
Effect of Solvents, Stabilizers and the Concentration of Stabilizers on the Physical Properties of Poly(d,l-lactide-co-glycolide) Nanoparticles: Encapsulation, In Vitro Release of Indomethacin and Cytotoxicity against HepG2-Cell





Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chromatographic Analysis of Indomethacin
2.3. Preparation of PLGA Nanoparticles
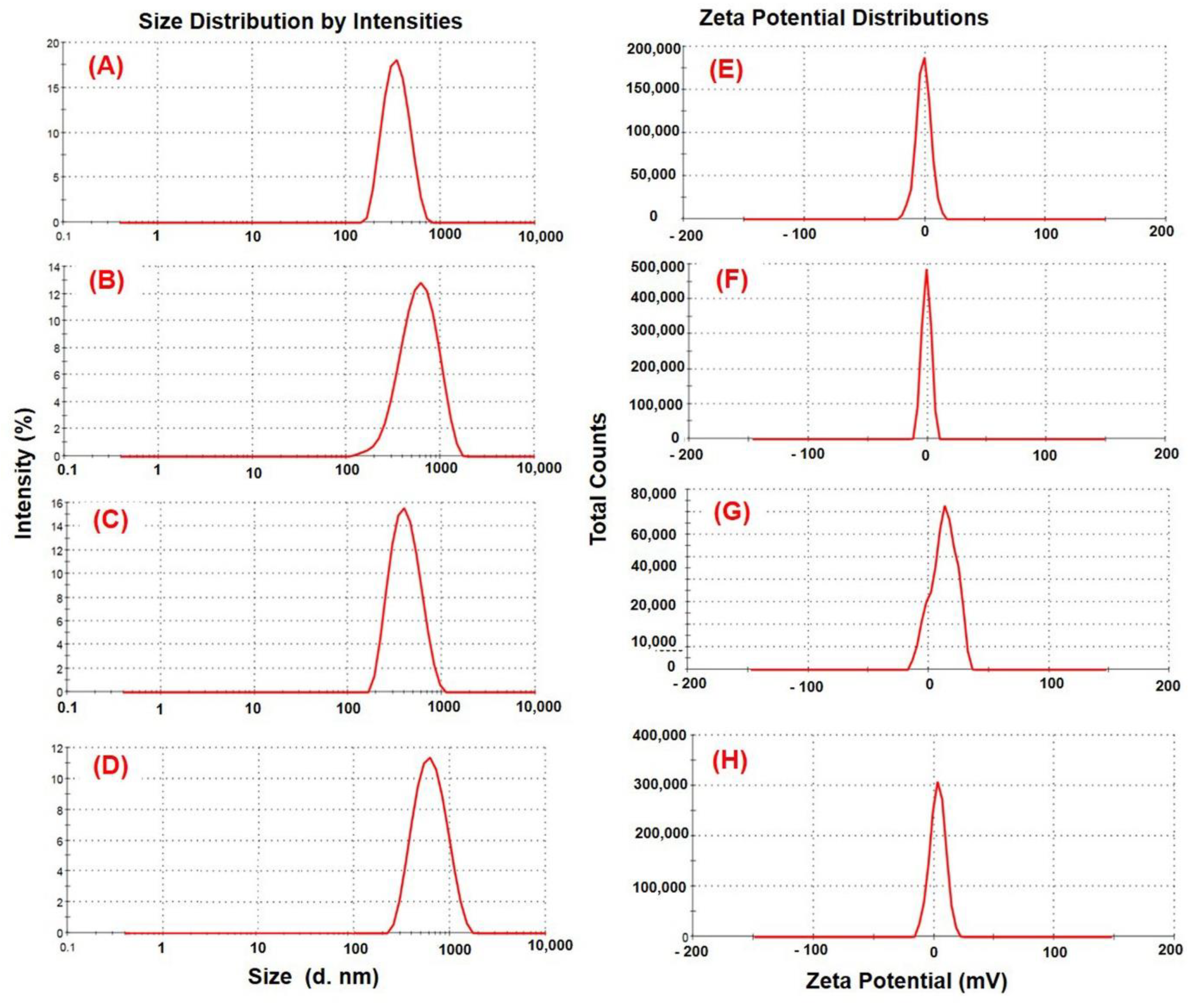
2.4. Particle Size and Zeta Potential Measurement
2.5. Morphological Characterization of PLGA-NPs
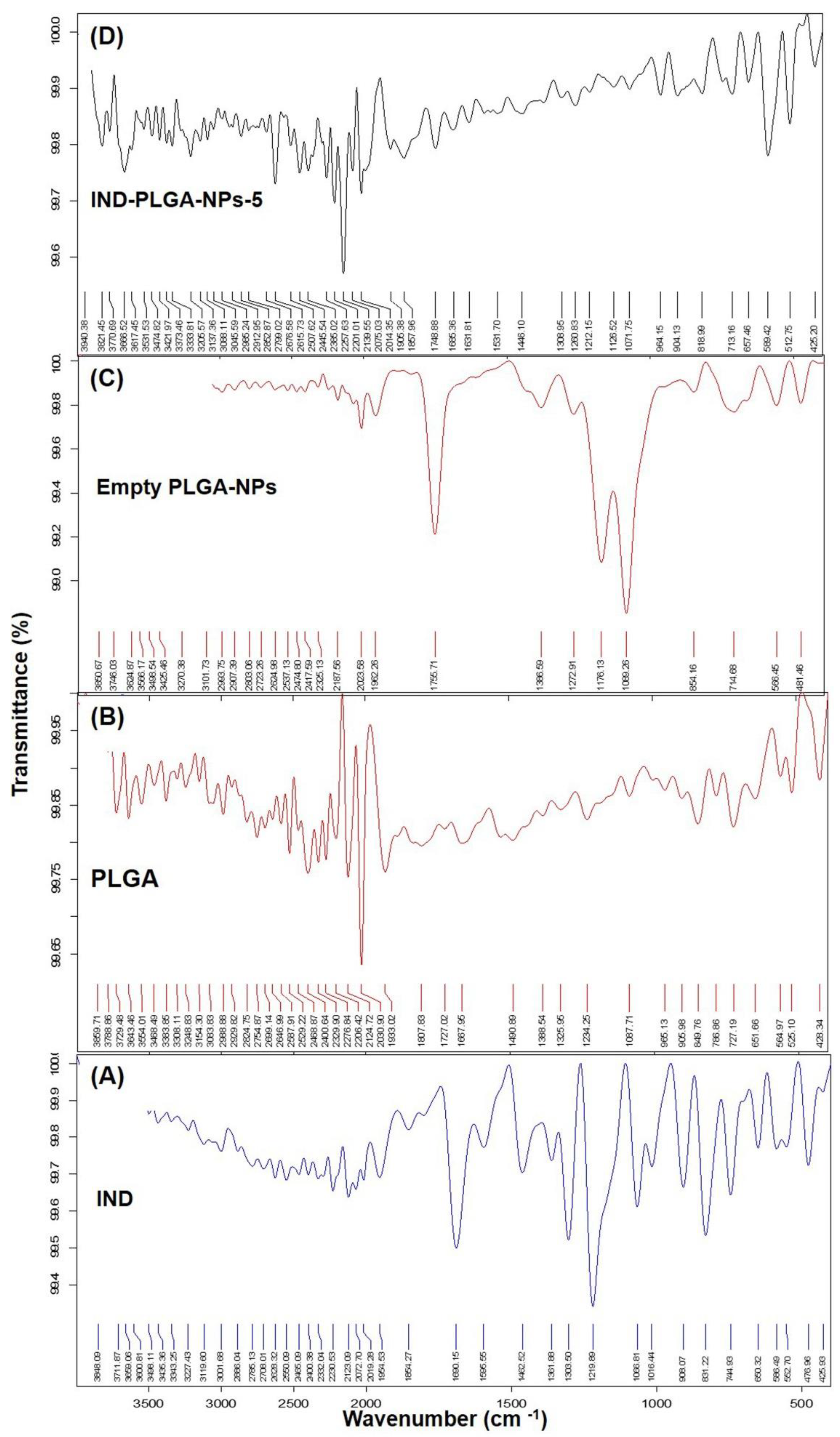
2.6. FTIR Spectral Analysis
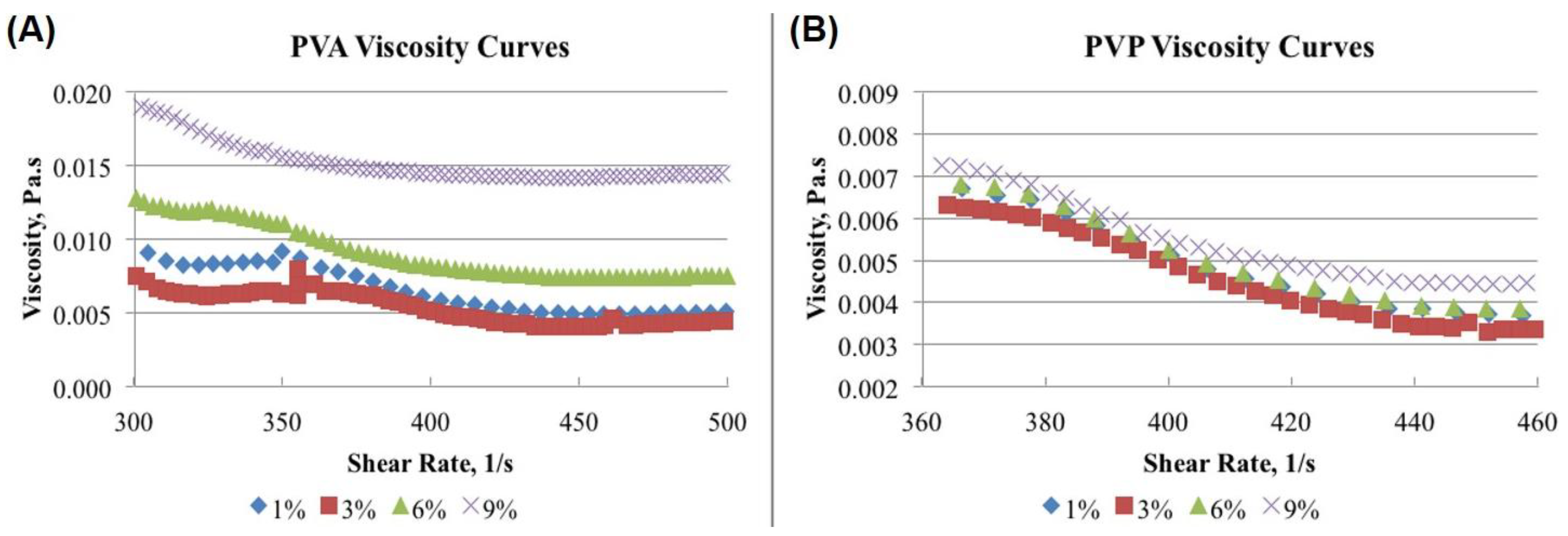
2.7. Viscosity Evaluation of the Surfactant Solutions
2.8. Encapsulation Efficiency and Drug-Loading Capacity
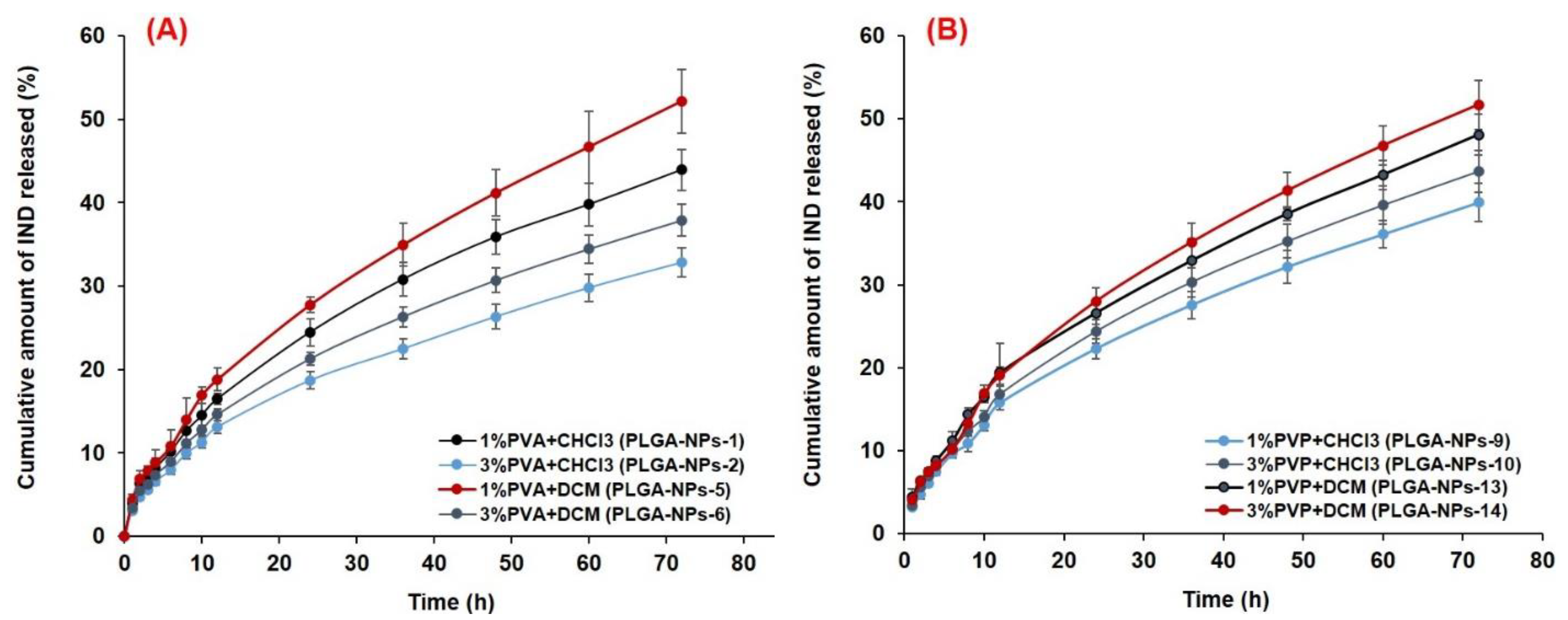
2.9. In Vitro Release and Kinetics Study
2.10. Differential Scanning Calorimetry (DSC)
2.11. Stability of IND-Loaded PLGA-NPs
2.12. In Vitro Cytotoxicity Studies
2.12.1. Maintenance and Growth of Cell Line
2.12.2. MTT Assay
2.13. Statistical Analysis
3. Results and Discussion
3.1. Preparation of PLGA-NPs
3.2. Effect of Formulation Factors on Particle Size and Zeta Potential
3.3. Effect of the Two Stabilizers on the Morphology of PLGA-NPs
3.4. Fourier Transform Infrared (FTIR) Analysis
3.5. Viscosity-Shear Rate Relationship of Different Concentrations of PVA and PVP
3.6. Loading of Drug (IND) and In Vitro Release
3.6.1. Encapsulation Efficiency and Drug Loading Capacity
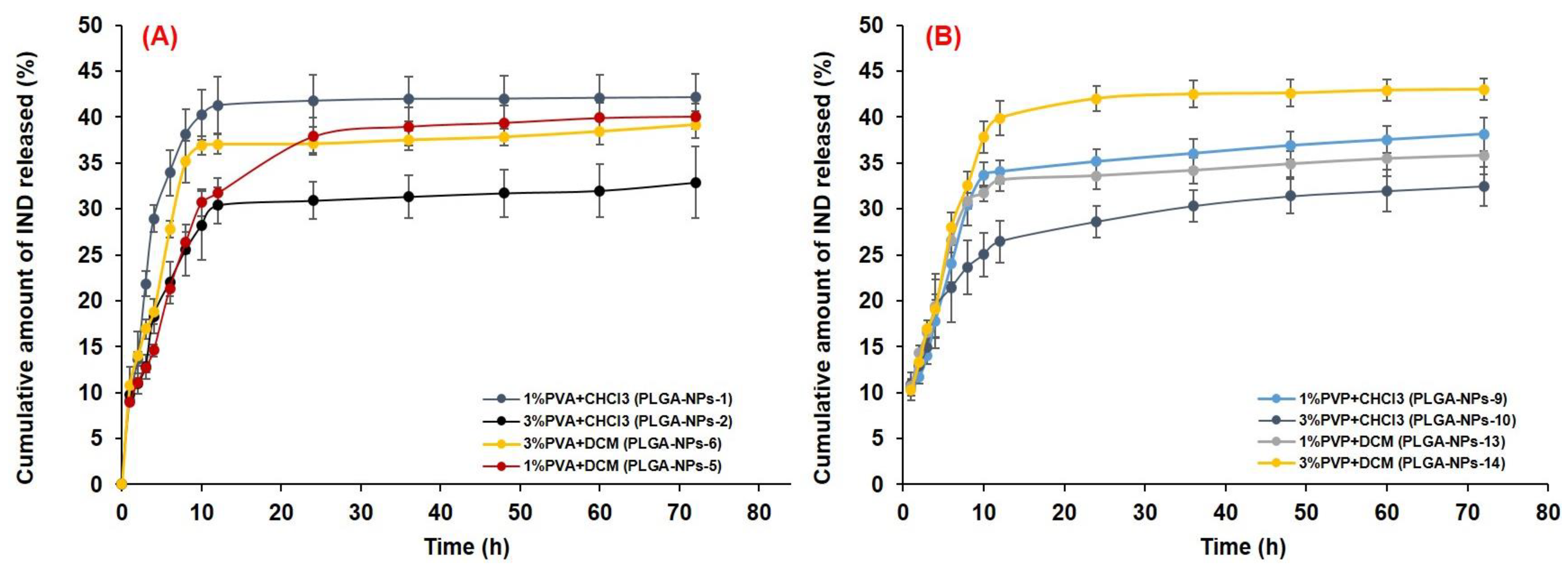
3.6.2. In Vitro Drug Release and Release Kinetics
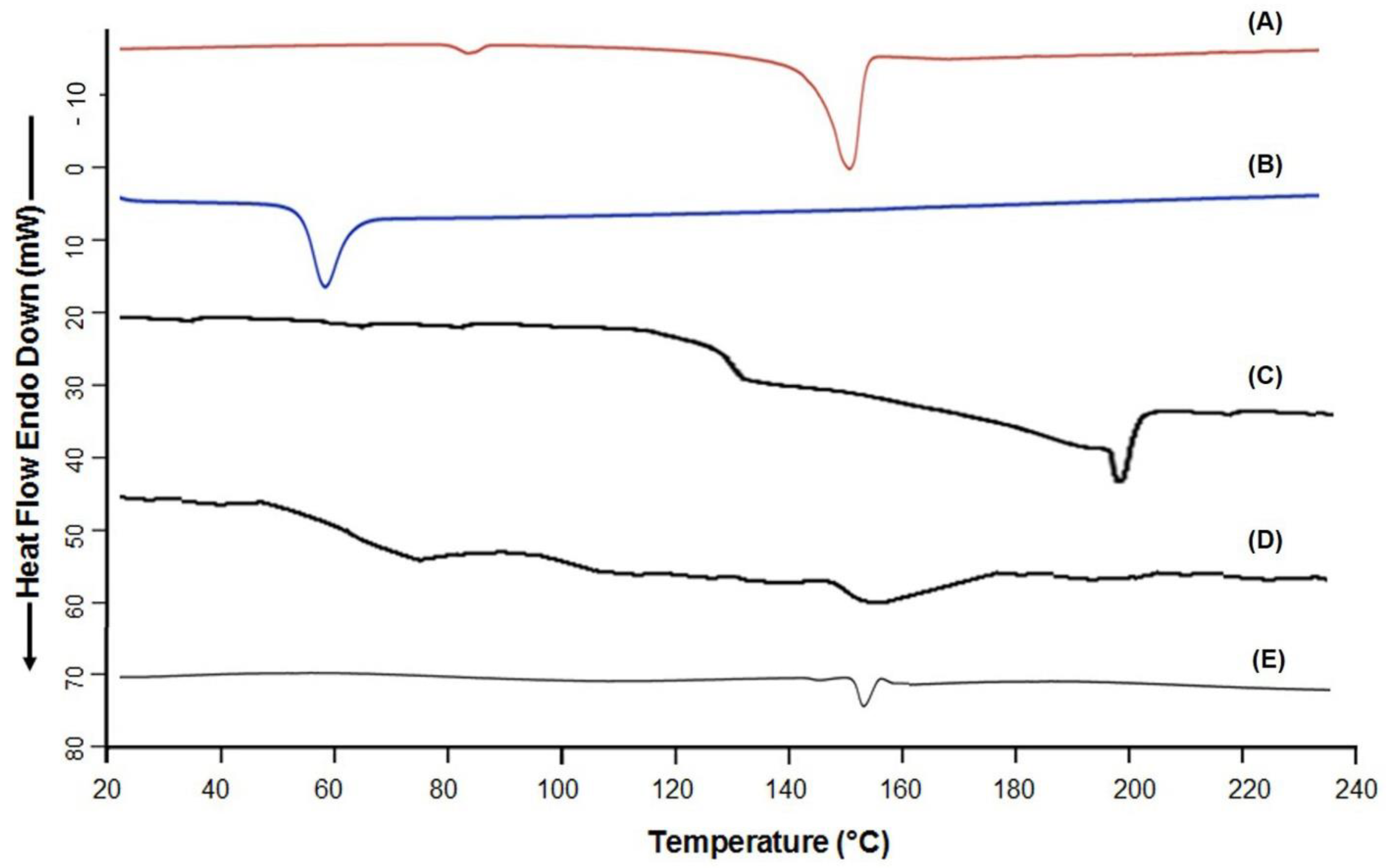
3.7. DSC Analysis
3.8. Stability Studies
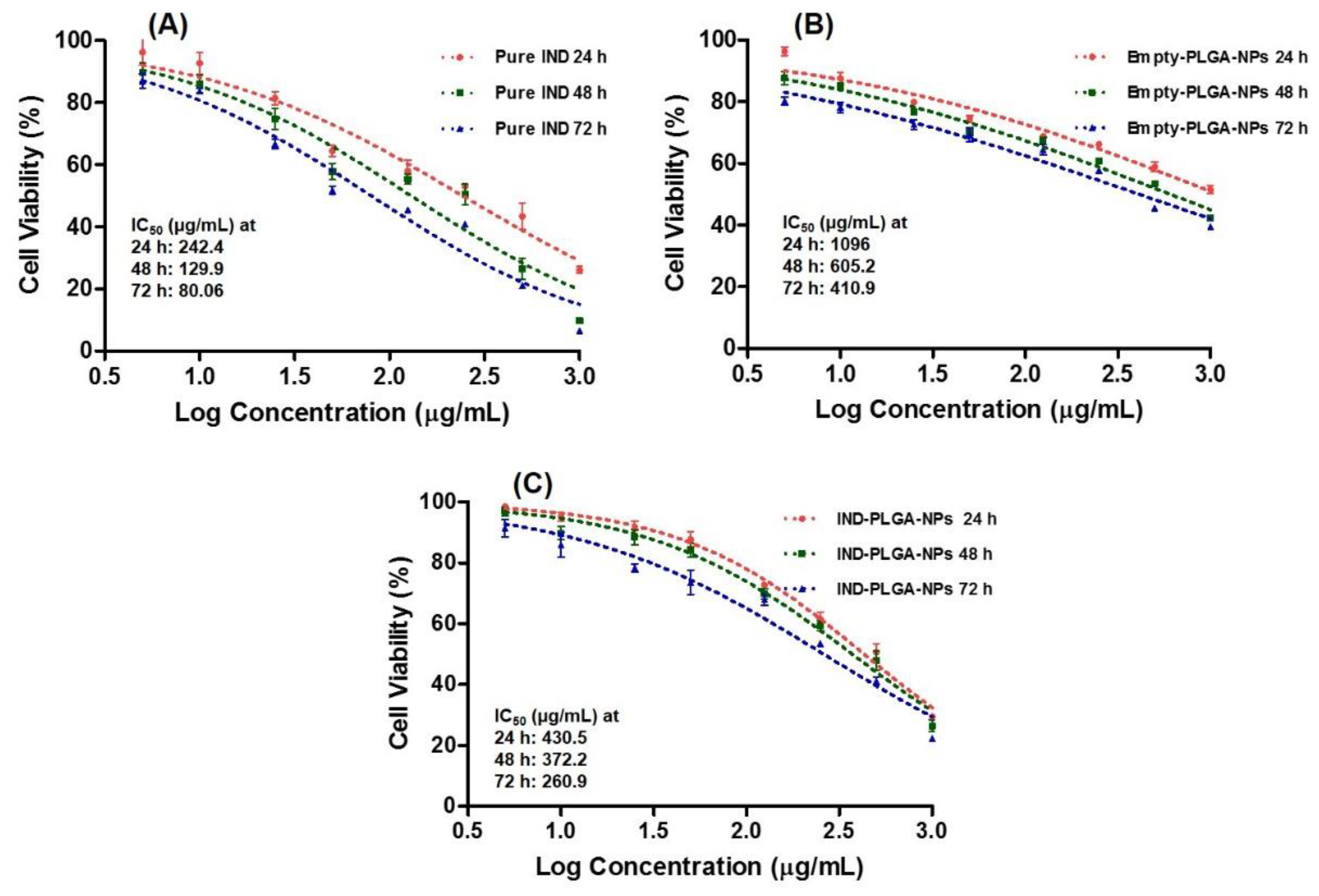
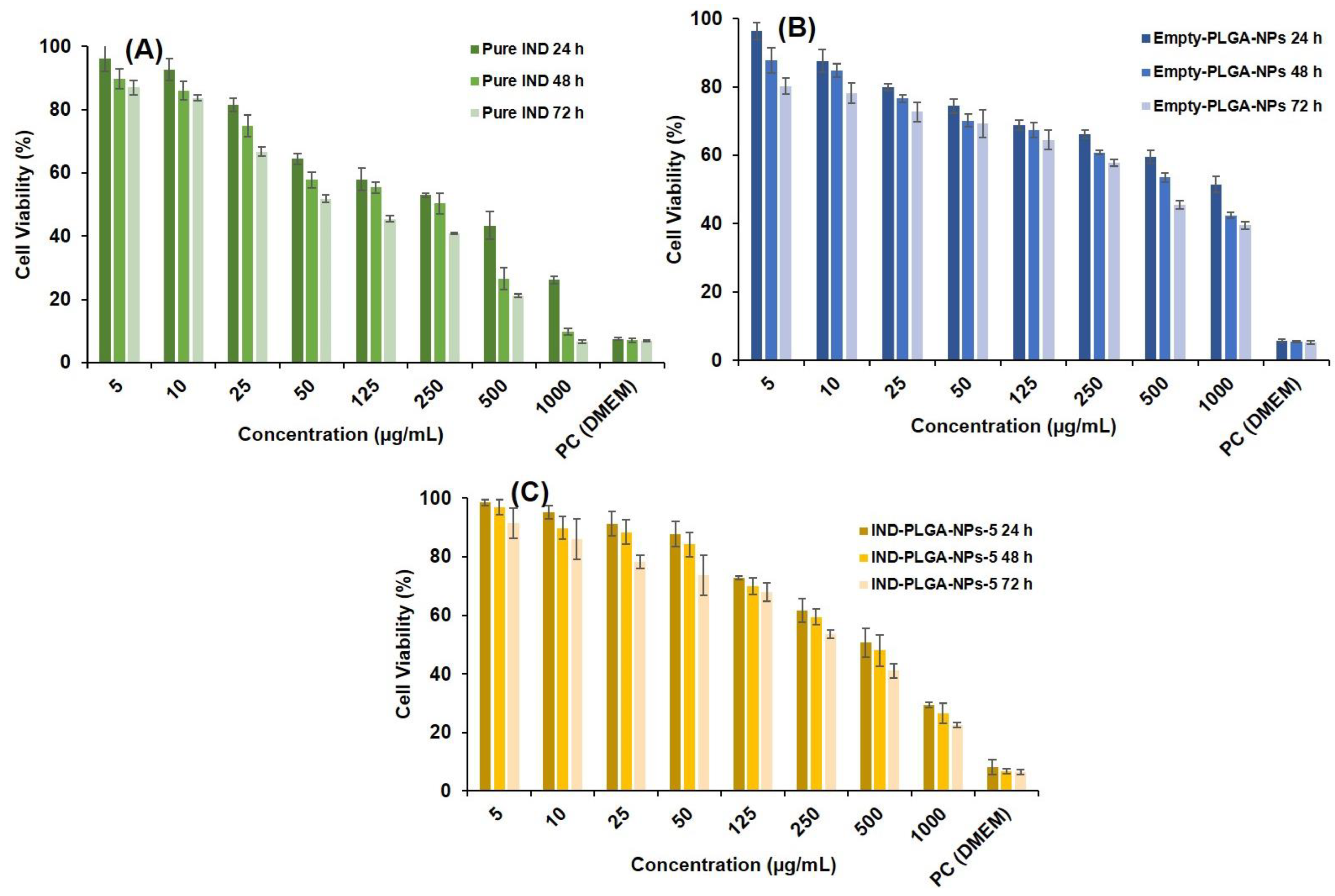
3.9. Cytotoxicity Study by MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developmentsi and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, Y.K.; Kim, S.W. Recent advances in polymeric drug delivery systems. Biomater. Res. 2020, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-X.; Lin, D.-Q.; Yao, S.-J. Design of Chitosan and Its Water Soluble Derivatives-Based Drug Carriers with Polyelectrolyte Complexes. Mar. Drugs 2014, 12, 6236–6253. [Google Scholar] [CrossRef] [PubMed]
- Martinho, N.; Damgé, C.; Reis, C.P. Recent Advances in Drug Delivery Systems. J. Biomater. Nanobiotechnol. 2011, 2, 510–526. [Google Scholar] [CrossRef] [Green Version]
- Dinarvand, R.; Sepehri, N.; Manouchehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [Green Version]
- Astete, C.E.; Sabliov, C.M. Synthesis and characterization of PLGA nanoparticles. J. Biomater. Sci. Polym. Ed. 2006, 17, 247–289. [Google Scholar] [CrossRef]
- Jain, R.A. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) As biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Albericio, F. Polymers and drug delivery systems. Curr. Drug Deliv. 2012, 9, 367–394. [Google Scholar] [CrossRef]
- Fathi-Achachelouei, M.; Keskin, D.; Bat, E.; Vrana, N.E.; Tezcaner, A. Dual growth factor delivery using PLGA nanoparticles in silk fibroin/PEGDMA hydrogels for articular cartilage tissue engineering. J. Biomed. Mater. Res. Part B Appl. Biomater. 2020, 108, 2041–2062. [Google Scholar] [CrossRef]
- Gu, P.; Wusiman, A.; Zhang, Y.; Liu, Z.; Bo, R.; Hu, Y.; Liu, J.; Wang, D. Rational Design of PLGA Nanoparticle Vaccine Delivery Systems To Improve Immune Responses. Mol. Pharm. 2019, 16, 5000–5012. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.F.; Zhu, Y.L.; Sun, B.; Hu, F.H.; Tian, T.; Li, S.C.; Xiao, Z.D. PLGA-based gene delivering nanoparticle enhance suppression effect of miRNA in HePG2 cells. Nanoscale Res. Lett. 2011, 6, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramezani, M.; Ebrahimian, M.; Hashemi, M. Current Strategies in the Modification of PLGA-based Gene Delivery System. Curr. Med. Chem. 2017, 24, 728–739. [Google Scholar] [CrossRef]
- Petrizzo, A.; Conte, C.; Tagliamonte, M.; Napolitano, M.; Bifulco, K.; Carriero, V.; De Stradis, A.; Tornesello, M.L.; Buonaguro, F.M.; Quaglia, F.; et al. Functional characterization of biodegradable nanoparticles as antigen delivery system. J. Exp. Clin. Cancer Res. 2015, 34, 114. [Google Scholar] [CrossRef] [Green Version]
- Cegnar, M.; Kos, J.; Kristl, J. Cystatin incorporated in poly(lactide-co-glycolide) nanoparticles: Development and fundamental studies on preservation of its activity. Eur. J. Pharm. Sci. 2004, 22, 357–364. [Google Scholar] [CrossRef]
- Bisht, R.; Rupenthal, I.D. PLGA nanoparticles for intravitreal peptide delivery: Statistical optimization, characterization and toxicity evaluation. Pharm. Dev. Technol. 2018, 23, 324–333. [Google Scholar] [CrossRef]
- Mundargi, R.C.; Babu, V.R.; Rangaswamy, V.; Patel, P.; Aminabhavi, T.M. Nano/micro technologies for delivering macromolecular therapeutics using poly(d,l-lactide-co-glycolide) and its derivatives. J. Control. Release 2008, 125, 193–209. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Freitas, S.; Merkle, H.P.; Gander, B. Microencapsulation by solvent extraction/evaporation: Reviewing the state of the art of microsphere preparation process technology. J. Control. Release 2005, 102, 313–332. [Google Scholar] [CrossRef]
- Martins, C.; Sousa, F.; Araújo, F.; Sarmento, B. Functionalizing PLGA and PLGA Derivatives for Drug Delivery and Tissue Regeneration Applications. Adv. Health Mater. 2018, 7, 1701035. [Google Scholar] [CrossRef]
- Bala, I.; Hariharan, S.; Kumar, M.N.V.R. PLGA Nanoparticles in Drug Delivery: The State of the Art. Crit. Rev. Ther. Drug Carr. Syst. 2004, 21, 387–422. [Google Scholar] [CrossRef] [PubMed]
- McCall, R.L.; Sirianni, R.W. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J. Vis. Exp. 2013, 82, e51015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Uskoković, D.; Stevanovic, M. Poly(lactide-co-glycolide)-based Micro and Nanoparticles for the Controlled Drug Delivery of Vitamins. Curr. Nanosci. 2009, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Alshamsan, A. Nanoprecipitation is more efficient than emulsion solvent evaporation method to encapsulate cucurbitacin I in PLGA nanoparticles. Saudi Pharm. J. 2014, 22, 219–222. [Google Scholar] [CrossRef]
- McCarron, P.A.; Donnelly, R.F.; Marouf, W. Celecoxib-loaded poly(d,l-lactide-co-glycolide) nanoparticles prepared using a novel and controllable combination of diffusion and emulsification steps as part of the salting-out procedure. J. Microencapsul. 2006, 23, 480–498. [Google Scholar] [CrossRef]
- Alkholief, M.; Albasit, H.; Alhowyan, A.; Alshehri, S.; Raish, M.; Kalam, A.; Alshamsan, A. Employing a PLGA-TPGS based nanoparticle to improve the ocular delivery of Acyclovir. Saudi Pharm. J. 2018, 27, 293–302. [Google Scholar] [CrossRef]
- Kalam, A.; Alshamsan, A. Poly (d,l-lactide-co-glycolide) nanoparticles for sustained release of tacrolimus in rabbit eyes. Biomed. Pharmacother. 2017, 94, 402–411. [Google Scholar] [CrossRef]
- Beck-Broichsitter, M.; Rytting, E.; Lebhardt, T.; Wang, X.; Kissel, T. Preparation of nanoparticles by solvent displacement for drug delivery: A shift in the “ouzo region” upon drug loading. Eur. J. Pharm. Sci. 2010, 41, 244–253. [Google Scholar] [CrossRef]
- Sahana, D.; Mittal, G.; Bhardwaj, V.; Kumar, M. PLGA Nanoparticles for Oral Delivery of Hydrophobic Drugs: Influence of Organic Solvent on Nanoparticle Formation and Release Behavior In Vitro and In Vivo Using Estradiol as a Model Drug. J. Pharm. Sci. 2008, 97, 1530–1542. [Google Scholar] [CrossRef]
- Caputo, F.; Clogston, J.; Calzolai, L.; Rösslein, M.; Prina-Mello, A. Measuring particle size distribution of nanoparticle enabled medicinal products, the joint view of EUNCL and NCI-NCL. A step by step approach combining orthogonal measurements with increasing complexity. J. Control. Release 2019, 299, 31–43. [Google Scholar] [CrossRef]
- Lucas, S. The Pharmacology of Indomethacin. Headache J. Head Face Pain 2016, 56, 436–446. [Google Scholar] [CrossRef]
- Dannenfelser, R.-M.; Yalkowsky, S.H. Data base of aqueous solubility for organic non-electrolytes. Sci. Total Environ. 1991, 109–110, 625–628. [Google Scholar] [CrossRef]
- Williams, C.M. Using medications appropriately in older adults. Am. Fam. Physician 2002, 66, 1917. [Google Scholar]
- Kalam, M.A.; Khan, A.A.; Khan, S.; Almalik, A.; Alshamsan, A. Optimizing indomethacin-loaded chitosan nanoparticle size, encapsulation, and release using Box–Behnken experimental design. Int. J. Biol. Macromol. 2016, 87, 329–340. [Google Scholar] [CrossRef]
- Boon, V.; Glass, B.; Nimmo, A. High-Performance Liquid Chromatographic Assay of Indomethacin in Porcine Plasma with Applicability to Human Levels. J. Chromatogr. Sci. 2006, 44, 41–44. [Google Scholar] [CrossRef] [Green Version]
- Novakova, L.; Matysová, L.; Havlíková, L.C.; Solich, P. Development and validation of HPLC method for determination of indomethacin and its two degradation products in topical gel. J. Pharm. Biomed. Anal. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Kızılbey, K. Optimization of Rutin-Loaded PLGA Nanoparticles Synthesized by Single-Emulsion Solvent Evaporation Method. ACS Omega 2019, 4, 555–562. [Google Scholar] [CrossRef]
- Niu, L.; Panyam, J. Freeze concentration-induced PLGA and polystyrene nanoparticle aggregation: Imaging and rational design of lyoprotection. J. Control. Release 2017, 248, 125–132. [Google Scholar] [CrossRef]
- Alhowyan, A.A.; Altamimi, M.A.; Kalam, M.A.; Khan, A.A.; Badran, M.; Binkhathlan, Z.; Alkholief, M.; Alshamsan, A. Antifungal efficacy of Itraconazole loaded PLGA-nanoparticles stabilized by vitamin-E TPGS: In vitro and ex vivo studies. J. Microbiol. Methods 2019, 161, 87–95. [Google Scholar] [CrossRef]
- Guettari, M.; Gomati, R. Influence of polyvinylpyrrolidone on the interaction between water and methanol. Braz. J. Chem. Eng. 2013, 30, 677–682. [Google Scholar] [CrossRef]
- Kalam, M.A.; Raish, M.; Ahmed, A.; Alkharfy, K.M.; Mohsin, K.; Alshamsan, A.; Al-Jenoobi, F.I.; Al-Mohizea, A.M.; Shakeel, F. Oral bioavailability enhancement and hepatoprotective effects of thymoquinone by self-nanoemulsifying drug delivery system. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 76, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, F.; Haq, N.; El-Badry, M.; Alanazi, F.K.; Alsarra, I. Ultra fine super self-nanoemulsifying drug delivery system (SNEDDS) enhanced solubility and dissolution of indomethacin. J. Mol. Liq. 2013, 180, 89–94. [Google Scholar] [CrossRef]
- Ahnfelt, E.; Sjögren, E.; Hansson, P.; Lennernäs, H. In Vitro Release Mechanisms of Doxorubicin From a Clinical Bead Drug-Delivery System. J. Pharm. Sci. 2016, 105, 3387–3398. [Google Scholar] [CrossRef] [Green Version]
- Alkholief, M. Optimization of Lecithin-Chitosan nanoparticles for simultaneous encapsulation of doxorubicin and piperine. J. Drug Deliv. Sci. Technol. 2019, 52, 204–214. [Google Scholar] [CrossRef]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V.; et al. Delivery of doxorubicin-loaded PLGA nanoparticles into U87 human glioblastoma cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef]
- Chourasiya, V.; Bohrey, S.; Pandey, A. Formulation, optimization, characterization and in-vitro drug release kinetics of atenolol loaded PLGA nanoparticles using 3 3 factorial design for oral delivery. Mater. Discov. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Kalam, M.A.; Humayun, M.; Parvez, N.; Yadav, S.; Garg, A.; Amin, S.; Sultana, Y.; Ali, A. Release kinetics of modified pharmaceutical dosage forms: A review. Cont. J. Pharm. Sci. 2007, 1, 30–35. [Google Scholar]
- Wei, X.; Sun, N.; Wu, B.; Yin, C.; Wu, W. Sigmoidal release of indomethacin from pectin matrix tablets: Effect of in situ crosslinking by calcium cations. Int. J. Pharm. 2006, 318, 132–138. [Google Scholar] [CrossRef]
- Altamimi, M.A.; Neau, S.H. Use of the Flory–Huggins theory to predict the solubility of nifedipine and sulfamethoxazole in the triblock, graft copolymer Soluplus. Drug Dev. Ind. Pharm. 2016, 42, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tao, W.; Zhang, D.; Wu, C.; Song, B.; Wang, S.; Wang, T.; Hu, M.; Liu, X.; Wang, Y.; et al. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J. Pharm. Sci. 2017, 12, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Raish, M.; Kalam, M.A.; Ahmad, A.; Shahid, M.; Ansari, M.A.; Ahad, A.; Ali, R.; Bin Jardan, Y.A.; Alshamsan, A.; Alkholief, M.; et al. Eudragit-Coated Sporopollenin Exine Microcapsules (SEMC) of Phoenix dactylifera L. of 5-Fluorouracil for Colon-Specific Drug Delivery. Pharmaceutics 2021, 13, 1921. [Google Scholar] [CrossRef] [PubMed]
- Alshememry, A.; Kalam, M.A.; Almoghrabi, A.; Alzahrani, A.; Shahid, M.; Khan, A.A.; Haque, A.; Ali, R.; Alkholief, M.; Binkhathlan, Z.; et al. Chitosan-coated poly (lactic-co-glycolide) nanoparticles for dual delivery of doxorubicin and naringin against MCF-7 cells. J. Drug Deliv. Sci. Technol. 2022, 68, 103036. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Evangelista, R.C. PLGA nanoparticles containing praziquantel: Effect of formulation variables on size distribution. Int. J. Pharm. 2005, 290, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Kobayashi, M.; Takeuchi, H.; Kawashima, Y. Preparation of poly(dl-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int. J. Pharm. 1999, 187, 143–152. [Google Scholar] [CrossRef]
- Kwon, H.-Y.; Lee, J.-Y.; Choi, S.-W.; Jang, Y.; Kim, J.-H. Preparation of PLGA nanoparticles containing estrogen by emulsification–diffusion method. Colloids Surfaces A Physicochem. Eng. Asp. 2001, 182, 123–130. [Google Scholar] [CrossRef]
- Quintanar-Guerrero, D.; Fessi, H.; Allémann, E.; Doelker, E. Influence of stabilizing agents and preparative variables on the formation of poly (D, L-lactic acid) nanoparticles by an emulsification-diffusion technique. Int. J. Pharm. 1996, 143, 133–141. [Google Scholar] [CrossRef]
- Feczkó, T.; Tóth, J.; Gyenis, J.J.C.; Physicochemical, S.A.; Aspects, E. Comparison of the preparation of PLGA–BSA nano-and microparticles by PVA, poloxamer and PVP. Colloids Surf. A 2008, 319, 188–195. [Google Scholar] [CrossRef]
- Coombes, A.; Yeh, M.-K.; Lavelle, E.; Davis, S. The control of protein release from poly(dl-lactide co-glycolide) microparticles by variation of the external aqueous phase surfactant in the water-in oil-in water method. J. Control. Release 1998, 52, 311–320. [Google Scholar] [CrossRef]
- Shakeri, S.; Roghanian, R.; Emtiazi, G.; Errico, C.; Chiellini, F.; Chiellini, E. Preparation of protein-loaded PLGA-PVP blend nanoparticles by nanoprecipitation method: Entrapment, Initial burst and drug release kinetic studies. Nanomed. J. 2015, 2, 175–186. [Google Scholar]
- Esmaeili, F.; Atyabi, F.; Dinarvand, R. Preparation of PLGA nanoparticles using TPGS in the spontaneous emulsification solvent diffusion method. J. Exp. Nanosci. 2007, 2, 183–192. [Google Scholar] [CrossRef]
- Jung, T.; Breitenbach, A.; Kissel, T. Sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide)s facilitate the preparation of small negatively charged biodegradable nanospheres. J. Control. Release 2000, 67, 157–169. [Google Scholar] [CrossRef]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Lehr, C.-M.; Maincent, P.; Kawashima, Y. Design of rolipram-loaded nanoparticles: Comparison of two preparation methods. J. Control. Release 2001, 71, 297–306. [Google Scholar] [CrossRef]
- Javadzadeh, Y.; Ahadi, F.; Davaran, S.; Mohammadi, G.; Sabzevari, A.; Adibkia, K. Preparation and physicochemical characterization of naproxen–PLGA nanoparticles. Colloids Surf. B Biointerfaces 2010, 81, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Auría-Soro, C.; Nesma, T.; Juanes-Velasco, P.; Landeira-Viñuela, A.; Fidalgo-Gomez, H.; Acebes-Fernandez, V.; Gongora, R.; Almendral Parra, M.J.; Manzano-Roman, R.; Fuentes, M. Interactions of Nanoparticles and Biosystems: Microenvironment of Nanoparticles and Biomolecules in Nanomedicine. Nanomaterials 2019, 9, 1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-Y.; Chia, H.-H.; Chung, T.-S. Effect of preparation temperature on the characteristics and release profiles of PLGA microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. J. Control. Release 2000, 69, 81–96. [Google Scholar] [CrossRef]
- Yang, Y.Y. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials 2001, 22, 231–241. [Google Scholar] [CrossRef]
- Vanderhoff, J.W.; El-Aasser, M.S.; Ugelstad, J. Polymer Emulsification Process. U.S. Patent US4177177A, 4 December 1979. [Google Scholar]
- Adibkia, K.; Omidi, Y.; Siahi, M.R.; Javadzadeh, A.R.; Barzegar-Jalali, M.; Barar, J.; Maleki, N.; Mohammadi, G.; Nokhodchi, A.; Javadzadeh, A. Inhibition of Endotoxin-Induced Uveitis by Methylprednisolone Acetate Nanosuspension in Rabbits. J. Ocul. Pharmacol. Ther. 2007, 23, 421–432. [Google Scholar] [CrossRef]
- Alshamsan, A.; Binkhathlan, Z.; Kalam, M.A.; Qamar, W.; Kfouri, H.; Alghonaim, M.; Lavasanifar, A. Mitigation of Tacrolimus-Associated Nephrotoxicity by PLGA Nanoparticulate Delivery Following Multiple Dosing to Mice while Maintaining its Immunosuppressive Activity. Sci. Rep. 2020, 10, 6675. [Google Scholar] [CrossRef] [Green Version]
- Adibkia, K.; Shadbad, M.R.S.; Nokhodchi, A.; Javadzedeh, A.; Barzegar-Jalali, M.; Barar, J.; Mohammadi, G.; Omidi, Y. Piroxicam nanoparticles for ocular delivery: Physicochemical characterization and implementation in endotoxin-induced uveitis. J. Drug Target. 2007, 15, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Prabha, S.; Labhasetwar, V. Critical determinants in PLGA/PLA nanoparticle-mediated gene expression. Pharm. Res. 2004, 21, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.-M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y.; Lammers, T.; Kiessling, F. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Pandit, J.K. Preparation and Characterization and Biodistribution Studies of Lomustine Loaded PLGA Nanoparticles by Interfacial Deposition Method. J. Nanomed. Biother. Discov. 2015, 5, 4. [Google Scholar] [CrossRef]
- Turk, C.T.S.; Oz, U.C.; Serim, T.M.; Hascicek, C. Formulation and Optimization of Nonionic Surfactants Emulsified Nimesulide-Loaded PLGA-Based Nanoparticles by Design of Experiments. AAPS PharmSciTech. 2014, 15, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Panyam, J.; Dali, M.M.; Sahoo, S.K.; Ma, W.; Chakravarthi, S.S.; Amidon, G.L.; Levy, R.J.; Labhasetwar, V. Polymer degradation and in vitro release of a model protein from poly(d,l-lactide-co-glycolide) nano- and microparticles. J. Control. Release 2003, 92, 173–187. [Google Scholar] [CrossRef]
- Siepmann, J.; Faisant, N.; Akiki, J.; Richard, J.; Benoit, J. Effect of the size of biodegradable microparticles on drug release: Experiment and theory. J. Control. Release 2004, 96, 123–134. [Google Scholar] [CrossRef]
- Yeo, Y.; Basaran, O.A.; Park, K. A new process for making reservoir-type microcapsules using ink-jet technology and interfacial phase separation. J. Control. Release 2003, 93, 161–173. [Google Scholar] [CrossRef]
- Cartiera, M.S.; Johnson, K.M.; Rajendran, V.; Caplan, M.J.; Saltzman, W.M. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 2009, 30, 2790–2798. [Google Scholar] [CrossRef] [Green Version]
- Wischke, C.; Schwendeman, S.P. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. Int. J. Pharm. 2008, 364, 298–327. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Kawashima, Y.; Niwa, T.; Hino, T.; Takeuchi, H.; Kobayashi, M. Influence of the degrees of hydrolyzation and polymerization of poly(vinylalcohol) on the preparation and properties of poly(dl-lactide-co-glycolide) nanoparticle. Int. J. Pharm. 1997, 149, 43–49. [Google Scholar] [CrossRef]
- Lv, C.; Su, Y.; Wang, Y.; Ma, X.; Sun, Q.; Jiang, Z. Enhanced permeation performance of cellulose acetate ultrafiltration membrane by incorporation of Pluronic F127. J. Membr. Sci. 2007, 294, 68–74. [Google Scholar] [CrossRef]
- Jain, D.K.; Darwhekar, G.; Solanki, S.S.; Sharma, R. Osmotically regulated asymmetric capsular system for sustained delivery of indomethacin. J. Pharm. Investig. 2013, 43, 27–35. [Google Scholar] [CrossRef]
- GÜMÜŞDERELIOĞLU, M.; Deniz, G. Synthesis, characterization and in vitro degradation of poly (dl-lactide)/poly (dl-lactide-co-glycolide) films. Turk. J. Chem. 1999, 23, 153–162. [Google Scholar]
- Shams, T.; Parhizkar, M.; Illangakoon, U.; Orlu, M.; Edirisinghe, M. Core/shell microencapsulation of indomethacin/paracetamol by co-axial electrohydrodynamic atomization. Mater. Des. 2017, 136, 204–213. [Google Scholar] [CrossRef]
- Singh, G.; Kaur, T.; Kaur, R.; Kaur, A. Recent biomedical applications and patents on biodegradable polymer-PLGA. Int. J. Pharmacol. Pharm. Sci. 2014, 1, 30–42. [Google Scholar]
- Panda, S.; Mohanty, G.C.; Samal, R.; Roy, G.S. Viscosity studies of polyvinyl alcohol (PVA, Mw = 1,25,000) in solvent distilled water and aqueous solution of urea. Mater. Sci. Res. India 2010, 7, 443–448. [Google Scholar] [CrossRef]
- Shimizu, Y.; Tanabe, T.; Yoshida, H.; Kasuya, M.; Matsunaga, T.; Haga, Y.; Kurihara, K.; Ohta, M. Viscosity measurement of Xanthan–Poly(vinyl alcohol) mixture and its effect on the mechanical properties of the hydrogel for 3D modeling. Sci. Rep. 2018, 8, 16538. [Google Scholar] [CrossRef]
- Augusto, P.; Ibarz, A.; Cristianini, M. Effect of high pressure homogenization (HPH) on the rheological properties of tomato juice: Time-dependent and steady-state shear. J. Food Eng. 2012, 111, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-S.; Senak, L.; Bonilla, J.; Cullen, J. Comparison of relative viscosity measurement of polyvinylpyrrolidone in water by glass capillary viscometer and differential dual-capillary viscometer. J. Appl. Polym. Sci. 2002, 86, 1312–1315. [Google Scholar] [CrossRef]
- Tóthová, J.; Lisý, V. Intrinsic viscosity of PVP polymers in extremely diluted solutions. e-Polymers 2013, 13, 22. [Google Scholar] [CrossRef]
- Maggio, T.; Scales, C.; Romo-Uribe, A.; Liang, B. The shear rate and concentration dependence of viscosity in concentrated solutions of PVP and Bovine Mucin. Contact Lens Anterior Eye 2018, 41, S67. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Gouda, R.; Baishya, H.; Qing, Z. Application of mathematical models in drug release kinetics of carbidopa and levodopa ER tablets. J. Dev. Drugs 2017, 6, 1–8. [Google Scholar]
- Supramaniam, J.; Adnan, R.; Kaus, N.H.M.; Bushra, R. Magnetic nanocellulose alginate hydrogel beads as potential drug delivery system. Int. J. Biol. Macromol. 2018, 118, 640–648. [Google Scholar] [CrossRef]
- Singhvi, G.; Singh, M. Review: In vitro drug release characterization models. Int. J. Pharm. Stud. Res. 2011, 2, 77–84. [Google Scholar]
- Jing, Z.; Dai, X.; Xian, X.; Du, X.; Liao, M.; Hong, P.; Li, Y. Tough, stretchable and compressive alginate-based hydrogels achieved by non-covalent interactions. RSC Adv. 2020, 10, 23592–23606. [Google Scholar] [CrossRef]
- Gbureck, U.; Vorndran, E.; Barralet, J.E. Modeling vancomycin release kinetics from microporous calcium phosphate ceramics comparing static and dynamic immersion conditions. Acta Biomater. 2008, 4, 1480–1486. [Google Scholar] [CrossRef]
- Unagolla, J.M.; Jayasuriya, A.C. Drug transport mechanisms and in vitro release kinetics of vancomycin encapsulated chitosan-alginate polyelectrolyte microparticles as a controlled drug delivery system. Eur. J. Pharm. Sci. 2018, 114, 199–209. [Google Scholar] [CrossRef]
- Hung, H.-I.; Klein, O.J.; Peterson, S.W.; Rokosh, R.; Osseiran, S.; Nowell, N.H.; Evans, C.L. PLGA nanoparticle encapsulation reduces toxicity while retaining the therapeutic efficacy of EtNBS-PDT in vitro. Sci. Rep. 2016, 6, 33234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; George, S.; Yu, H.; Damoiseaux, R.; France, B.; Ng, K.W.; Loo, J.S.-C. Size influences the cytotoxicity of poly (lactic-co-glycolic acid) (PLGA) and titanium dioxide (TiO2) nanoparticles. Arch. Toxicol. 2012, 87, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.I.; Samad, N.A.; Fang, L.; Lim, V. Cytotoxicity of targeted PLGA nanoparticles: A systematic review. RSC Adv. 2021, 11, 9433–9449. [Google Scholar] [CrossRef]
- Di-Wen, S.; Pan, G.-Z.; Hao, L.; Zhang, J.; Xue, Q.-Z.; Wang, P.; Yuan, Q.-Z. Improved antitumor activity of epirubicin-loaded CXCR4-targeted polymeric nanoparticles in liver cancers. Int. J. Pharm. 2016, 500, 54–61. [Google Scholar] [CrossRef]
- Miura, N.; Matsumoto, Y.; Miyairi, S.; Nishiyama, S.; Naganuma, A. Protective Effects of Triterpene Compounds Against the Cytotoxicity of Cadmium in HepG2 Cells. Mol. Pharmacol. 1999, 56, 1324–1328. [Google Scholar] [CrossRef]
- Louisa, M.; Suyatna, F.D.; Wanandi, S.I.; Asih, P.B.S.; Syafruddin, D. Differential expression of several drug transporter genes in HepG2 and Huh-7 cell lines. Adv. Biomed. Res. 2016, 5, 104. [Google Scholar] [CrossRef]
- Norouzzadeh, M.; Kalikias, Y.; Mohamadpur, Z.; Sharifi, L.; Mahmoudi, M. Determining population doubling time and the appropriate number of HepG2 cells for culturing in 6-well plate. Int. Res. Bas. Sci. 2016, 10, 299–303. [Google Scholar]
- Wen-Sheng, W.J.O. ERK signaling pathway is involved in p15INK4b/p16INK4a expression and HepG2 growth inhibition triggered by TPA and Saikosaponin a. Oncogene 2003, 22, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Sriuttha, P.; Sirichanchuen, B.; Permsuwan, U. Hepatotoxicity of Nonsteroidal Anti-Inflammatory Drugs: A Systematic Review of Randomized Controlled Trials. Int. J. Hepatol. 2018, 2018, 5253623. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Organic Solvents * | Type of Stabilizer, Its Concentration * | Mean ± SD *, n = 3 | ||
|---|---|---|---|---|---|
| Particle Size (nm) | Polydispersity Index | Zeta-Potential (mV) | |||
| Batch 1 | |||||
| PLGA-NPs-1 | CHCl3 | PVA (1%, w/v) | 328.1 ± 24.4 | 0.096 ± 0.079 | −1.90 ± 0.91 |
| PLGA-NPs-2 | CHCl3 | PVA (3%, w/v) | 345.2 ± 8.3 | 0.061 ± 0.036 | −1.12 ± 0.06 |
| PLGA-NPs-3 | CHCl3 | PVA (6%, w/v) | 588.1 ± 23.9 | 0.096 ± 0.028 | −0.74 ± 0.12 |
| PLGA-NPs-4 | CHCl3 | PVA (9%, w/v) | 317.6 ± 2.7 | 0.285 ± 0.088 | −0.68 ± 0.11 |
| Batch 2 | |||||
| PLGA-NPs-5 | DCM | PVA (1%, w/v) | 273.2 ± 3.3 | 0.087 ± 0.064 | −0.92 ± 0.21 |
| PLGA-NPs-6 | DCM | PVA (3%, w/v) | 406.3 ± 8.5 | 0.213 ± 0.044 | −0.89 ± 0.28 |
| PLGA-NPs-7 | DCM | PVA (6%, w/v) | 554.4 ± 23.8 | 0.239 ± 0.045 | −0.76 ± 0.06 |
| PLGA-NPs-8 | DCM | PVA (9%, w/v) | 563.9 ± 54.4 | 0.170 ± 0.015 | −0.62 ± 0.58 |
| Batch 3 | |||||
| PLGA-NPs-9 | CHCl3 | PVP (1%, w/v) | 456.9 ± 61.4 | 0.152 ± 0.069 | 17.73 ± 3.45 |
| PLGA-NPs-10 | CHCl3 | PVP (3%, w/v) | 448.8 ± 22.4 | 0.142 ± 0.035 | 14.05 ± 2.05 |
| PLGA-NPs-11 | CHCl3 | PVP (6%, w/v) | 466.6 ± 90.5 | 0.256 ± 0.181 | 3.86 ± 1.26 |
| PLGA-NPs-12 | CHCl3 | PVP (9%, w/v) | 381.4 ± 83.5 | 0.255 ± 0.179 | 2.67 ± 1.41 |
| Batch 4 | |||||
| PLGA-NPs-13 | DCM | PVP (1%, w/v) | 287.8 ± 12.0 | 0.035 ± 0.029 | 10.24 ± 3.93 |
| PLGA-NPs-14 | DCM | PVP (3%, w/v) | 566.9 ± 64.1 | 0.268 ± 0.042 | 5.82 ± 2.65 |
| PLGA-NPs-15 | DCM | PVP (6%, w/v) | 609.3 ± 26.9 | 0.512 ± 0.055 | 5.37 ± 1.06 |
| PLGA-NPs-16 | DCM | PVP (9%, w/v) | 684.2 ± 43.1 | 0.846 ± 0.016 | 4.91 ± 2.37 |
| Formulations | Mean ± SD, n = 3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Particle Size (nm) | Polydispersity Index | Zeta Potential (mV) | Encapsulation Efficiency (%) | Drug Loading (%) | Cumulative Drug Release (%) at pH 7.4 | Cumulative Drug Release (%) at pH 1.2 | |||
| At 12 h | At 72 h | At 12 h | At 72 h | ||||||
| PVA Group formulations | |||||||||
| Batch-1 | |||||||||
| 1%, PVA with CHCl3 (PLGA-NPs-1) | 331.8 ± 8.4 | 0.174 ± 0.035 | −2.89 ± 0.95 | 68.74 ± 5.47 | 6.24 ± 0.51 | 16.46 ± 0.63 | 43.94 ± 2.43 | 41.28 ± 3.09 | 42. 16 ± 2.49 |
| 3%, PVA with CHCl3 (PLGA-NPs-2) | 547.4 ± 10.3 | 0.288 ± 0.078 | −0.81 ± 0.14 | 69.59 ± 6.15 | 6.32 ± 0.56 | 13.09 ± 0.78 | 32.83 ± 1.76 | 30.38 ± 2.01 | 32.89 ± 3.88 |
| Batch-2 | |||||||||
| 1%, PVA with DCM (PLGA-NPs-5) | 275.4 ± 8.5 | 0.157 ± 0.048 | −1.13 ± 0.29 | 72.96 ± 4.59 | 6.63 ± 0.42 | 18.82 ± 1.38 | 52.16 ± 3.83 | 31.70 ± 2.03 | 40.06 ± 1.32 |
| 3%, PVA with DCM (PLGA-NPs-6) | 410.9 ± 11.3 | 0.205 ± 0.061 | −1.14 ± 0.58 | 74.86 ± 4.78 | 6.81 ± 0.45 | 14.58 ± 0.81 | 37.88 ± 2.11 | 37.04 ± 1.07 | 39. 17 ± 1.49 |
| PVP Group formulations | |||||||||
| Batch-3 | |||||||||
| 1%, PVP with CHCl3 (PLGA-NPs-9) | 439.3 ± 15.1 | 0.179 ± 0.033 | 16.1 ± 3.5 | 54.94 ± 6.34 | 4.99 ± 0.58 | 15.76 ± 0.86 | 39.90 ± 2.28 | 34.05 ± 1.27 | 38.14 ± 1.83 |
| 3%, PVP with CHCl3 (PLGA-NPs-10) | 379.5 ± 10.5 | 0.177 ± 0.065 | 15.5 ± 2.6 | 58.91 ± 4.84 | 5.36 ± 0.44 | 16.83 ± 0.95 | 43.64 ± 2.53 | 26.46 ± 2.28 | 32.48 ± 2.12 |
| Batch-4 | |||||||||
| 1%, PVP with DCM (PLGA-NPs-13) | 286.9 ± 13.2 | 0.066 ± 0.002 | 10.4 ± 3.94 | 63.33 ± 6.77 | 5.76 ± 0.62 | 19.42 ± 3.47 | 48.07 ± 2.47 | 33.13 ± 1.18 | 35.81 ± 2.08 |
| 3%, PVP with DCM (PLGA-NPs-14) | 569.6 ± 15.6 | 0.276 ± 0.055 | 7.6 ± 2.37 | 65.37 ± 5.36 | 5.94 ± 0.49 | 19.05 ± 1.08 | 51.68 ± 2.99 | 39.89 ± 1.86 | 43.01 ± 1.17 |
| Formulations | Release Kinetic Models | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Zero Order | First Order | Higuchi Matrix | Korsmeyer–Peppas | Hixon–Crowell | |||||||||||
| R2 | *k0 (10−2) | n-Value | R2 | *k1 (10−1) | n-Value | R2 | *kHM (10−2) | n-Value | R2 | *kKP (10−1) | n-Value | R2 | *kHC (10−0) | n-Value | |
| PLGA-NPs-1 | 0.966 | 1.66 | 0.0024 | 0.985 | 9.31 | 0.0014 | 0.998 | 4.64 | 0.2366 | 0.996 | 1.78 | 0.2461 | 0.979 | 1.38 | 0.0009 |
| PLGA-NPs-2 | 0.964 | 1.28 | 0.0018 | 0.979 | 9.35 | 0.0009 | 0.999 | 3.54 | 0.0173 | 0.998 | 1.35 | 0.2412 | 0.974 | 1.39 | 0.0006 |
| PLGA-NPs-5 | 0.968 | 1.85 | 0.0029 | 0.989 | 9.28 | 0.0018 | 0.997 | 5.26 | 0.0282 | 0.994 | 2.08 | 0.2563 | 0.984 | 1.44 | 0.0012 |
| PLGA-NPs-6 | 0.965 | 1.44 | 0.0021 | 0.982 | 9.33 | 0.0012 | 0.999 | 4.02 | 0.0203 | 0.998 | 1.58 | 0.2462 | 0.976 | 1.42 | 0.0008 |
| PLGA-NPs-9 | 0.963 | 1.43 | 0.0022 | 0.982 | 9.33 | 0.0013 | 0.998 | 4.12 | 0.0216 | 0.997 | 1.65 | 0.2579 | 0.976 | 1.27 | 0.0009 |
| PLGA-NPs-10 | 0.964 | 1.58 | 0.0024 | 0.984 | 9.32 | 0.0014 | 0.841 | 4.51 | 0.0236 | 0.998 | 1.81 | 0.2562 | 0.978 | 1.30 | 0.0009 |
| PLGA-NPs-13 | 0.936 | 1.81 | 0.0077 | 0.958 | 9.29 | 0.0058 | 0.991 | 5.07 | 0.0822 | 0.990 | 2.01 | 0.0947 | 0.951 | 1.34 | 0.0037 |
| PLGA-NPs-14 | 0.967 | 1.76 | 0.0029 | 0.959 | 9.30 | 0.0022 | 0.997 | 5.11 | 0.0284 | 0.994 | 2.02 | 0.2649 | 0.983 | 1.36 | 0.0012 |
| Characterization Parameters | Storage Time (Days) | ||
|---|---|---|---|
| Initial (0 Day) | At 15th Day | At 30th Day | |
| At 4 °C | |||
| Particle Size (nm) | 275.4 ± 8.5 | 275.8 ± 8.67 | 276.9 ± 7.81 |
| Polydispersity-index | 0.157 ± 0.048 | 0.158 ± 0.051 | 0.163 ± 0.054 |
| Zeta-Potential (mV) | −1.13 ± 0.29 | −1.12 ± 0.27 | −1.11 ± 0.28 |
| Encapsulation efficiency (%) | 72.96 ± 4.59 | 72.54 ± 4.51 | 71.02 ± 4.63 |
| Drug loading (%) | 6.63 ± 0.42 | 6.50 ± 0.43 | 6.46 ± 0.45 |
| At 30 °C | |||
| Particle Size (nm) | 275.4 ± 8.5 | 277.2 ± 8.65 | 279.0 ± 8.81 |
| Polydispersity-index | 0.157 ± 0.048 | 0.158 ± 0.049 | 0.159 ± 0.047 |
| Zeta-Potential (mV) | −1.13 ± 0.29 | −1.12 ± 0.27 | −1.11 ± 0.31 |
| Encapsulation efficiency (%) | 72.96 ± 4.59 | 71.87 ± 4.54 | 71.0 ± 4.64 |
| Drug loading (%) | 6.63 ± 0.42 | 6.53 ± 0.41 | 6.46 ± 0.42 |
| At 37 °C | |||
| Particle Size (nm) | 275.4 ± 8.5 | 277.8 ± 9.36 | 279.8 ± 6.95 |
| Polydispersity-index | 0.157 ± 0.048 | 0.159 ± 0.049 | 0.161 ± 0.048 |
| Zeta-Potential (mV) | −1.13 ± 0.29 | −1.12 ± 0.28 | −1.11 ± 0.29 |
| Encapsulation efficiency (%) | 72.96 ± 4.59 | 71.38 ± 4.72 | 70.28 ± 4.22 |
| Drug loading (%) | 6.63 ± 0.42 | 6.44 ± 0.39 | 6.42 ± 0.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkholief, M.; Kalam, M.A.; Anwer, M.K.; Alshamsan, A. Effect of Solvents, Stabilizers and the Concentration of Stabilizers on the Physical Properties of Poly(d,l-lactide-co-glycolide) Nanoparticles: Encapsulation, In Vitro Release of Indomethacin and Cytotoxicity against HepG2-Cell. Pharmaceutics 2022, 14, 870. https://doi.org/10.3390/pharmaceutics14040870
Alkholief M, Kalam MA, Anwer MK, Alshamsan A. Effect of Solvents, Stabilizers and the Concentration of Stabilizers on the Physical Properties of Poly(d,l-lactide-co-glycolide) Nanoparticles: Encapsulation, In Vitro Release of Indomethacin and Cytotoxicity against HepG2-Cell. Pharmaceutics. 2022; 14(4):870. https://doi.org/10.3390/pharmaceutics14040870
Chicago/Turabian StyleAlkholief, Musaed, Mohd Abul Kalam, Md Khalid Anwer, and Aws Alshamsan. 2022. "Effect of Solvents, Stabilizers and the Concentration of Stabilizers on the Physical Properties of Poly(d,l-lactide-co-glycolide) Nanoparticles: Encapsulation, In Vitro Release of Indomethacin and Cytotoxicity against HepG2-Cell" Pharmaceutics 14, no. 4: 870. https://doi.org/10.3390/pharmaceutics14040870
APA StyleAlkholief, M., Kalam, M. A., Anwer, M. K., & Alshamsan, A. (2022). Effect of Solvents, Stabilizers and the Concentration of Stabilizers on the Physical Properties of Poly(d,l-lactide-co-glycolide) Nanoparticles: Encapsulation, In Vitro Release of Indomethacin and Cytotoxicity against HepG2-Cell. Pharmaceutics, 14(4), 870. https://doi.org/10.3390/pharmaceutics14040870