
Microgels Formed by Spontaneous Click Chemistries Utilizing Microfluidic Flow Focusing for Cargo Release in Response to Endogenous or Exogenous Stimuli


Abstract
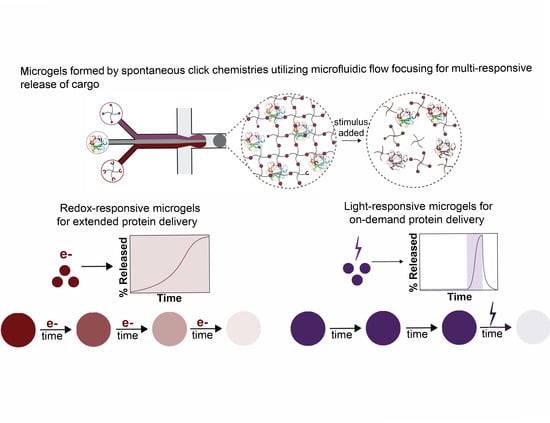
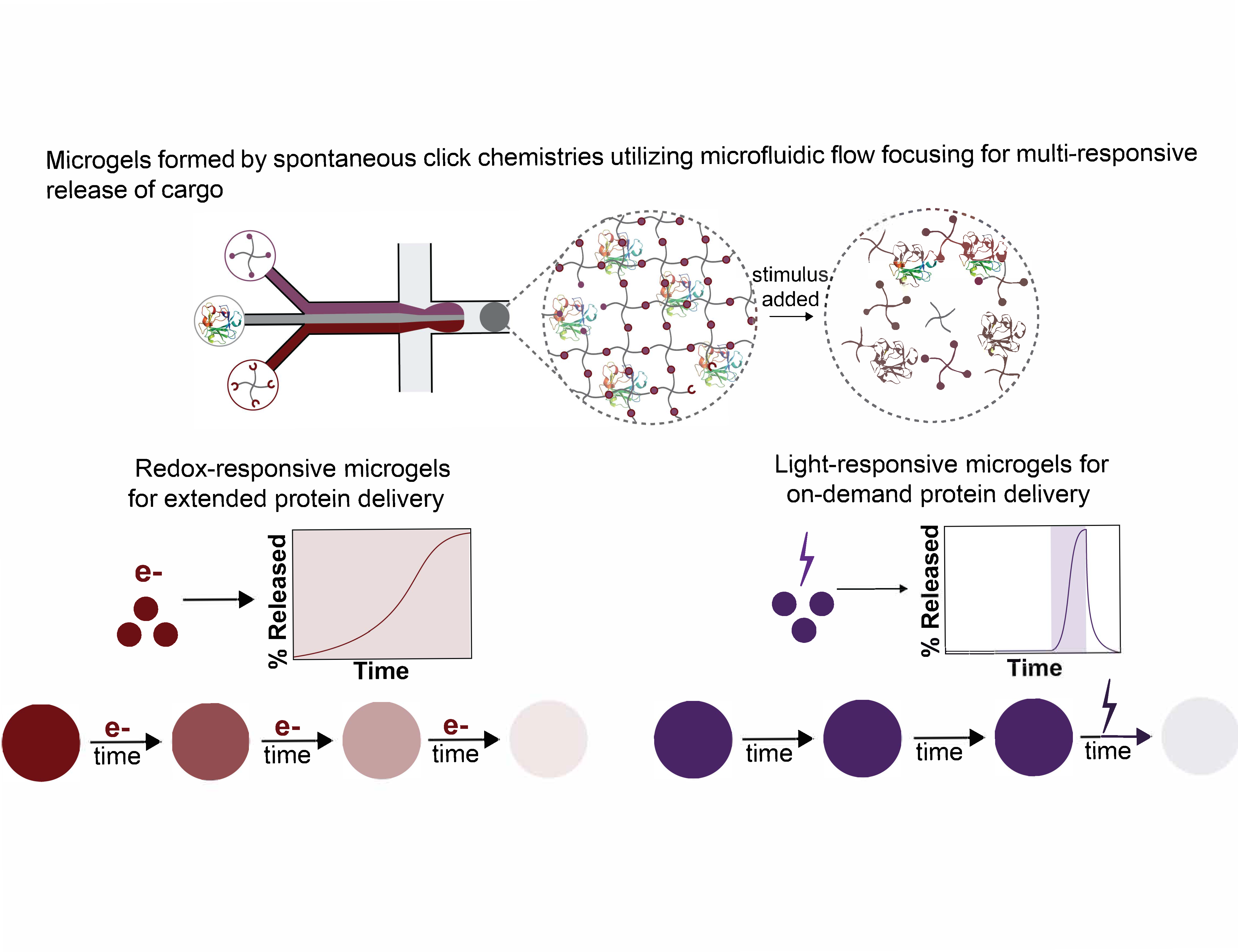
:
{kind=link}
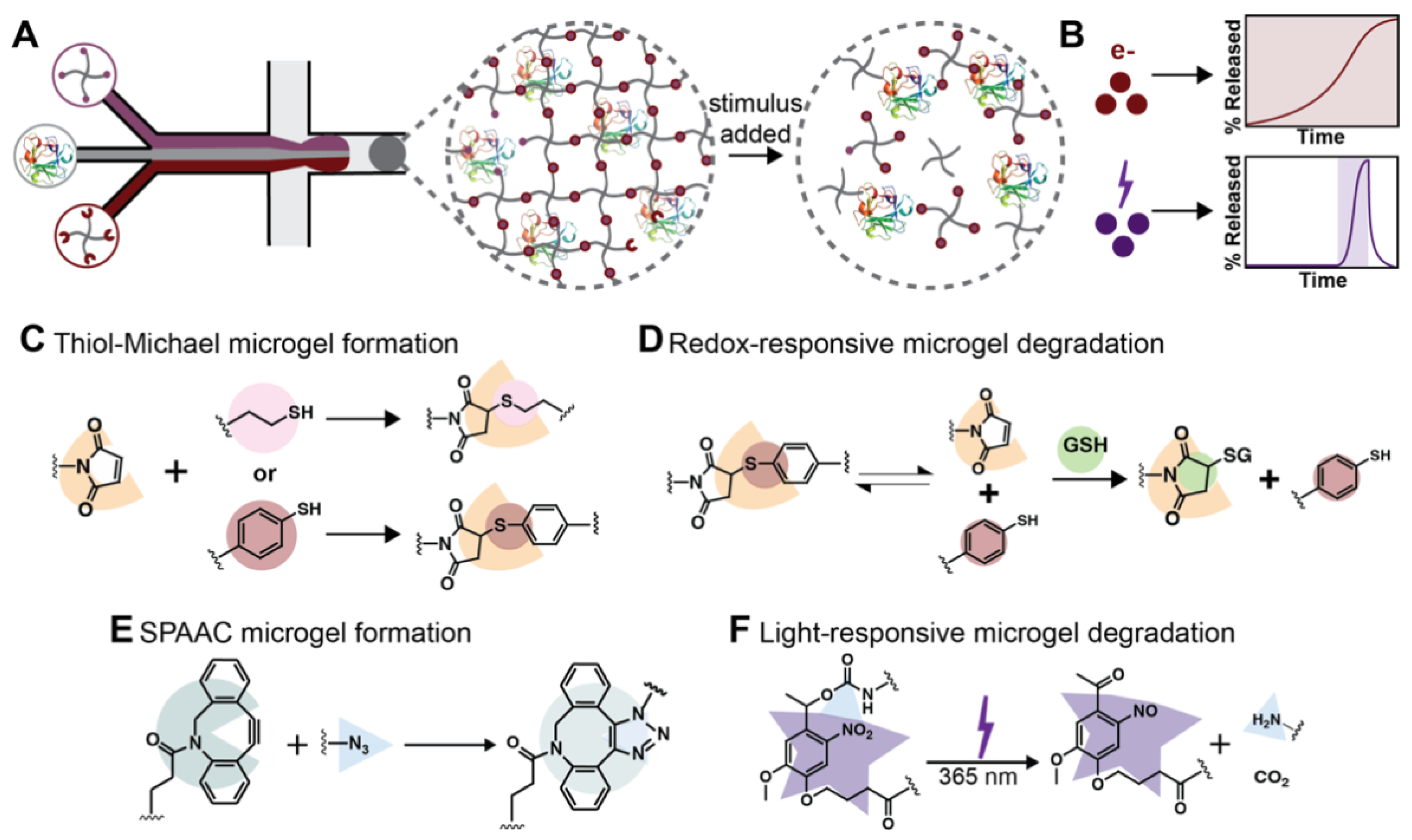{kind=link}
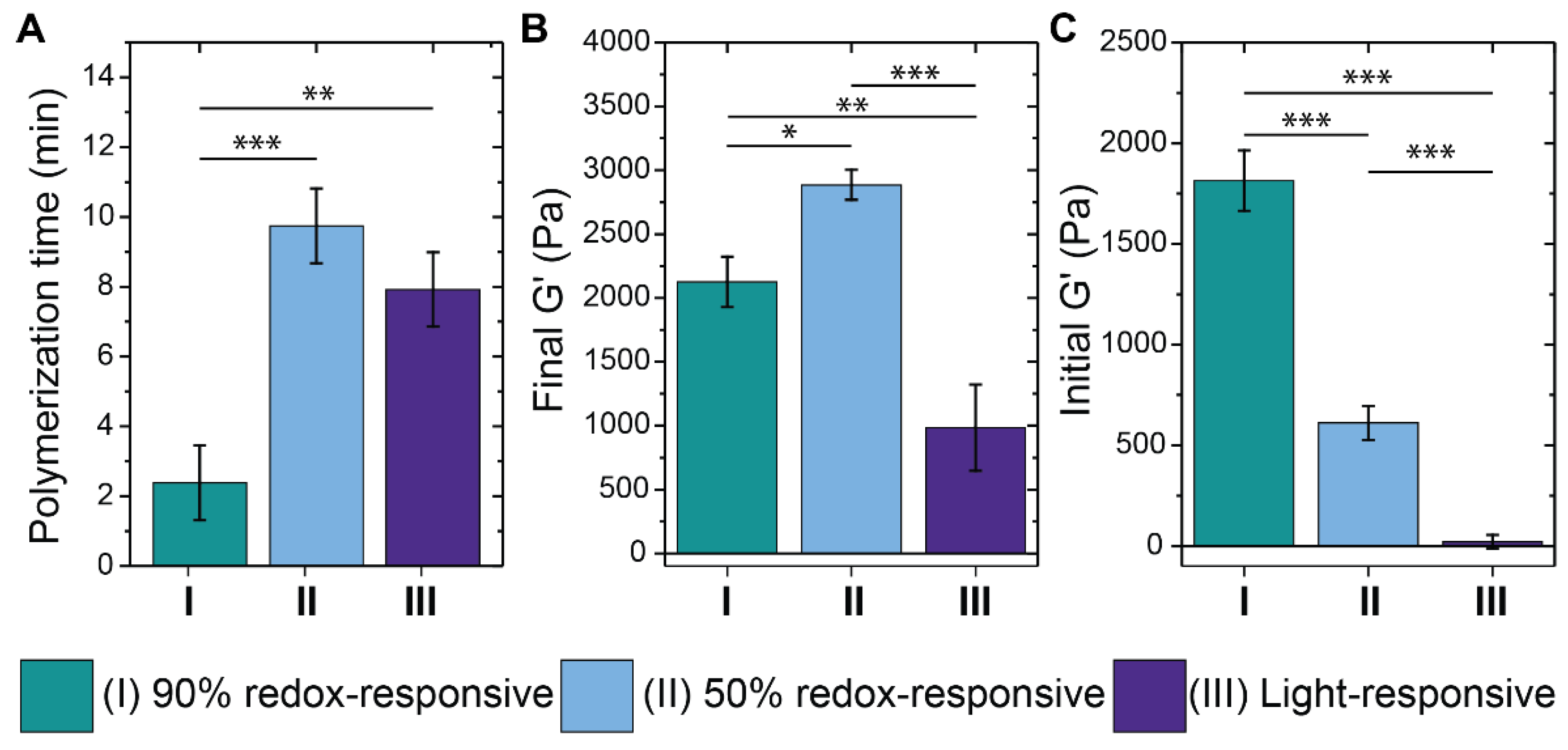{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Bulk Hydrogel Formation and Mechanical Properties
3.2. Formation of Thiol-Michael PEG Microgels Using Flow Focusing Microfluidics
3.3. Degradation of Redox-Sensitive Microgels
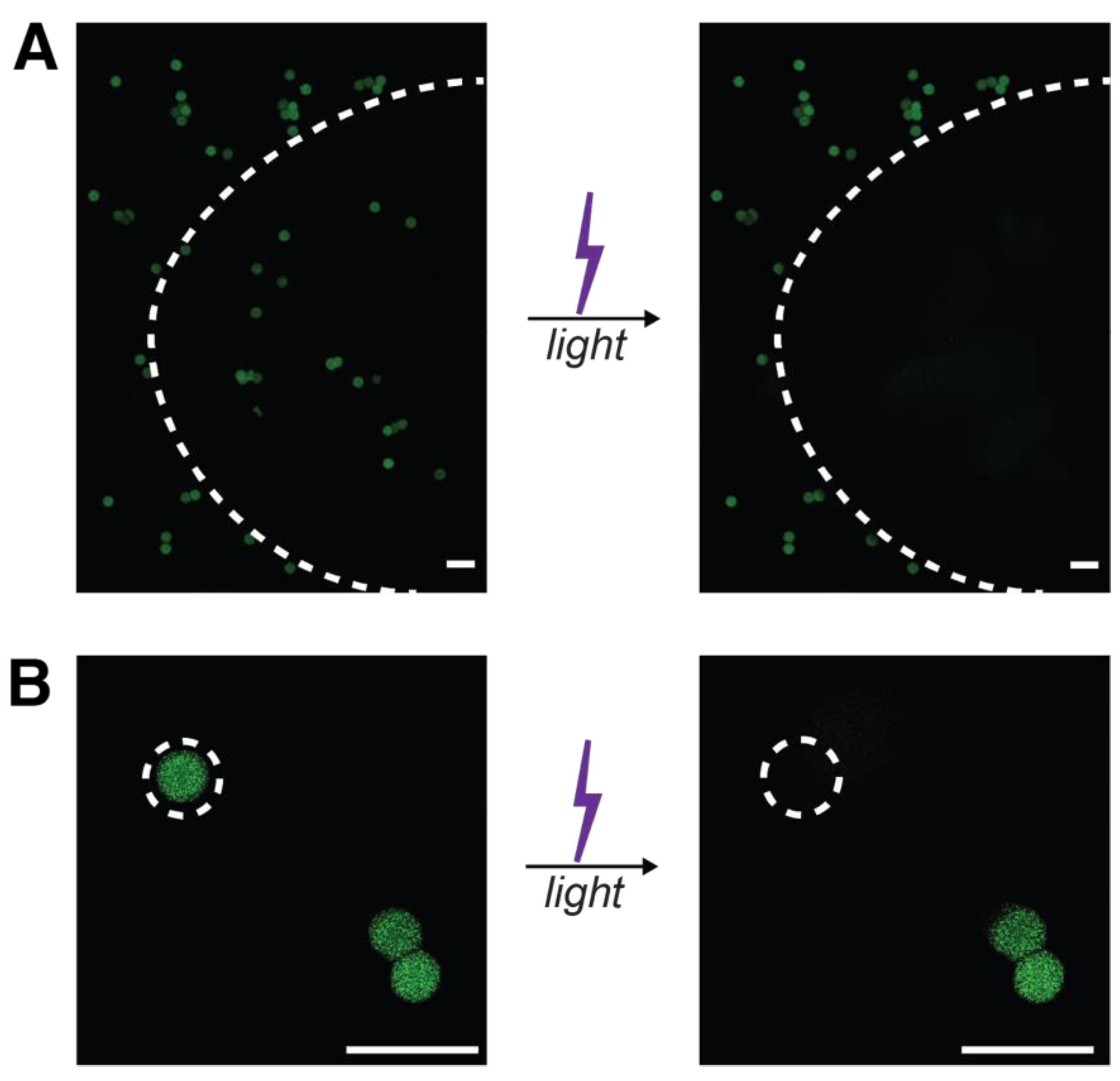
3.4. Formation and Degradation of Light-Sensitive Microgels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mullard, A. 2018 FDA drug approvals. Nat. Rev. Drug Discov. 2019, 18, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of Therapeutic Proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Y.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.K.; Coffin, M.V.; Manceva, S.D.; Chichester, J.A.; Jones, R.M.; Kiick, K.L. Controlled release of an anthrax toxin-neutralizing antibody from hydrolytically degradable polyethylene glycol hydrogels. J. Biomed. Mater. Res. Part A 2016, 104, 113–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Debroy, D.; Liu, J.; Li-Oakey, K.; Oakey, J. Structured Hydrogel Particles With Nanofabricated Interfaces via Controlled Oxygen Inhibition. IEEE Trans. NanoBioscience 2019, 18, 253–256. [Google Scholar] [CrossRef]
- Singh, S.K.; Luisi, D.L.; Pak, R.H. Antibody-Drug Conjugates: Design, Formulation and Physicochemical Stability. Pharm. Res. 2015, 32, 3541–3571. [Google Scholar] [CrossRef]
- Kharkar, P.M.; Scott, R.A.; Olney, L.P.; LeValley, P.J.; Maverakis, E.; Kiick, K.L.; Kloxin, A.M. Controlling the Release of Small, Bioactive Proteins via Dual Mechanisms with Therapeutic Potential. Adv. Healthc. Mater. 2017, 6, 1700713. [Google Scholar] [CrossRef]
- Daly, A.C.; Riley, L.; Segura, T.; Burdick, J.A. Hydrogel microparticles for biomedical applications. Nat. Rev. Mater. 2020, 5, 20–43. [Google Scholar] [CrossRef]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem. Soc. Rev. 2013, 42, 7335–7372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimatteo, R.; Darling, N.J.; Segura, T. In situ forming injectable hydrogels for drug delivery and wound repair. Adv. Drug Deliv. Rev. 2018, 127, 167–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.M.; Sun, P.J. pH-Sensitive Polyampholyte Microgels of Poly(Acrylic Acid-co-Vinylamine) as Injectable Hydrogel for Controlled Drug Release. Polymers 2019, 11, 285. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.R.; Weaver, W.M.; Scumpia, P.O.; Di Carlo, D.; Segura, T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nat. Mater. 2015, 14, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, V.G.; Qazi, T.H.; Shan, J.; Groll, J.; Burdick, J.A. Influence of Microgel Fabrication Technique on Granular Hydrogel Properties. ACS Biomater. Sci. Eng. 2021, 7, 4269–4281. [Google Scholar] [CrossRef]
- Alemán, J.V.; Chadwick, A.V.; He, J.; Hess, M.; Horie, K.; Jones, R.G.; Kratochvíl, P.; Meisel, I.; Mita, I.; Moad, G.; et al. Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic-organic hybrid materials (IUPAC Recommendations 2007). Pure Appl. Chem. 2007, 79, 1801–1829. [Google Scholar] [CrossRef]
- Debroy, D.; Li-Oakey, K.D.; Oakey, J. Engineering functional hydrogel microparticle interfaces by controlled oxygen-inhibited photopolymerization. Colloids Surf. B Biointerfaces 2019, 180, 371–375. [Google Scholar] [CrossRef]
- Mealy, J.E.; Chung, J.J.; Jeong, H.H.; Issadore, D.; Lee, D.; Atluri, P.; Burdick, J.A. Injectable Granular Hydrogels with Multifunctional Properties for Biomedical Applications. Adv. Mater. 2018, 30, 1705912. [Google Scholar] [CrossRef]
- Headen, D.M.; Aubry, G.; Lu, H.; Garcia, A.J. Microfluidic-Based Generation of Size-Controlled, Biofunctionalized Synthetic Polymer Microgels for Cell Encapsulation. Adv. Mater. 2014, 26, 3003–3008. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Zou, Q.; Zuo, Y.; Mei, Q.; Ma, J.; Lin, L.; Chen, L.; Li, Y. Injectable Gel Constructs with Regenerative and Anti-Infective Dual Effects Based on Assembled Chitosan Microspheres. ACS Appl. Mater. Interfaces 2018, 10, 25099–25112. [Google Scholar] [CrossRef]
- Hu, Z.; Ma, C.; Rong, X.; Zou, S.; Liu, X. Immunomodulatory ECM-like Microspheres for Accelerated Bone Regeneration in Diabetes Mellitus. ACS Appl Mater Interfaces 2018, 10, 2377–2390. [Google Scholar] [CrossRef] [PubMed]
- Switacz, V.K.; Wypysek, S.K.; Degen, R.; Crassous, J.J.; Spehr, M.; Richtering, W. Influence of Size and Cross-Linking Density of Microgels on Cellular Uptake and Uptake Kinetics. Biomacromolecules 2020, 21, 4532–4544. [Google Scholar] [CrossRef] [PubMed]
- Karg, M.; Pich, A.; Hellweg, T.; Hoare, T.; Lyon, L.A.; Crassous, J.J.; Suzuki, D.; Gumerov, R.A.; Schneider, S.; Potemkin, I.I.; et al. Nanogels and Microgels: From Model Colloids to Applications, Recent Developments, and Future Trends. Langmuir 2019, 35, 6231–6255. [Google Scholar] [CrossRef] [PubMed]
- Mejías, J.C.; Roy, K. In-vitro and in-vivo characterization of a multi-stage enzyme-responsive nanoparticle-in-microgel pulmonary drug delivery system. J. Control. Release 2019, 316, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Shahid, M.; Farooqi, Z.H.; Begum, R.; Arif, M.; Wu, W.; Irfan, A. Hybrid Microgels for Catalytic and Photocatalytic Removal of Nitroarenes and Organic Dyes From Aqueous Medium: A Review. Crit. Rev. Anal. Chem. 2020, 50, 513–537. [Google Scholar] [CrossRef]
- de Carvalho, B.G.; Taketa, T.B.; Garcia, B.B.M.; Han, S.W.; de la Torre, L.G. Hybrid microgels produced via droplet microfluidics for sustainable delivery of hydrophobic and hydrophilic model nanocarriers. Mater. Sci. Eng. C 2021, 118, 111467. [Google Scholar] [CrossRef]
- Xie, X.; Hu, Y.; Ye, T.; Chen, Y.; Zhou, L.; Li, F.; Xi, X.; Wang, S.; He, Y.; Gao, X.; et al. Therapeutic vaccination against leukaemia via the sustained release of co-encapsulated anti-PD-1 and a leukaemia-associated antigen. Nat. Biomed. Eng. 2021, 5, 414–428. [Google Scholar] [CrossRef]
- Yu, L.; Sun, Q.; Hui, Y.; Seth, A.; Petrovsky, N.; Zhao, C.-X. Microfluidic formation of core-shell alginate microparticles for protein encapsulation and controlled release. J. Colloid Interface Sci. 2019, 539, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhu, Y.; Wang, F.; Deng, L.; Xu, X.; Cui, W. Microfluidic liposomes-anchored microgels as extended delivery platform for treatment of osteoarthritis. Chem. Eng. J. 2020, 400, 126004. [Google Scholar] [CrossRef]
- Guerzoni, L.P.B.; Bohl, J.; Jans, A.; Rose, J.C.; Koehler, J.; Kuehne, A.J.C.; De Laporte, L. Microfluidic fabrication of polyethylene glycol microgel capsules with tailored properties for the delivery of biomolecules. Biomater. Sci. 2017, 5, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- De Geest, B.G.; Urbanski, J.P.; Thorsen, T.; Demeester, J.; De Smedt, S.C. Synthesis of Monodisperse Biodegradable Microgels in Microfluidic Devices. Langmuir 2005, 21, 10275–10279. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L. Liquid-liquid two-phase systems for the production of porous hydrogels and hydrogel microspheres for biomedical applications: A tutorial review. Acta Biomater. 2011, 7, 31–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalko, E.; Huang, K.; Sproul, E.; Cheng, K.; Brown, A.C. Targeted Treatment of Ischemic and Fibrotic Complications of Myocardial Infarction Using a Dual-Delivery Microgel Therapeutic. ACS Nano 2018, 12, 7826–7837. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Kloxin, A.M.; Kiick, K.L. Dually degradable click hydrogels for controlled degradation and protein release. J. Mater. Chem. B 2014, 2, 5511–5521. [Google Scholar] [CrossRef] [PubMed]
- LeValley, P.J.; Sutherland, B.P.; Jaje, J.; Gibbs, S.; Jones, R.M.; Gala, R.P.; Kloxin, C.J.; Kiick, K.L.; Kloxin, A.M. On-Demand and Tunable Dual Wavelength Release of Antibodies Using Light-Responsive Hydrogels. ACS Appl. Bio Mater. 2020, 3, 6944–6958. [Google Scholar] [CrossRef] [PubMed]
- LeValley, P.J.; Neelarapu, R.; Sutherland, B.; Dasgupta, S.; Kloxin, C.J.; Kloxin, A.M. Photolabile linkers: Exploiting labile bond chemistry to control mode and rate of hydrogel degradation and protein release. J. Am. Chem. Soc. 2020, 142, 4671–4679. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Whitesides, G.M. Soft lithography. Annu. Rev. Mater. Sci. 1998, 28, 153–184. [Google Scholar] [CrossRef]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Steinhilber, D.; Rossow, T.; Wedepohl, S.; Paulus, F.; Seiffert, S.; Haag, R. A Microgel Construction Kit for Bioorthogonal Encapsulation and pH-Controlled Release of Living Cells. Angew. Chem.-Int. Ed. 2013, 52, 13538–13543. [Google Scholar] [CrossRef]
- Darling, N.J.; Hung, Y.S.; Sharma, S.; Segura, T. Controlling the kinetics of thiol-maleimide Michael-type addition gelation kinetics for the generation of homogenous poly(ethylene glycol) hydrogels. Biomaterials 2016, 101, 199–206. [Google Scholar] [CrossRef]
- Jansen, L.E.; Negron-Pineiro, L.J.; Galarza, S.; Peyton, S.R. Control of thiol-maleimide reaction kinetics in PEG hydrogel networks. Acta Biomater. 2018, 70, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Tunable Degradation of Maleimide-Thiol Adducts in Reducing Environments. Bioconjugate Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, K.L.; Sutherland, B.P.; Ogunnaike, B.A.; Kloxin, A.M. Rational Design of Hydrogel Networks with Dynamic Mechanical Properties to Mimic Matrix Remodeling. Adv. Healthc. Mater. 2022, 11, 2101947. [Google Scholar] [CrossRef] [PubMed]
- Zander, Z.K.; Hua, G.; Wiener, C.G.; Vogt, B.D.; Becker, M.L. Control of Mesh Size and Modulus by Kinetically Dependent Cross-Linking in Hydrogels. Adv. Mater. 2015, 27, 6283–6288. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Wang, R.; Kawamoto, K.; Olsen Bradley, D.; Johnson Jeremiah, A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264–1268. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Xia, B.; McBride, R.; Oakey, J. A microfluidic-based cell encapsulation platform to achieve high long-term cell viability in photopolymerized PEGNB hydrogel microspheres. J. Mater. Chem. B 2017, 5, 173–180. [Google Scholar] [CrossRef]
- Liu, A.L.; García, A.J. Methods for Generating Hydrogel Particles for Protein Delivery. Ann. Biomed. Eng. 2016, 44, 1946–1958. [Google Scholar] [CrossRef]
- Rehmann, M.S.; Skeens, K.M.; Kharkar, P.M.; Ford, E.M.; Maverakis, E.; Lee, K.H.; Kloxin, A.M. Tuning and Predicting Mesh Size and Protein Release from Step Growth Hydrogels. Biomacromolecules 2017, 18, 3131–3142. [Google Scholar] [CrossRef]
- Jiang, Z.L.; Shaha, R.; McBride, R.; Jiang, K.; Tang, M.C.; Xu, B.; Goroncy, A.K.; Frick, C.; Oakey, J. Crosslinker length dictates step-growth hydrogel network formation dynamics and allows rapid on-chip photoencapsulation. Biofabrication 2020, 12, 035006. [Google Scholar] [CrossRef]
- Gill, H.S.; Prausnitz, M.R. Does Needle Size Matter? J. Diabetes Sci. Technol. 2007, 1, 725–729. [Google Scholar] [CrossRef] [Green Version]
- Bertz, A.; Wohl-Bruhn, S.; Miethe, S.; Tiersch, B.; Koetz, J.; Hust, M.; Bunjes, H.; Menzel, H. Encapsulation of proteins in hydrogel carrier systems for controlled drug delivery: Influence of network structure and drug size on release rate. J. Biotechnol. 2013, 163, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Rep. 2015, 93, 1–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.C.; LeValley, P.J.; Luo, T.Z.; Kloxin, A.M.; Kiick, K.L. Manipulation of Glutathione-Mediated Degradation of Thiol Maleimide Conjugates. Bioconjugate Chem. 2018, 29, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Tibbitt, M.W.; Han, B.W.; Kloxin, A.M.; Anseth, K.S. Synthesis and application of photodegradable microspheres for spatiotemporal control of protein delivery. J. Biomed. Mater. Res. Part A 2012, 100A, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Finnson, K.W.; McLean, S.; Di Guglielmo, G.M.; Philip, A. Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring. Adv. Wound Care 2013, 2, 195–214. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Saulis, A.S.; Liu, W.R.; Roy, N.K.; Chao, J.D.; Ledbetter, S.; Mustoe, T.A. The Temporal Effects of Anti-TGF-β1, 2, and 3 Monoclonal Antibody on Wound Healing and Hypertrophic Scar Formation. J. Am. Coll. Surg. 2005, 201, 391–397. [Google Scholar] [CrossRef]
- Reid, R.R.; Roy, N.; Mogford, J.E.; Zimmerman, H.; Lee, C.; Mustoe, T.A. Reduction of hypertrophic scar via retroviral delivery of a dominant negative TGF-β receptor II. J. Plast. Reconstr. Aesthetic Surg. 2007, 60, 64–72. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeValley, P.J.; Parsons, A.L.; Sutherland, B.P.; Kiick, K.L.; Oakey, J.S.; Kloxin, A.M. Microgels Formed by Spontaneous Click Chemistries Utilizing Microfluidic Flow Focusing for Cargo Release in Response to Endogenous or Exogenous Stimuli. Pharmaceutics 2022, 14, 1062. https://doi.org/10.3390/pharmaceutics14051062
LeValley PJ, Parsons AL, Sutherland BP, Kiick KL, Oakey JS, Kloxin AM. Microgels Formed by Spontaneous Click Chemistries Utilizing Microfluidic Flow Focusing for Cargo Release in Response to Endogenous or Exogenous Stimuli. Pharmaceutics. 2022; 14(5):1062. https://doi.org/10.3390/pharmaceutics14051062
Chicago/Turabian StyleLeValley, Paige J., Amanda L. Parsons, Bryan P. Sutherland, Kristi L. Kiick, John S. Oakey, and April M. Kloxin. 2022. "Microgels Formed by Spontaneous Click Chemistries Utilizing Microfluidic Flow Focusing for Cargo Release in Response to Endogenous or Exogenous Stimuli" Pharmaceutics 14, no. 5: 1062. https://doi.org/10.3390/pharmaceutics14051062
APA StyleLeValley, P. J., Parsons, A. L., Sutherland, B. P., Kiick, K. L., Oakey, J. S., & Kloxin, A. M. (2022). Microgels Formed by Spontaneous Click Chemistries Utilizing Microfluidic Flow Focusing for Cargo Release in Response to Endogenous or Exogenous Stimuli. Pharmaceutics, 14(5), 1062. https://doi.org/10.3390/pharmaceutics14051062