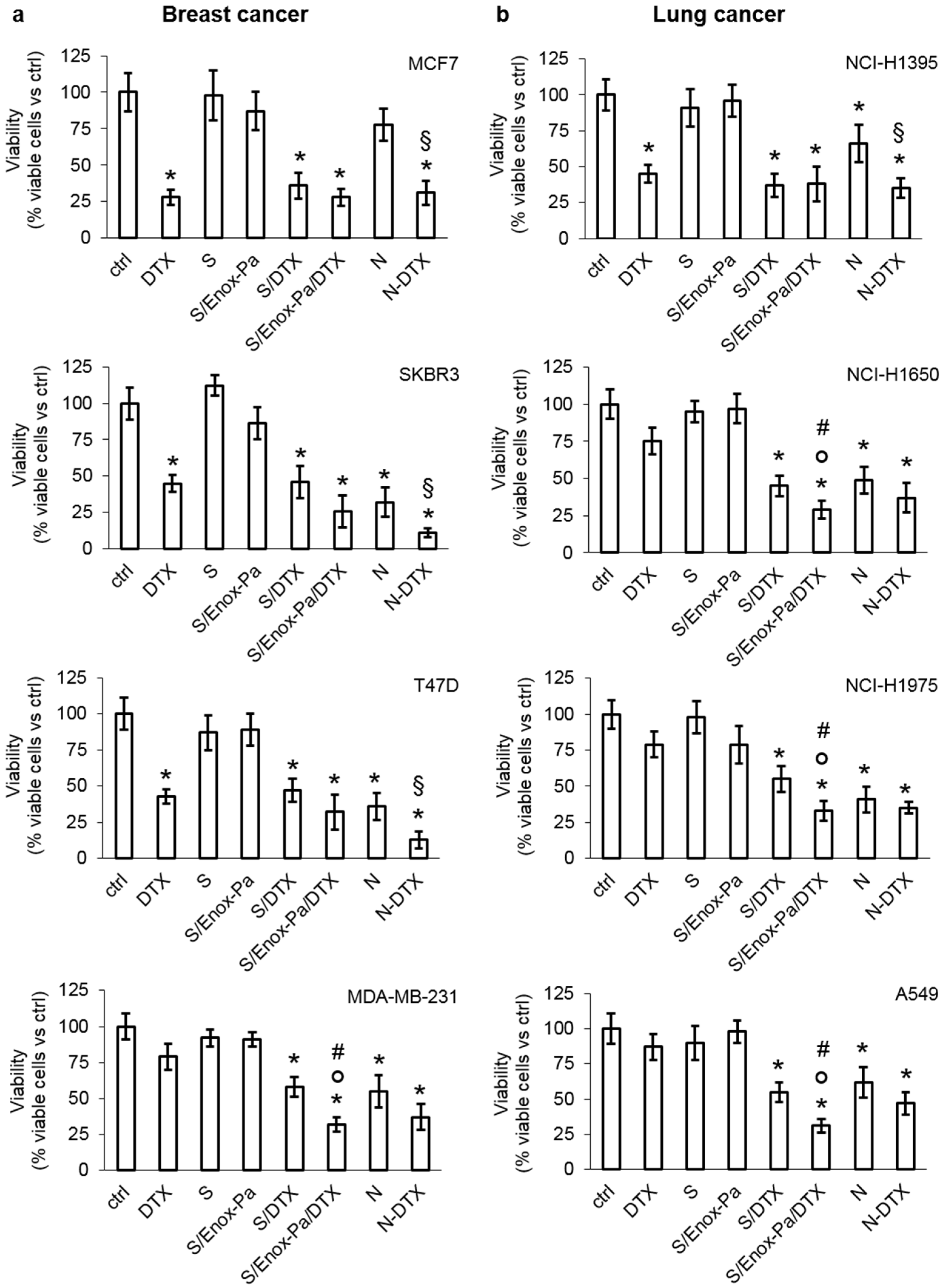
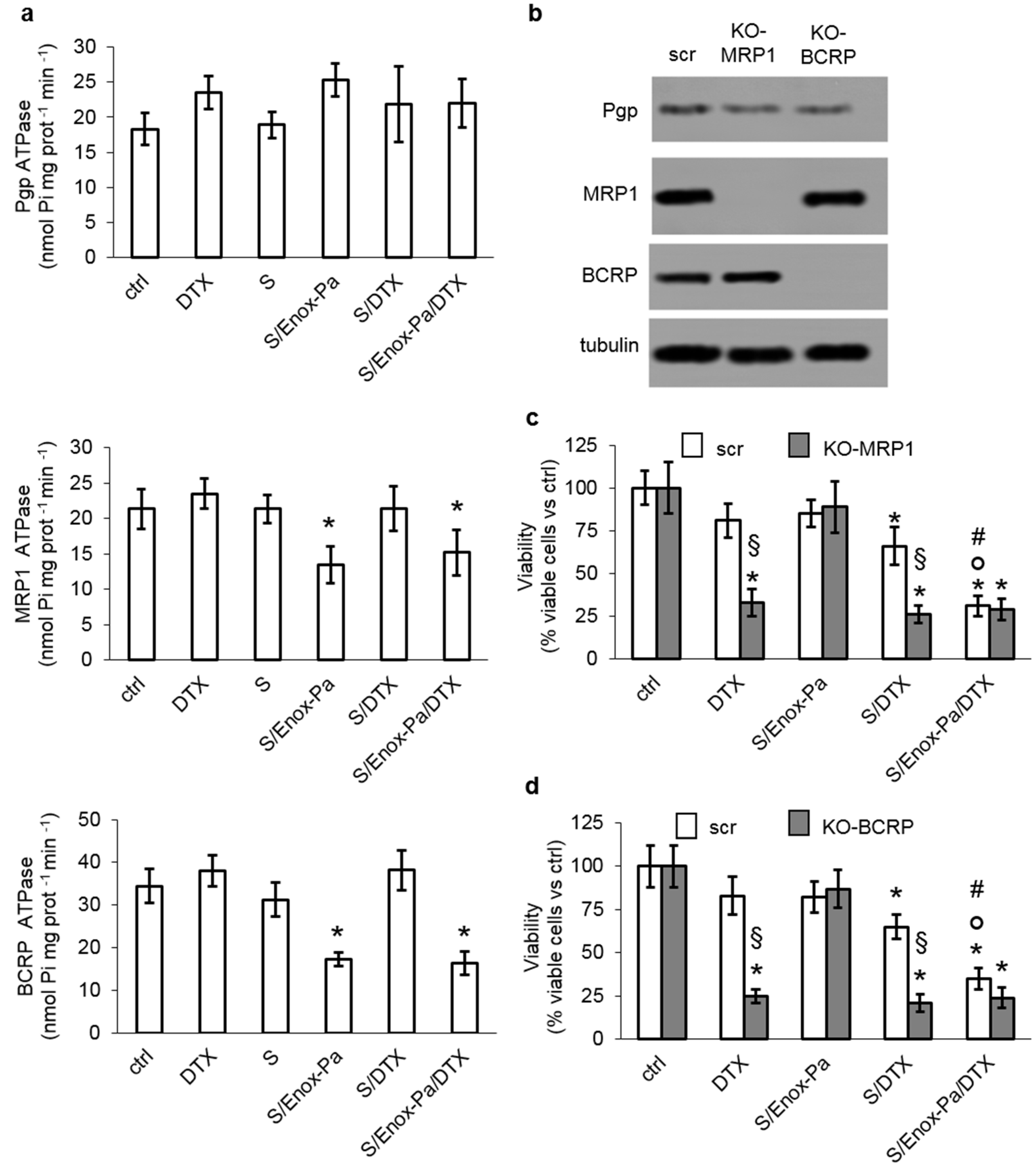
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Markman, J.L.; Rekechenetskiy, A.; Holler, E.; Ljubimova, J.Y. Nanomedicine Therapeutic Approaches to Overcome Cancer Drug Resistance. Adv. Drug Deliv. Rev. 2013, 65, 1866–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP–Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allikmets, R.; Gerrard, B.; Hutchinson, A.; Dean, M. Characterization of the Human ABC Superfamily: Isolation and Mapping of 21 New Genes Using the Expressed Sequence Tags Database. Hum. Mol. Genet. 1996, 5, 1649–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, Y.; Schuetz, J.D. ABC Transporters and Their Role in Nucleoside and Nucleotide Drug Resistance. Biochem. Pharmacol. 2012, 83, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Gligorov, J.; Lotz, J.P. Preclinical Pharmacology of the Taxanes: Implications of the Differences. Oncologist 2004, 9, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringel, I.; Horwitz, S.B. Studies with RP 56976 (Taxotere): A Semisynthetic Analogue of Taxol. JNCI J. Natl. Cancer Inst. 1991, 83, 288–291. [Google Scholar] [CrossRef]
- Hwang, C. Overcoming Docetaxel Resistance in Prostate Cancer: A Perspective Review. Ther. Adv. Med. Oncol. 2012, 4, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Galletti, E.; Magnani, M.; Renzulli, M.L.; Botta, M. Paclitaxel and Docetaxel Resistance: Molecular Mechanisms and Development of New Generation Taxanes. ChemMedChem 2007, 2, 920–942. [Google Scholar] [CrossRef]
- Murray, S.; Briasoulis, E.; Linardou, H.; Bafaloukos, D.; Papadimitriou, C. Taxane Resistance in Breast Cancer: Mechanisms, Predictive Biomarkers and Circumvention Strategies. Cancer Treat. Rev. 2012, 38, 890–903. [Google Scholar] [CrossRef]
- Iyer, A.K.; Singh, A.; Ganta, S.; Amiji, M.M. Role of Integrated Cancer Nanomedicine in Overcoming Drug Resistance. Adv. Drug Deliv. Rev. 2013, 65, 1784–1802. [Google Scholar] [CrossRef]
- Lepeltier, E.; Rijo, P.; Rizzolio, F.; Popovtzer, R.; Petrikaite, V.; Assaraf, Y.G.; Passirani, C. Nanomedicine to Target Multidrug Resistant Tumors. Drug Resist. Updates 2020, 52, 100704. [Google Scholar] [CrossRef]
- Neslihan Gursoy, R.; Benita, S. Self-Emulsifying Drug Delivery Systems (SEDDS) for Improved Oral Delivery of Lipophilic Drugs. Biomed. Pharmacother. 2004, 58, 173–182. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Pouton, C.W.; Cuine, J.F.; Charman, W.N. Enhancing Intestinal Drug Solubilisation Using Lipid-Based Delivery Systems. Adv. Drug Deliv. Rev. 2008, 60, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-Nano-Emulsifying Drug Delivery Systems: An Update of the Biopharmaceutical Aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, J.; Zhang, Q. Self-Emulsifying Drug Delivery System Improve Oral Bioavailability: Role of Excipients and Physico-Chemical Characterization. Pharm. Nanotechnol. 2020, 8, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Giarra, S.; Lupo, N.; Campani, V.; Carotenuto, A.; Mayol, L.; de Rosa, G.; Bernkop-Schnürch, A. In Vitro Evaluation of Tumor Targeting Ability of a Parenteral Enoxaparin-Coated Self-Emulsifying Drug Delivery System. J. Drug Deliv. Sci. Technol. 2019, 53, 101144. [Google Scholar] [CrossRef]
- Ingle, R.G.; Agarwal, A.S. A World of Low Molecular Weight Heparins (LMWHs) Enoxaparin as a Promising Moiety—A Review. Carbohydr. Polym. 2014, 106, 148–153. [Google Scholar] [CrossRef]
- Ihaddadene, R.; le Gal, G.; Delluc, A.; Carrier, M. Dose Escalation of Low Molecular Weight Heparin in Patients with Recurrent Cancer-Associated Thrombosis. Thromb. Res. 2014, 134, 93–95. [Google Scholar] [CrossRef]
- Chen, Y.; Scully, M.; Dawson, G.; Goodwin, C.; Xia, M.; Lu, X.; Kakkar, A. Perturbation of the Heparin/Heparin-Sulfate Interactome of Human Breast Cancer Cells Modulates pro-Tumourigenic Effects Associated with PI3K/Akt and MAPK/ERK Signalling. Thromb. Haemost. 2013, 109, 1148–1157. [Google Scholar] [CrossRef] [Green Version]
- Bouris, P.; Skandalis, S.S.; Piperigkou, Z.; Afratis, N.; Karamanou, K.; Aletras, A.J.; Moustakas, A.; Theocharis, A.D.; Karamanos, N.K. Estrogen Receptor Alpha Mediates Epithelial to Mesenchymal Transition, Expression of Specific Matrix Effectors and Functional Properties of Breast Cancer Cells. Matrix Biol. 2015, 43, 42–60. [Google Scholar] [CrossRef]
- Afratis, N.A.; Karamanou, K.; Piperigkou, Z.; Vynios, D.H.; Theocharis, A.D. The Role of Heparins and Nano-Heparins as Therapeutic Tool in Breast Cancer. Glycoconj. J. 2017, 34, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-Anticoagulant Heparins and Inhibition of Cancer. Pathophysiol. Haemost. Thromb. 2007, 36, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.E.; Cotter, M.J.; Stewart, J.B.; Abbitt, K.B.; Ali, M.; Wagner, B.E.; Wallace, W.A.H.; Forlow, S.B.; Hellewell, P.G. Combined Anticoagulant and Antiselectin Treatments Prevent Lethal Intravascular Coagulation. Blood 2003, 101, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Curigliano, G.; Thürlimann, B.; Weber, W.P.; Poortmans, P.; Regan, M.M.; Senn, H.J.; Winer, E.P.; Gnant, M.; Aebi, S.; et al. Customizing Local and Systemic Therapies for Women with Early Breast Cancer: The St. Gallen International Consensus Guidelines for Treatment of Early Breast Cancer 2021. Ann. Oncol. 2021, 32, 1216–1235. [Google Scholar] [CrossRef]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non–Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef]
- Griesser, J.; Hetényi, G.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Hydrophobic Ion Pairing: Key to Highly Payloaded Self-Emulsifying Peptide Drug Delivery Systems. Int. J. Pharm. 2017, 520, 267–274. [Google Scholar] [CrossRef]
- Shahzadi, I.; Dizdarević, A.; Efiana, N.A.; Matuszczak, B.; Bernkop-Schnürch, A. Trypsin Decorated Self-Emulsifying Drug Delivery Systems (SEDDS): Key to Enhanced Mucus Permeation. J. Colloid Interface Sci. 2018, 531, 253–260. [Google Scholar] [CrossRef]
- Nazir, I.; Asim, M.H.; Dizdarević, A.; Bernkop-Schnürch, A. Self-Emulsifying Drug Delivery Systems: Impact of Stability of Hydrophobic Ion Pairs on Drug Release. Int. J. Pharm. 2019, 561, 197–205. [Google Scholar] [CrossRef]
- Campani, V.; Zappavigna, S.; Scotti, L.; Abate, M.; Porru, M.; Leonetti, C.; Caraglia, M.; de Rosa, G. Hybrid Lipid Self-Assembling Nanoparticles for Brain Delivery of MicroRNA. Int. J. Pharm. 2020, 588, 119693. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Jalil, A. Do Drug Release Studies from SEDDS Make Any Sense? J. Control. Release 2018, 271, 55–59. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. “PubChem Compound Summary for CID 148124, Docetaxel” PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/docetaxel (accessed on 30 November 2021).
- Zhang, E.; Xing, R.; Liu, S.; Li, P. Current Advances in Development of New Docetaxel Formulations. Expert Opin. Drug Deliv. 2019, 16, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Scully, M.; Petralia, G.; Kakkar, A. Binding and Inhibition of Drug Transport Proteins by Heparin. Cancer Biol. Ther. 2014, 15, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuddanda, P.R.; Rajamanickam, V.M.; Yaspal, M.; Singh, S. Investigations on Agglomeration and Haemocompatibility of Vitamin E TPGS Surface Modified Berberine Chloride Nanoparticles. BioMed Res. Int. 2014, 2014, 951942. [Google Scholar] [CrossRef] [PubMed]
- Jansook, P.; Pichayakorn, W.; Ritthidej, G.C. Amphotericin B-Loaded Solid Lipid Nanoparticles (SLNs) and Nanostructured Lipid Carrier (NLCs): Effect of Drug Loading and Biopharmaceutical Characterizations. Drug Dev. Ind. Pharm. 2018, 44, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Lenaerts, V.; Nagelkerke, J.F.; van Berkel, T.J.C.; Couvreur, P.; Grislain, L.; Roland, M.; Speiser, P. In Vivo Uptake of Polyisobutyl Cyanoacrylate Nanoparticles by Rat Liver Kupffer, Endothelial, and Parenchymal Cells. J. Pharm. Sci. 1984, 73, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Leroux, J.-C.; de Jaeghere, F.; Anner, B.; Doelker, E.; Gurny, R. An Investigation on the Role of Plasma and Serum Opsonins on the Evternalization of Biodegradable Poly(D,L-Lactic Acid) Nanoparticles by Human Monocytes. Life Sci. 1995, 57, 695–703. [Google Scholar] [CrossRef]
- Owens, D.E.; Peppas, N.A. Opsonization, Biodistribution, and Pharmacokinetics of Polymeric Nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Caparica, R.; Lambertini, M.; de Azambuja, E. How I Treat Metastatic Triple-Negative Breast Cancer. ESMO Open 2019, 4, e000504. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Liu, Y.; Beenken, A.; Jiang, L.; Gao, X.; Huang, Z.; Hsu, A.; Gross, G.J.; Wang, Y.-G.; Mohammadi, M.; et al. A Novel Fibroblast Growth Factor-1 Ligand with Reduced Heparin Binding Protects the Heart against Ischemia-Reperfusion Injury in the Presence of Heparin Co-Administration. Cardiovasc. Res. 2017, 113, 1585–1602. [Google Scholar] [CrossRef]
- Hilberg, F.; Tontsch-Grunt, U.; Baum, A.; Le, A.T.; Doebele, R.C.; Lieb, S.; Gianni, D.; Voss, T.; Garin-Chesa, P.; Haslinger, C.; et al. Triple Angiokinase Inhibitor Nintedanib Directly Inhibits Tumor Cell Growth and Induces Tumor Shrinkage via Blocking Oncogenic Receptor Tyrosine Kinases. J. Pharmacol. Exp. Ther. 2018, 364, 494. [Google Scholar] [CrossRef]
- Capelletto, E.; Migliorino, M.R.; Morabito, A.; Chiari, R.; Grossi, F.; Tiseo, M.; Di Costanzo, F.; Delmonte, A.; Romano, G.; Galetta, D.; et al. Final Results of the SENECA (SEcond Line NintEdanib in Non-Small Cell Lung CAncer) Trial. Lung Cancer 2019, 134, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Marie, P.J. Fibroblast Growth Factor Signaling in Skeletal Development and Disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaroglio, I.; Gazzano, E.; Kopecka, J.; Chegaev, K.; Costamagna, C.; Fruttero, R.; Guglielmo, S.; Riganti, C. New Tetrahydroisoquinoline Derivatives Overcome Pgp Activity in Brain-Blood Barrier and Glioblastoma Multiforme in Vitro. Molecules 2018, 23, 1401. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, C.M.; Earle, K.A.; Fetter, J.; Xu, S.; Hartman, T.; Chan, D.C.; Zhao, T.L.M.; Piazza, G.; Klein-Szanto, A.J.P.; Pamukcu, R.; et al. Exisulind-Induced Apoptosis in a Non-Small Cell Lung Cancer Orthotopic Lung Tumor Model Augments Docetaxel Treatment and Contributes to Increased Survival. Mol. Cancer Ther. 2003, 2, 479. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Mean Diameter (nm ± SD) | PI (Mean ± SD) | ζ-Potential (mV ± SD) |
|---|---|---|---|
| S | 109.2 ± 1.5 | 0.21 ± 0.02 | −8.8 ± 0.7 |
| S/DTX | 113.9 ± 1.6 | 0.24 ± 0.01 | −11.2 ± 1.0 |
| S/Enox-Pa | 115.3 ± 1.3 | 0.23 ± 0.01 | −14.6 ± 0.8 |
| S/Enox-Pa/DTX | 144.8 ± 3.7 | 0.24 ± 0.05 | −15.0 ± 1.6 |
| BSA 37 °C | ||||
|---|---|---|---|---|
| Formulation | Mean Diameter (nm ± SD) | Mean Diameter (nm ± SD) | PI (Mean ± SD) | PI (Mean ± SD) |
| Time 0 | 4 h | Time 0 | 4 h | |
| S | 127.9 ± 8.2 | 125.5 ± 1.3 | 0.21 ± 0.04 | 0.21 ± 0.04 |
| S/DTX | 114.4 ± 0.5 | 124.8 ± 0.2 | 0.19 ± 0.07 | 0.21 ± 0.01 |
| S/Enox-Pa | 135.1 ± 4.1 | 135.2 ± 8.2 | 0.27 ± 0.04 | 0.23 ± 0.02 |
| S/Enox-Pa/DTX | 158.1 ± 2.2 | 159.6 ± 7.1 | 0.44 ± 0.16 | 0.49 ± 0.04 |
| Human Plasma 37 °C | ||||
| Formulation | Mean Diameter (nm ± SD) | Mean Diameter (nm ± SD) | PI (Mean ± SD) | PI (Mean ± SD) |
| Time 0 | 4 h | Time 0 | 4 h | |
| S | 114.7 ± 0.7 | 125.5 ± 0.6 | 0.16 ± 0.03 | 0.17 ± 0.01 |
| S/DTX | 118.1 ± 0.4 | 127.9 ± 1.8 | 0.16 ± 0.01 | 0.17 ± 0.01 |
| S/Enox-Pa | 113.2 ± 1.5 | 121.7 ± 5.3 | 0.25 ± 0.04 | 0.16 ± 0.04 |
| S/Enox-Pa/DTX | 146.7 ± 4.4 | 151.4 ± 10.6 | 0.41 ± 0.23 | 0.41 ± 0.23 |
| Formulation | Hemolysis (%) |
|---|---|
| S | 1.6 ± 0.1 |
| S/DTX | 2.3 ± 0.2 |
| S/Enox-Pa | 1.6 ± 0.0 |
| S/Enox-Pa/DTX | 1.6 ± 0.1 |
| Mean Diameter (nm ± SD) | PI (Mean ± SD) | |||
|---|---|---|---|---|
| Formulation | Before Filtration | After Filtration | Before Filtration | After Filtration |
| S | 101.62 ± 5.48 | 102.40 ± 0.19 | 0.18 ± 0.02 | 0.19 ± 0.03 |
| S/DTX | 115.18 ± 0.98 | 116.37 ± 0.27 | 0.17 ± 0.01 | 0.13 ± 0.01 |
| S/Enox-Pa | 116.10 ± 0.24 | 112.35 ± 2.57 | 0.23 ± 0.02 | 0.27 ± 0.03 |
| S/Enox-Pa/DTX | 141.13 ± 2.85 | 142.70 ± 4.22 | 0.23 ± 0.05 | 0.28 ± 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campani, V.; Salaroglio, I.C.; Nele, V.; Kopecka, J.; Bernkop-Schnürch, A.; Riganti, C.; De Rosa, G. Targeted Self-Emulsifying Drug Delivery Systems to Restore Docetaxel Sensitivity in Resistant Tumors. Pharmaceutics 2022, 14, 292. https://doi.org/10.3390/pharmaceutics14020292
Campani V, Salaroglio IC, Nele V, Kopecka J, Bernkop-Schnürch A, Riganti C, De Rosa G. Targeted Self-Emulsifying Drug Delivery Systems to Restore Docetaxel Sensitivity in Resistant Tumors. Pharmaceutics. 2022; 14(2):292. https://doi.org/10.3390/pharmaceutics14020292
Chicago/Turabian StyleCampani, Virginia, Iris Chiara Salaroglio, Valeria Nele, Joanna Kopecka, Andreas Bernkop-Schnürch, Chiara Riganti, and Giuseppe De Rosa. 2022. "Targeted Self-Emulsifying Drug Delivery Systems to Restore Docetaxel Sensitivity in Resistant Tumors" Pharmaceutics 14, no. 2: 292. https://doi.org/10.3390/pharmaceutics14020292
APA StyleCampani, V., Salaroglio, I. C., Nele, V., Kopecka, J., Bernkop-Schnürch, A., Riganti, C., & De Rosa, G. (2022). Targeted Self-Emulsifying Drug Delivery Systems to Restore Docetaxel Sensitivity in Resistant Tumors. Pharmaceutics, 14(2), 292. https://doi.org/10.3390/pharmaceutics14020292