
Timosaponin A3 Inhibits Palmitate and Stearate through Suppression of SREBP-1 in Pancreatic Cancer

 , ,
, ,  ,
, 
Abstract
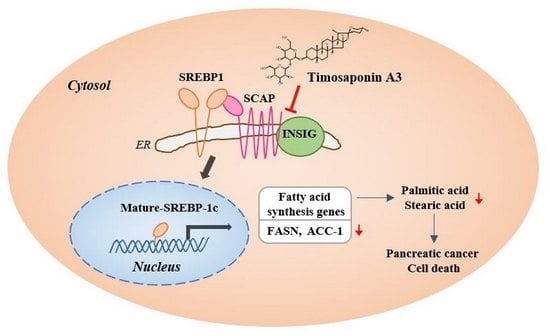
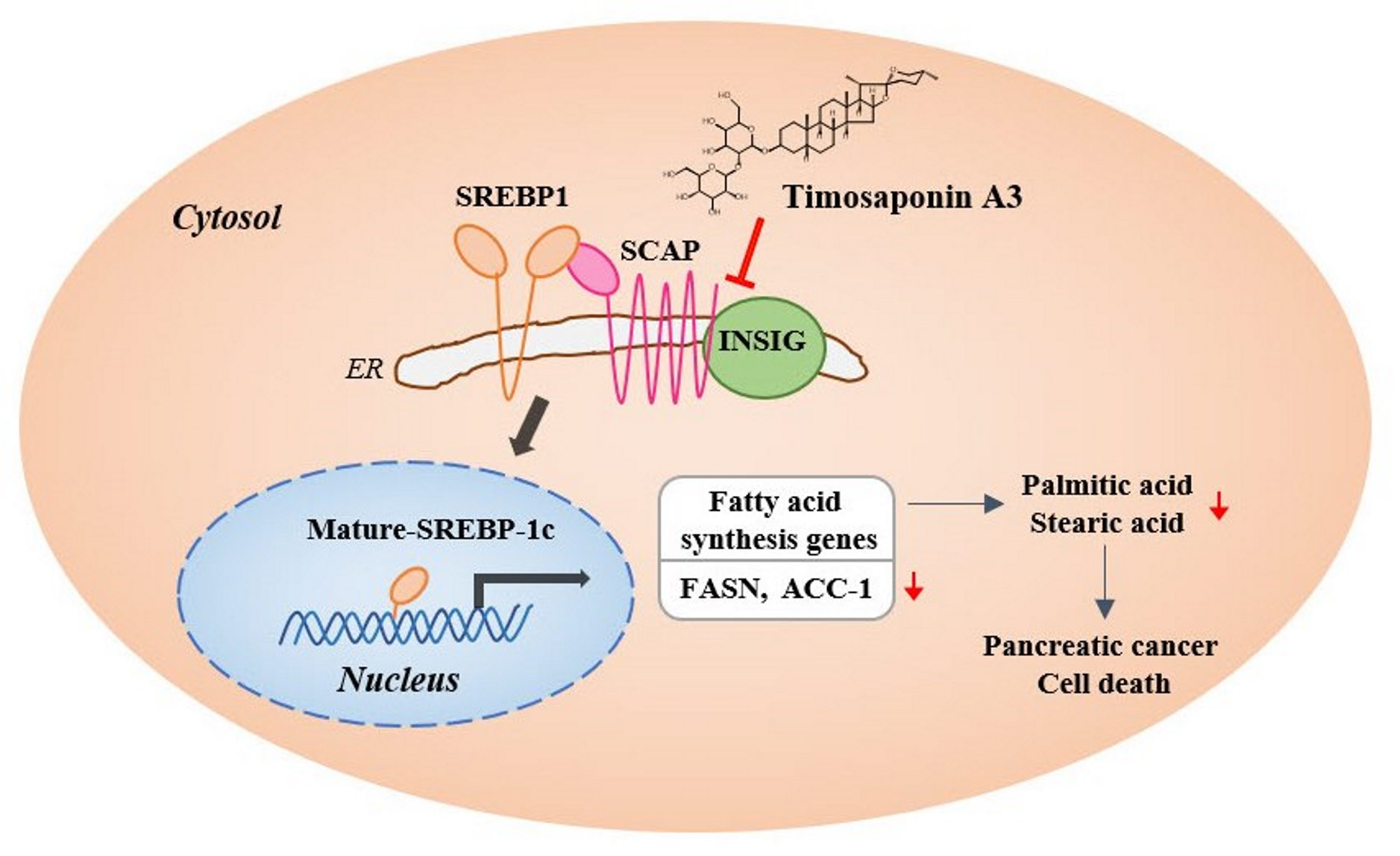
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and Treatment
2.3. Cell Viability Assay
2.4. Microarray Analysis
2.5. Oil Red O (ORO) Assay
2.6. Immunocytochemistry for SREBP-1 Localization
2.7. Cell Lysis and Immunoblotting
2.8. Cell Cycle Analysis
2.9. Apoptotic Assay
2.10. Quantitative Real Time-PCR
2.11. siRNA Transfections
2.12. Animals
2.13. Tumor Xenograft Experiments
2.14. IHC
2.15. Preparation of Sample and Fatty Acid Standards
2.16. Quantitative Analysis of Fatty Acids in Tumor Tissue Using UPLC/Q-TOF-MS
2.17. Statistical Analysis
3. Results
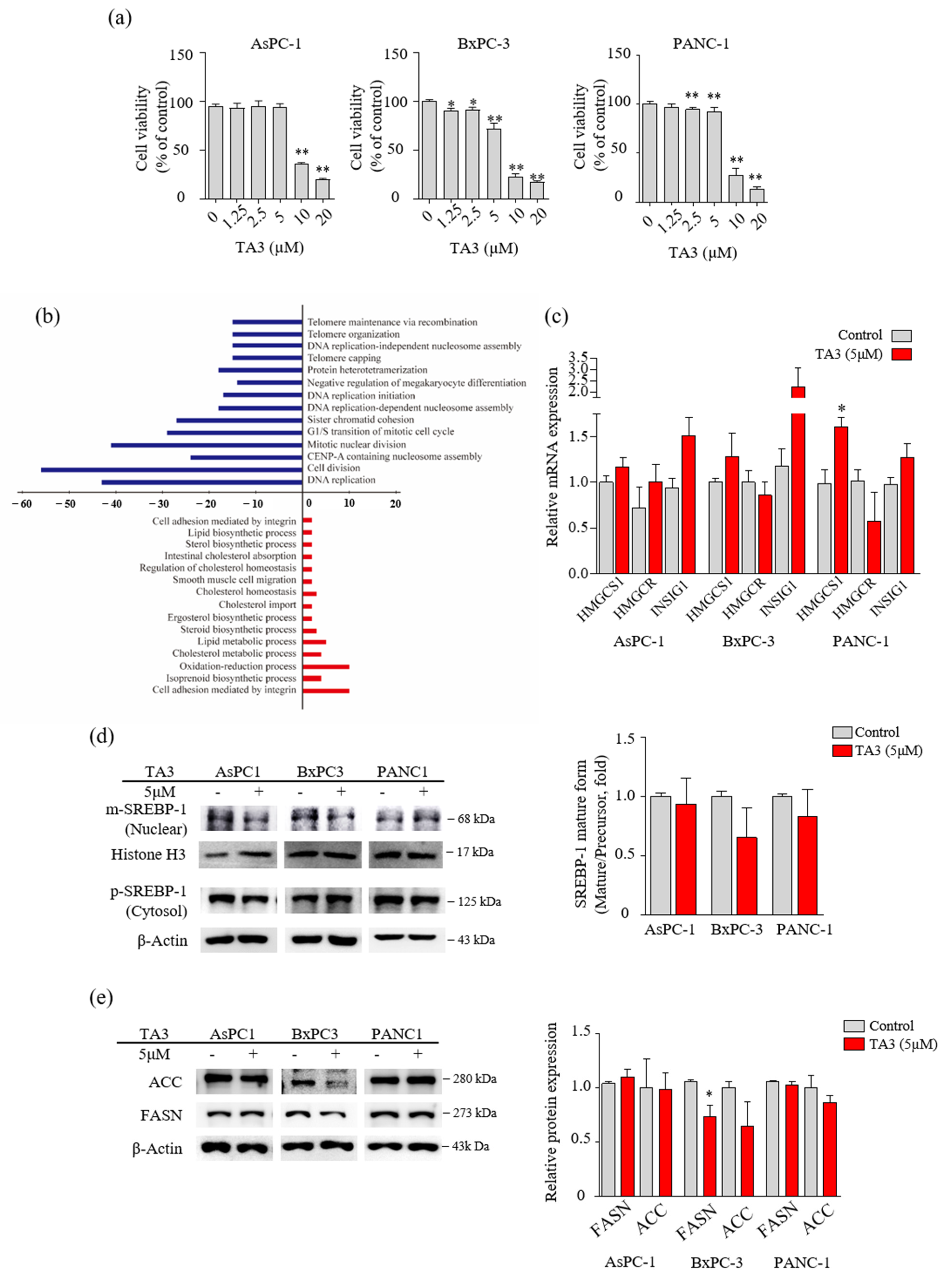
3.1. TA3 Regulated Lipid Metabolism in Pancreatic Cancer Cells
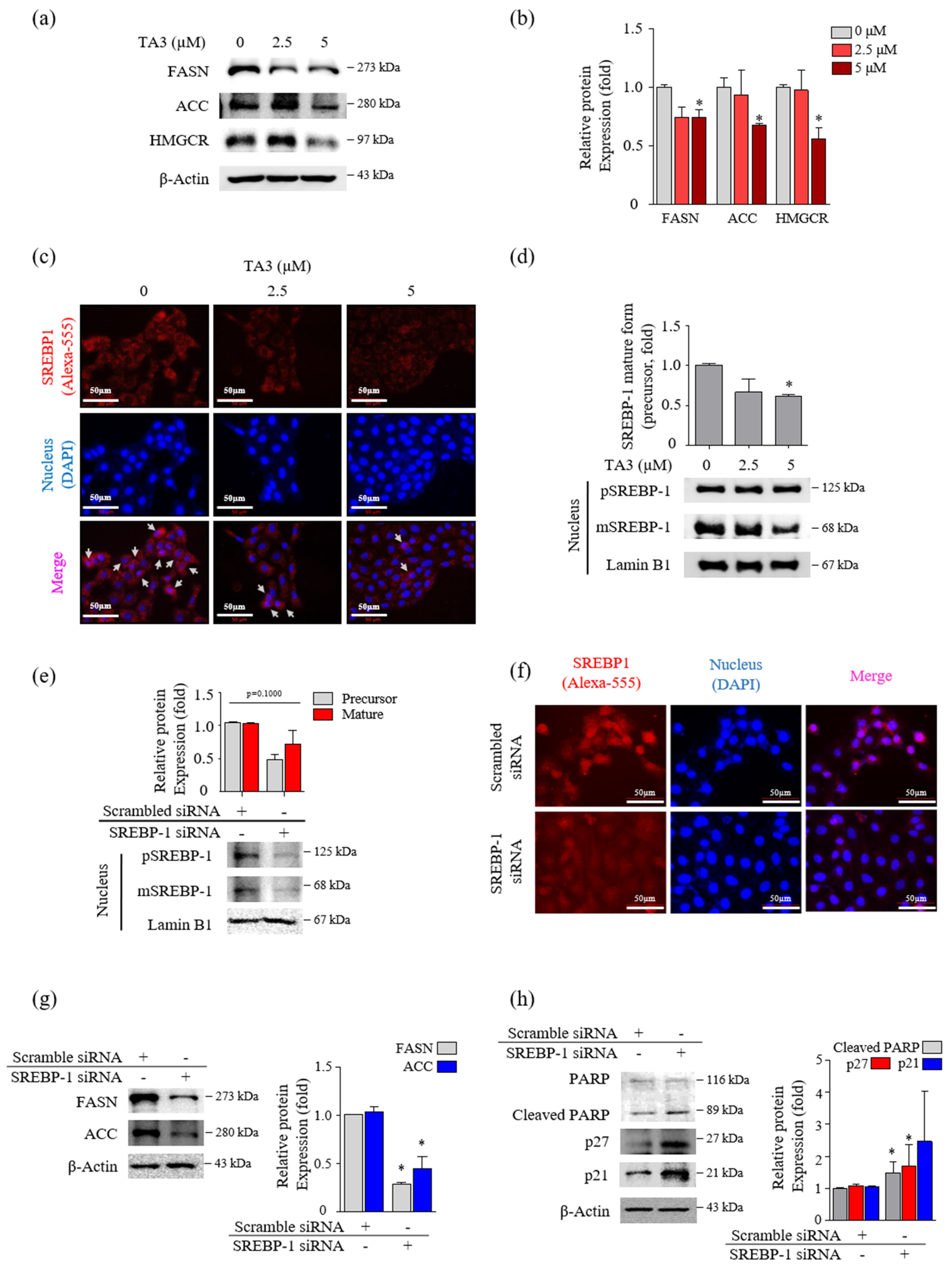
3.2. TA3 Reduced the Mature SREBPs in BxPC-3 Cells
3.3. TA3 Increased Cell Cycle Arrest in the G0/G1 Phase and Regulated the Expression of Various Genes Involved in Apoptosis
3.4. The Inhibition of SREBP-1 by TA3 Appeared Independent of the Akt/Gsk3β Mechanism
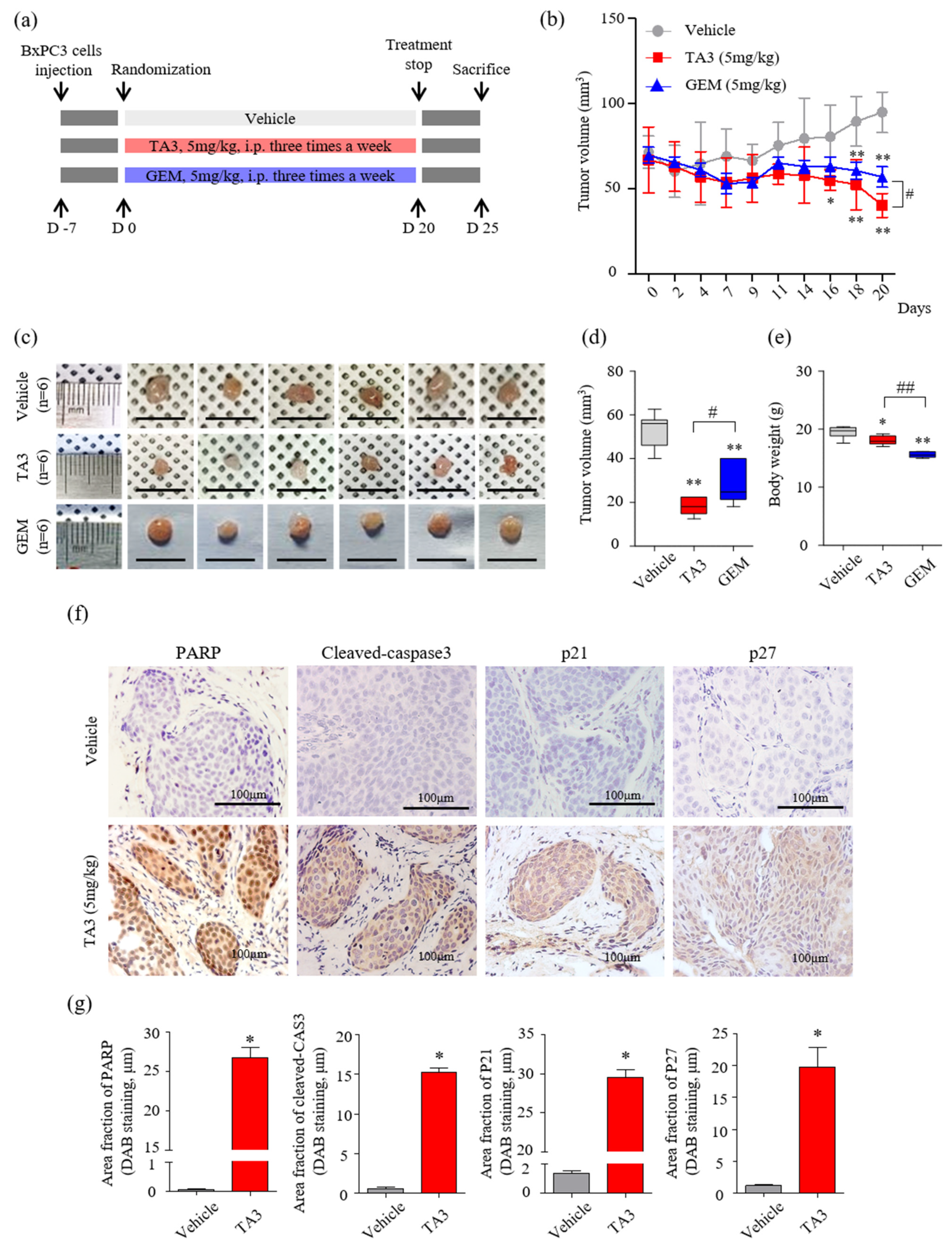
3.5. The Effect of TA3 on Tumor Growth Reduction Was Greater Than That of GEM in a Xenograft Model
3.6. Downregulation of Palmitate and Stearate Levels in the Pancreatic Tumor by TA3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Lin, A.N.; Lin, S.; Lin, T.; Liu, Y.X.; Reddy, M. A Silent Asymptomatic Solid Pancreas Tumor in a Nonsmoking Athletic Female: Pancreatic Ductal Adenocarcinoma. Case Rep. Gastroenterol. 2017, 11, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Boreddy, S.R.; Srivastava, S.K. Pancreatic cancer chemoprevention by phytochemicals. Cancer Lett. 2013, 334, 86–94. [Google Scholar] [CrossRef]
- Han, F.Y.; Song, X.Y.; Chen, J.J.; Yao, G.D.; Song, S.J. Timosaponin AIII: A novel potential anti-tumor compound from Anemarrhena asphodeloides. Steroids 2018, 140, 125–130. [Google Scholar] [CrossRef]
- Lin, Y.; Zhao, W.R.; Shi, W.T.; Zhang, J.; Zhang, K.Y.; Ding, Q.; Chen, X.L.; Tang, J.Y.; Zhou, Z.Y. Pharmacological Activity, Pharmacokinetics, and Toxicity of Timosaponin AIII, a Natural Product Isolated From Anemarrhena asphodeloides Bunge: A Review. Front. Pharm. 2020, 11, 764. [Google Scholar] [CrossRef]
- Nho, K.J.; Chun, J.M.; Kim, H.K. Induction of mitochondria-dependent apoptosis in HepG2 human hepatocellular carcinoma cells by timosaponin A-III. Env. Toxicol. Pharm. 2016, 45, 295–301. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, K.H.; Lee, I.S.; Park, J.Y.; Na, Y.C.; Chung, W.S.; Jang, H.J. Apoptosis and G2/M cell cycle arrest induced by a timosaponin A3 from Anemarrhena asphodeloides Bunge on AsPC-1 pancreatic cancer cells. Phytomed. Int. J. Phytother. Phytopharm. 2019, 56, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.J.; Nie, X.Q.; Liu, D.; Bian, K. Effects of four kinds of Chinese medicine monomer on growth of PANC-1 xenograft tumor and studying of molecular mechanism. Zhongguo Zhong Yao Za Zhi 2013, 38, 245–248. [Google Scholar] [PubMed]
- MarElia, C.B.; Sharp, A.E.; Shemwell, T.A.; Clare Zhang, Y.; Burkhardt, B.R. Anemarrhena asphodeloides Bunge and its constituent timosaponin-AIII induce cell cycle arrest and apoptosis in pancreatic cancer cells. FEBS Open Bio 2018, 8, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Espenshade, P.J. Expanding roles for SREBP in metabolism. Cell Metab. 2012, 16, 414–419. [Google Scholar] [CrossRef] [Green Version]
- Misawa, K.; Horiba, T.; Arimura, N.; Hirano, Y.; Inoue, J.; Emoto, N.; Shimano, H.; Shimizu, M.; Sato, R. Sterol regulatory element-binding protein-2 interacts with hepatocyte nuclear factor-4 to enhance sterol isomerase gene expression in hepatocytes. J. Biol. Chem. 2003, 278, 36176–36182. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Yecies, J.L.; Zhang, H.H.; Menon, S.; Liu, S.; Yecies, D.; Lipovsky, A.I.; Gorgun, C.; Kwiatkowski, D.J.; Hotamisligil, G.S.; Lee, C.H.; et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011, 14, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.-J.; Huh, S.-E.; Kim, Y.; Jang, H.-J. Anti-cancer Activity of Boswellia Carterii Extract Alters the Stress Functional Gene Expression in the Pancreatic Cancer Cell. BioChip J. 2019, 13, 191–201. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Amemiya-Kudo, M.; Shimano, H.; Hasty, A.H.; Yahagi, N.; Yoshikawa, T.; Matsuzaka, T.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Ohashi, K.; et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 2002, 43, 1220–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, W.; Dauner, K.; Courtney, K.C.; Gagnon, A.; Parks, R.J.; Sorisky, A.; Zha, X. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl. Acad. Sci. USA 2017, 114, 7999–8004. [Google Scholar] [CrossRef] [Green Version]
- Lin, V.C.; Tsai, Y.C.; Lin, J.N.; Fan, L.L.; Pan, M.H.; Ho, C.T.; Wu, J.Y.; Way, T.D. Activation of AMPK by pterostilbene suppresses lipogenesis and cell-cycle progression in p53 positive and negative human prostate cancer cells. J. Agric. Food Chem. 2012, 60, 6399–6407. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Sundqvist, A.; Bengoechea-Alonso, M.T.; Ye, X.; Lukiyanchuk, V.; Jin, J.; Harper, J.W.; Ericsson, J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005, 1, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296 Pt 1, 15–19. [Google Scholar] [CrossRef]
- Inoue, J.; Sato, R. New insights into the activation of sterol regulatory element-binding proteins by proteolytic processing. Biomol. Concepts 2013, 4, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; So, J.S.; Park, J.G.; Lee, A.H. Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin. Liver Dis. 2013, 33, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vock, C.; Doring, F.; Nitz, I. Transcriptional regulation of HMG-CoA synthase and HMG-CoA reductase genes by human ACBP. Cell Physiol. Biochem. 2008, 22, 515–524. [Google Scholar] [CrossRef]
- Ouyang, S.; Mo, Z.; Sun, S.; Yin, K.; Lv, Y. Emerging role of Insig-1 in lipid metabolism and lipid disorders. Clin. Chim. Acta 2020, 508, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Fritz, V.; Hellerbrand, C.; Bosserhoff, A.; Dietrich, P. The potent modulator of cholesterol biosynthesis insulin-induced gene 1 (INSIG1) is a novel hypoxia-regulated tumor suppressor in hepatocellular carcinoma. Z. Für Gastroenterol. 2019, 57, 16. [Google Scholar]
- Gong, Y.; Lee, J.N.; Lee, P.C.; Goldstein, J.L.; Brown, M.S.; Ye, J. Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 2006, 3, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; He, W.; Luo, M.; Zhou, Y.; Chang, G.; Ren, W.; Wu, K.; Li, X.; Shen, J.; Zhao, X.; et al. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 4133–4141. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Bengoechea-Alonso, M.T.; Ericsson, J. The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth. Cell Cycle 2016, 15, 2753–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tai, Y.; Zhou, J.; Gu, W.; Bai, Z.; Zhou, T.; Zhong, Z.; McCue, P.A.; Sang, N.; Ji, J.Y.; et al. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell Cycle 2012, 11, 2348–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Sande, T.; De Schrijver, E.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in the overexpression of fatty acid synthase in LNCaP prostate cancer cells. Cancer Res. 2002, 62, 642–646. [Google Scholar] [PubMed]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev. 2009, 23, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [Green Version]
- Greenwell, M.; Rahman, P.K. Medicinal Plants: Their Use in Anticancer Treatment. Int. J. Pharm. Sci. Res. 2015, 6, 4103–4112. [Google Scholar]
- Chen, L.; Xie, B.; Li, L.; Jiang, W.; Zhang, Y.; Fu, J.; Guan, G.; Qiu, Y. Rapid and Sensitive LC–MS/MS Analysis of Fatty Acids in Clinical Samples. Chromatographia 2014, 77, 1241–1247. [Google Scholar] [CrossRef]
- Griffiths, B.; Lewis, C.A.; Bensaad, K.; Ros, S.; Zhang, Q.; Ferber, E.C.; Konisti, S.; Peck, B.; Miess, H.; East, P.; et al. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Kwan, H.Y.; Yang, Z.; Fong, W.F.; Hu, Y.M.; Yu, Z.L.; Hsiao, W.L. The anticancer effect of oridonin is mediated by fatty acid synthase suppression in human colorectal cancer cells. J. Gastroenterol. 2013, 48, 182–192. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, X.; Zhao, L.; Li, J.; Yang, H.; Zhu, Z.; Liu, J.; Huang, G. Arginine Methylation of SREBP1a via PRMT5 Promotes De Novo Lipogenesis and Tumor Growth. Cancer Res. 2016, 76, 1260–1272. [Google Scholar] [CrossRef] [Green Version]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| CDK6 | TCTTCATTCACACCGAGTAGTGC | TGAGGTTAGAGCCATCTGGAAA |
| CDK4 | ATGGCTACCTCTCGATATGAGC | CATTGGGGACTCTCACACTCT |
| CDK2 | AAAGCCAGAAACAAGTTGACG | GAGATCTCTCGGATGGCAGT |
| Cyclin D1 | TTCGATGATTGGAATAGC | TGTGAGCTGCTCATTGAG |
| Cyclin E1 | GAAATGGCCAAAATCGACAG | TGTCAGGTGTGGGGATCA |
| Bcl-2 | GATGGCAAATGACCAGCAGA | GCAGGATAGCAGCACAGGAT |
| Bid | ATGGACTGTGAGGTCAACAACGG | CACGTAGGTGCGTAGGTTCTGGTTA |
| Caspase-3 | AGCAAACCTCAGGGAAACATT | GTCTCAATGCCACAGTCCAGT |
| Caspase-9 | GGTTCTGGAGGATTTGGTGA | GACAGCCGTGAGAGAGAATGA |
| HMGCS1 | CTCCCTGACGTGGAATGTCT | GAACTGTCTGCCCAGGTGAT |
| HMGCR | CTTGCCGAGCCTAATGAAAG | TGACCCCCTGAGAAAGCTAA |
| INSIG1 | CAACACCTGGCATCATCG | CTCGGGGAAGAGAGTGACAT |
| Probe Set ID | Gene Accession | Gene Description | Gene Symbol | Log Ratio (TA vs. Ctr) | Fold Change (TA vs. Ctr) |
|---|---|---|---|---|---|
| Lipid metabolic process | |||||
| 16995890 | NM_001098272 | 3-Hydroxy-3-methylglutaryl-CoA synthase 1 (soluble) | HMGCS1 | 2.175 | 4.51 |
| 17053892 | NM_005542 | Insulin-induced gene 1 | INSIG1 | 2.043 | 4.12 |
| 16708249 | NM_005063 | Stearoyl-CoA desaturase (delta-9-desaturase) | SCD | 1.917 | 3.78 |
| 16972155 | ENST00000261507 | Methylsterol monooxygenase 1 | MSMO1 | 1.729 | 3.32 |
| 16741501 | ENST00000355527 | 7-Dehydrocholesterol reductase | DHCR7 | 1.616 | 3.07 |
| Cell cycle | |||||
| 16677201 | NM_016448 | Denticleless E3 ubiquitin protein ligase homolog (Drosophila) | DTL | −2.009 | 4.03 |
| 16702571 | NM_182751 | Minichromosome maintenance complex component 10 | MCM10 | −1.885 | 3.69 |
| 16685165 | NM_022111 | Claspin | CLSPN | −1.727 | 3.31 |
| 16850477 | NM_001071 | Thymidylate synthetase | TYMS | −1.727 | 3.31 |
| 17067332 | ENST00000305188 | Establishment of cohesion 1 homolog 2 (S. cerevisiae) | ESCO2 | −1.540 | 2.91 |
| 16877019 | ENST00000360566 | Ribonucleotide reductase M2 | RRM2 | −1.536 | 2.90 |
| 16703478 | NM_001172303 | Microtubule-associated serine/threonine kinase-like | MASTL | −1.527 | 2.88 |
| 16965346 | NM_022346 | Non-SMC condensin I complex, subunit G | NCAPG | −1.519 | 2.87 |
| 16844312 | NM_001067 | Topoisomerase (DNA) II alpha 170 kDa | TOP2A | −1.519 | 2.87 |
| 17079293 | NM_057749 | cyclin E2 | CCNE2 | −1.514 | 2.86 |
| Cell proliferation | |||||
| 17053892 | NM_005542 | Insulin-induced gene 1 | INSIG1 | 2.043 | 4.12 |
| Apoptosis | |||||
| 16685165 | NM_022111 | Claspin | CLSPN | −1.727 | 3.31 |
| 16844312 | NM_001067 | Topoisomerase (DNA) II alpha 170 kDa | TOP2A | −1.519 | 2.87 |
| Cell division | |||||
| 16703478 | NM_001172303 | Microtubule-associated serine/threonine kinase-like | MASTL | −1.527 | 2.88 |
| 16965346 | NM_022346 | Non-SMC condensin I complex, subunit G | NCAPG | −1.519 | 2.87 |
| 16844312 | NM_001067 | Topoisomerase (DNA) II alpha 170 kDa | TOP2A | −1.519 | 2.87 |
| 17079293 | NM_057749 | cyclin E2 | CCNE2 | −1.514 | 2.86 |
| Compounds | Formula | RT (min) | Equation |
|---|---|---|---|
| Palmitic acid (C16:0) | C16H32O2 | 15.18 | y = 0.001675x + 1.983431 |
| Stearic acid (C18:0) | C18H36O2 | 16.24 | y = 0.002015x + 2.559123 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Jee, W.; An, E.-J.; Ko, H.M.; Jung, J.H.; Na, Y.-C.; Jang, H.-J. Timosaponin A3 Inhibits Palmitate and Stearate through Suppression of SREBP-1 in Pancreatic Cancer. Pharmaceutics 2022, 14, 945. https://doi.org/10.3390/pharmaceutics14050945
Kim Y, Jee W, An E-J, Ko HM, Jung JH, Na Y-C, Jang H-J. Timosaponin A3 Inhibits Palmitate and Stearate through Suppression of SREBP-1 in Pancreatic Cancer. Pharmaceutics. 2022; 14(5):945. https://doi.org/10.3390/pharmaceutics14050945
Chicago/Turabian StyleKim, Yumi, Wona Jee, Eun-Jin An, Hyun Min Ko, Ji Hoon Jung, Yun-Cheol Na, and Hyeung-Jin Jang. 2022. "Timosaponin A3 Inhibits Palmitate and Stearate through Suppression of SREBP-1 in Pancreatic Cancer" Pharmaceutics 14, no. 5: 945. https://doi.org/10.3390/pharmaceutics14050945
APA StyleKim, Y., Jee, W., An, E. -J., Ko, H. M., Jung, J. H., Na, Y. -C., & Jang, H. -J. (2022). Timosaponin A3 Inhibits Palmitate and Stearate through Suppression of SREBP-1 in Pancreatic Cancer. Pharmaceutics, 14(5), 945. https://doi.org/10.3390/pharmaceutics14050945