4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease

 ,
,  , , , , and
, , , , and
Abstract
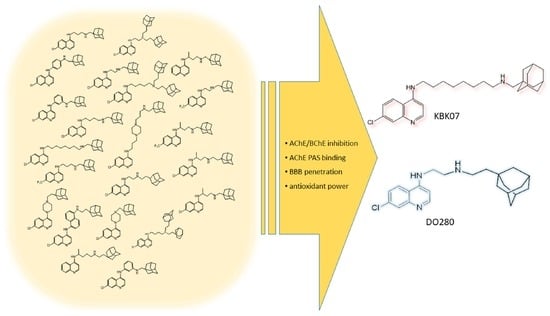
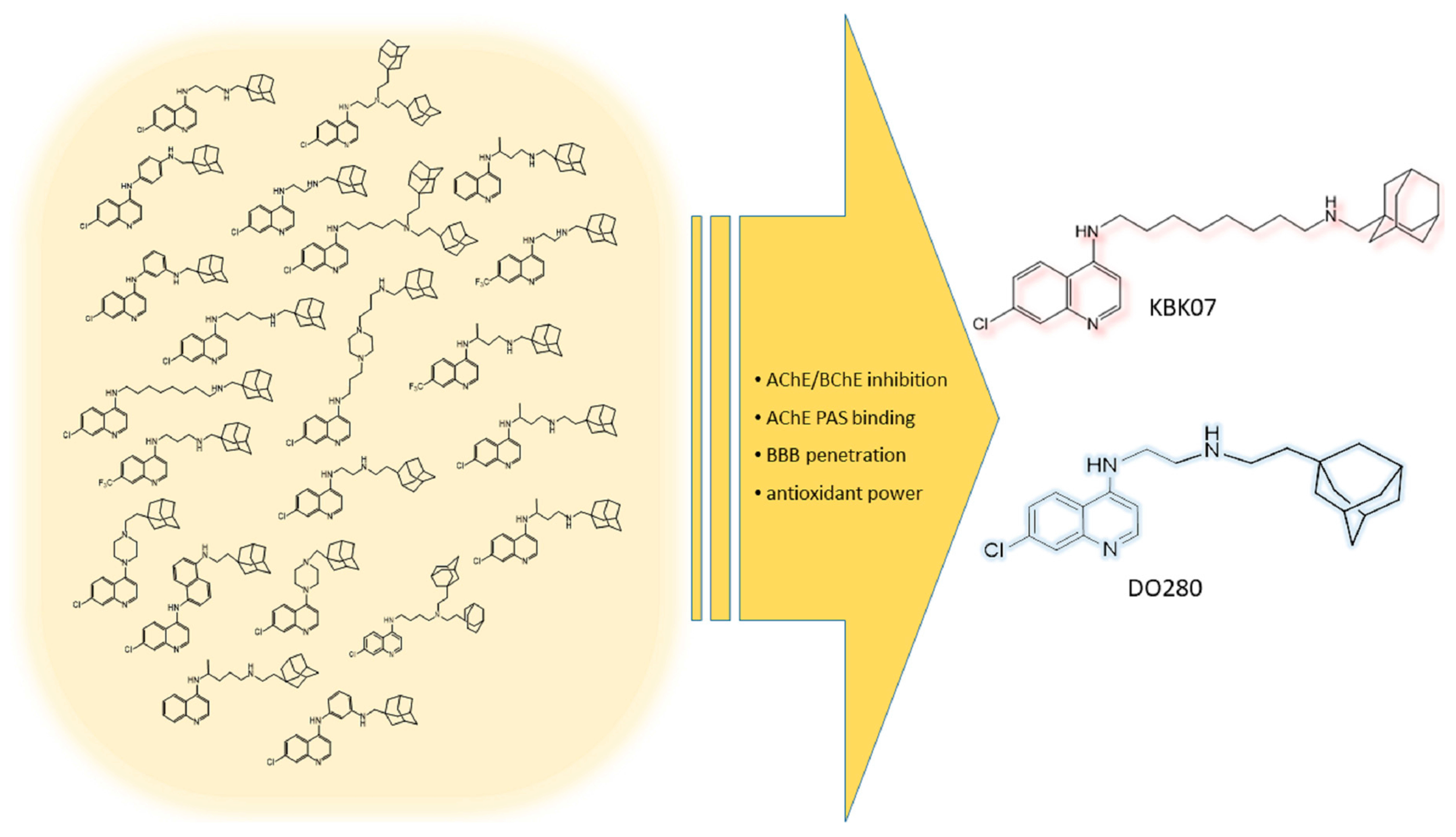
:
1. Introduction
2. Materials and Methods
2.1. Synthesis of Compounds
2.2. Inhibition of AChE and BChE
2.2.1. Chemicals
2.2.2. Enzyme Sources
2.2.3. Evaluation of Inhibition Constants
2.2.4. Inhibition Selectivity Evaluation
2.3. pKa Calculation
2.4. Docking Studies
2.5. Antioxidant Activity
2.6. In Silico Prediction of Druglikeness
2.7. In Silico Prediction of Blood–Brain Barrier (BBB) Penetration
2.8. Chromatographic Determination of Lipophilicity
2.9. Multivariate Statistical Analysis and Modelling
2.10. Molecular Descriptors Calculation
3. Results and Discussion
3.1. Inhibition of Cholinesterases
3.2. pKa and Distribution of Protonated Species
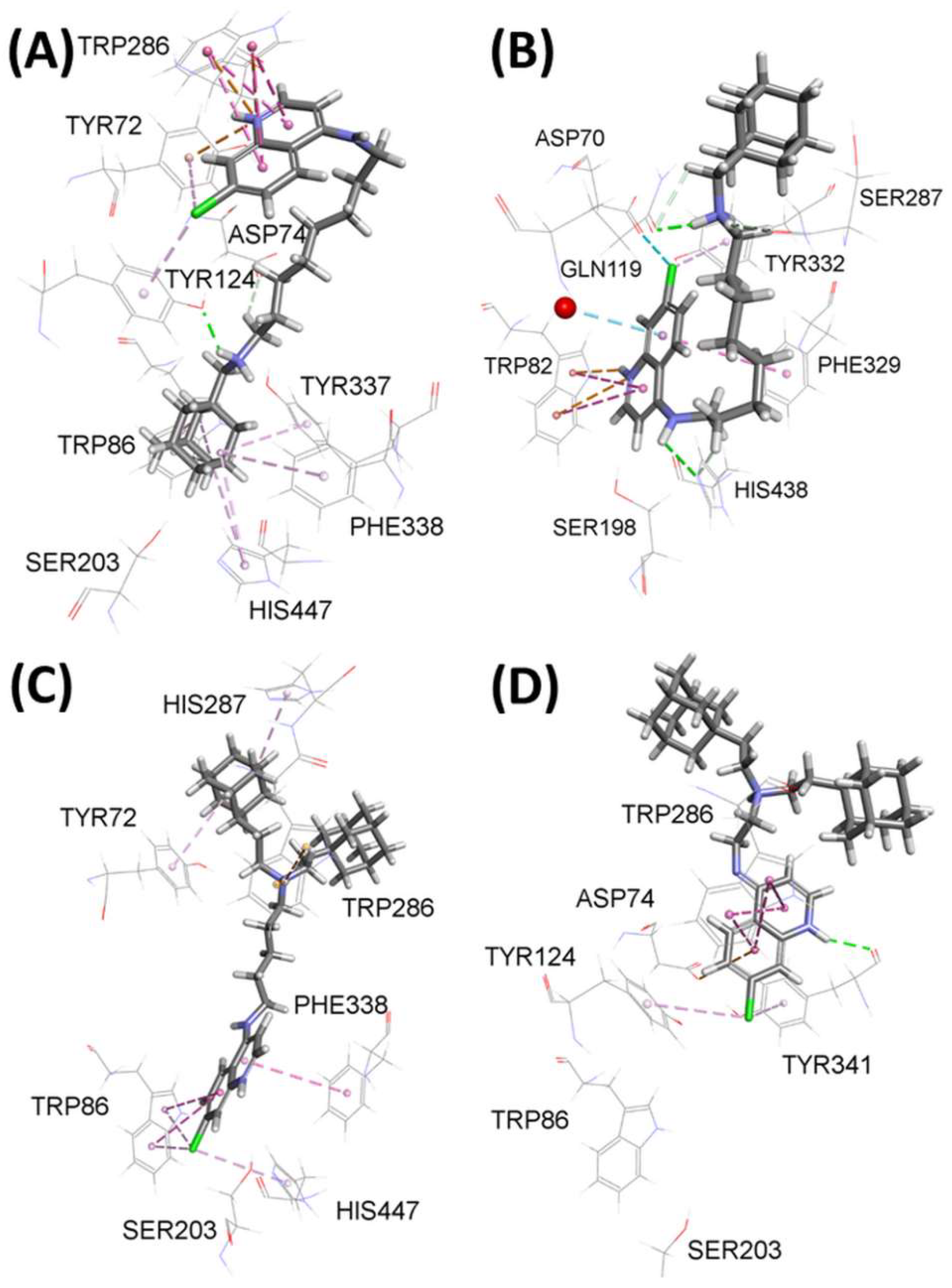
3.3. Docking Results
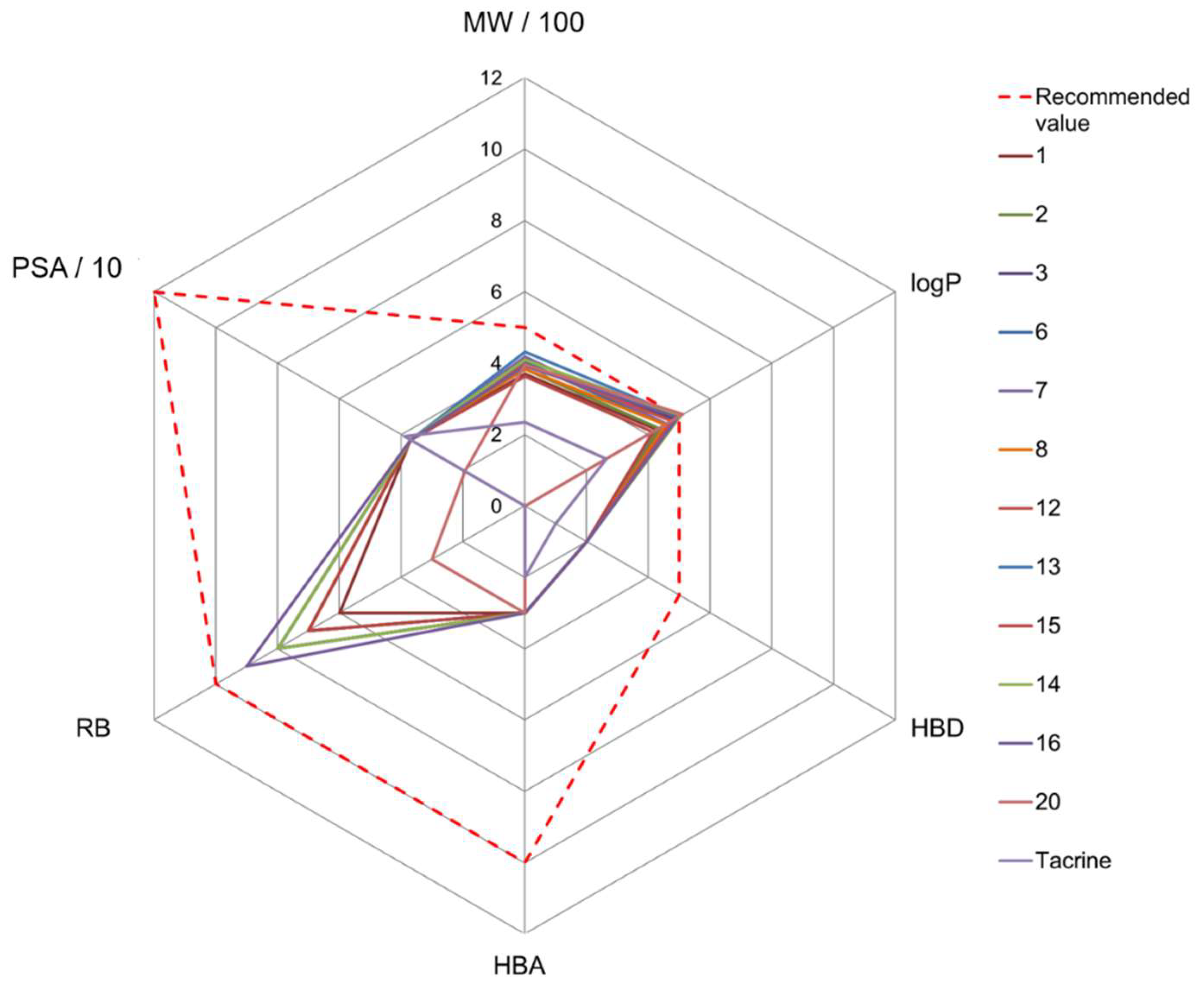
3.4. Predicted Druglikeness and Blood–Brain Barrier (BBB) Penetration
3.5. Lipophilicity and Quantitative Structure-Property Relationship (QSPR)
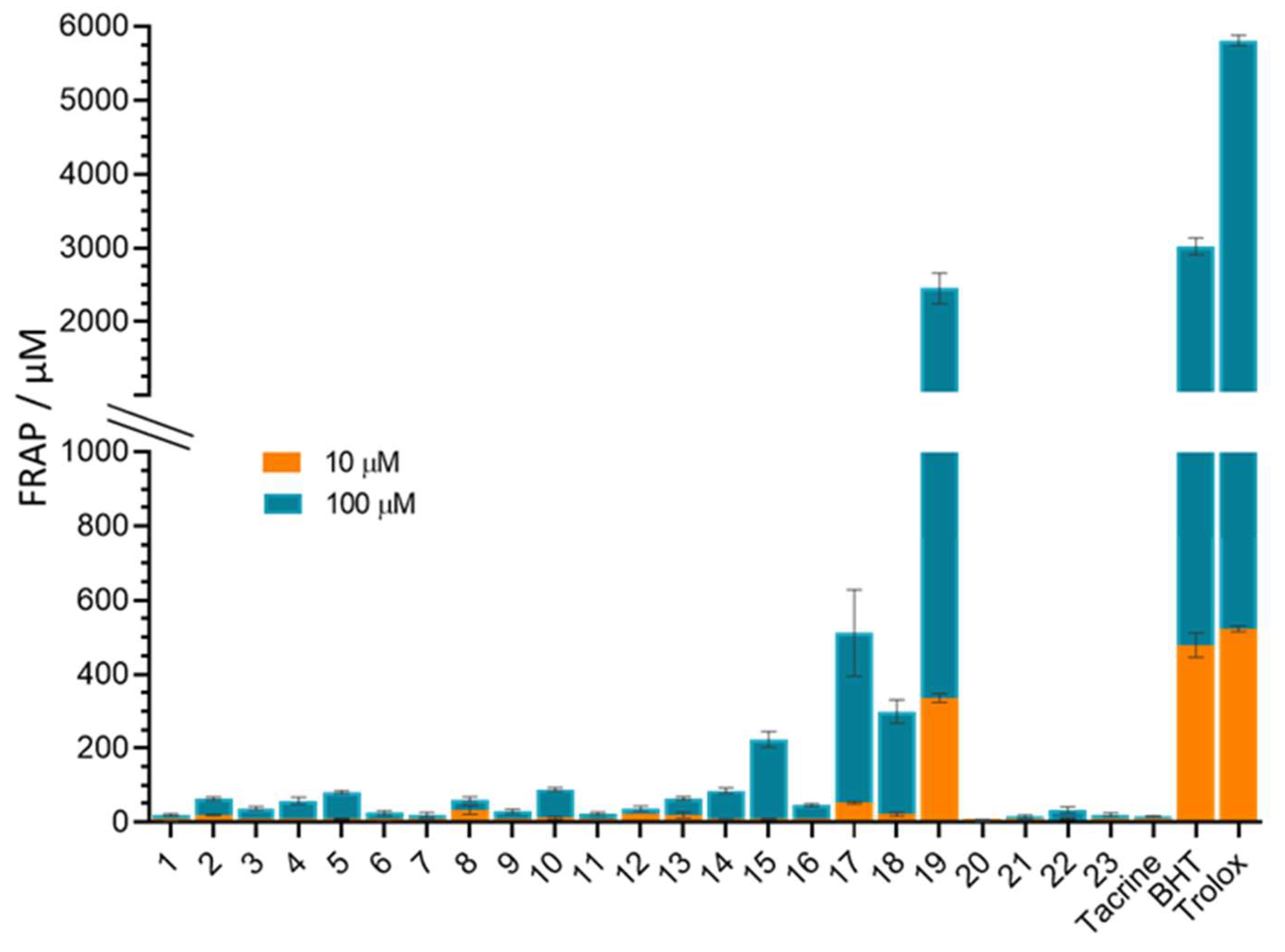
3.6. In Vitro Antioxidative Potential of 4-Aminoquinolines
3.7. Quantitative Structure–Activity Relationship (QSAR)
4. General Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P.N.; Banerjee, A.G.; Shrivastava, S.K. Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53–89. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 321–387. [Google Scholar]
- Szeto, J.Y.Y.; Lewis, S.J.G. Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef]
- BBC News, Alzheimer’s Drug Aducanumab Not Approved for Use in EU. Available online: https://www.bbc.com/news/health-59699907 (accessed on 24 January 2022).
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed drug design strategy: From a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef] [Green Version]
- Reiner, E.; Radić, Z. Mechanism of Action of Cholinesterase Inhibitors. In Cholinesterase’s and Cholinesterase Inhibitors, 3rd ed.; Giaccobini, E., Dunitz, M., Eds.; Informa Healthcare: London, UK, 2000; pp. 103–144. [Google Scholar]
- Giacobini, E. Butyrylcholinesterase: Its Role in Brain Function, 1st ed.; Giacobini, E., Ed.; Informa Healthcare: London, UK, 2003. [Google Scholar]
- Guillozet, A.L.; Smiley, J.F.; Mash, D.C.; Mesulam, M.M. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997, 42, 909–918. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, T.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [Green Version]
- Sussman, J.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [Green Version]
- Nachon, F.; Masson, P.; Nicolet, Y.; Lockridge, O.; Fontecilla-Camps, J.C. Comparison of structures of butyrylcholinesterase and acetylcholinesterase. In Butyrylcholinesterase, Its Function and Inhibitors, 3rd ed.; Giaccobini, E., Dunitz, M., Eds.; Informa Healthcare: London, UK, 2003; pp. 39–45. [Google Scholar]
- Rosenberry, T.; Brazzolotto, X.; Macdonald, I.; Wandhammer, M.; Trovaslet-Leroy, M.; Darvesh, S.; Nachon, F. Comparison of the binding of reversible inhibitors to human butyrylcholinesterase and acetylcholinesterase: A crystallographic, kinetic and calorimetric study. Molecules 2017, 22, 2098. [Google Scholar] [CrossRef] [Green Version]
- Bosak, A.; Gazić, I.; Vinković, V.; Kovarik, Z. Amino acid residues involved in stereoselective inhibition of cholinesterases with bambuterol. Arch. Biochem. Biophys. 2008, 471, 72–76. [Google Scholar] [CrossRef]
- Kovarik, Z.; Bosak, A.; Šinko, G.; Latas, T. Exploring the active sites of cholinesterases by inhibition with bambuterol and haloxon. Croat. Chem. Acta 2003, 76, 63–67. [Google Scholar]
- Bosak, A.; Smilovic, I.G.; Šinko, G.; Vinković, V.; Kovarik, Z. Metaproterenol, isoproternol and their bisdimethylcarbaamte derivates as human cholinesterase inhibitors. J. Med. Chem. 2012, 55, 6716–6723. [Google Scholar] [CrossRef]
- Bosak, A.; Knežević, A.; Gazić Smilović, I.; Šinko, G.; Kovarik, Z. Resorcinol-, catechol- and saligenin-based bronchodilating beta2-agonists as inhibitors of human cholinesterase activity. J. Enzym. Inhib. Med. Chem. 2017, 32, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Matošević, A.; Radman Kastelic, A.; Mikelić, A.; Zandona, A.; Katalinić, M.; Primožič, I.; Bosak, A.; Hrenar, T. Quinuclidine-based carbamates as potential CNS active compounds. Pharmaceutics 2021, 13, 420. [Google Scholar] [CrossRef]
- Korczyn, A.D. The amyloid cascade hypothesis. Alzheimer’s Dement. 2008, 4, 176–178. [Google Scholar] [CrossRef]
- Bayer, T.A.; Wirths, O. Focusing the amyloid cascade hypothesis on N-truncated Abeta peptides as drug targets against Alzheimer’s disease. Acta Neuropathol. 2014, 127, 787–801. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Urra, S.; Colombres, M. Actylcholinesterase (AChE)-amyloid-beta-peptide complexes in Alzheimer’s disease: The Wnt signalling pathway. Curr. Alzheimer Res. 2004, 1, 249–254. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. AChE and the amyloid precursor protein (APP)—Cross-talk in Alzheimer’s disease. Chem. Biol. Interact. 2016, 259, 301–306. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Christen, Y. Oxidative stress and Alzheimer disease. Am. J. Clin. Nutr. 2000, 71, 621–629. [Google Scholar] [CrossRef]
- Xie, S.-S.; Wang, X.-B.; Li, J.-Y.; Yang, L.; Kong, L.-Y. Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur. J. Med. Chem. 2013, 64, 540–553. [Google Scholar] [CrossRef]
- Hamulakova, S.; Janovec, L.; Soukup, O.; Jun, D.; Janockova, J.; Hrabinova, M.; Sepsova, V.; Kuca, K. Tacrine-coumarin and tacrine-7-chloroquinoline hybrids with thiourea linkers: Cholinesterase inhibition properties, kinetic study, molecular docking and permeability assay for blood-brain barrier. Curr. Alzheimer Res. 2018, 15, 1096–1105. [Google Scholar] [CrossRef]
- Rydberg, E.H.; Brumshtein, B.; Greenblatt, H.M.; Wong, D.M.; Shaya, D.; Williams, L.D.; Carlier, P.R.; Pang, Y.P.; Silman, I.; Sussman, J.L. Complexes of alkylene-linked tacrine dimers with Torpedo californica acetylcholinesterase: Binding of Bis5-tacrine produces a dramatic rearrangement in the active-site gorge. J. Med. Chem. 2006, 49, 5491–5500. [Google Scholar] [CrossRef]
- Bosak, A.; Ramic, A.; Smidlehner, T.; Hrenar, T.; Primozic, I.; Kovarik, Z. Design and evaluation of selective butyrylcholinesterase inhibitors based on Cinchona alkaloid scaffold. PLoS ONE 2018, 13, e0205193. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, D.; Fallarero, A.; Shinde, P.; Anju, C.P.; Busygin, I.; Leino, R.; Mohan, C.G.; Vuorela, P. Chemical modifications of cinchona alkaloids lead to enhanced inhibition of human butyrylcholinesterase. Nat. Prod. Commun. 2014, 9, 455–458. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, S.A.; Ayaz, M.; Brandt, W.; Wessjohann, L.A.; Westermann, B. Cation–π and π–π stacking interactions allow selective inhibition of butyrylcholinesterase by modified quinine and cinchonidine alkaloids. Biochem. Biophys. Res. Commun. 2011, 404, 935–940. [Google Scholar] [CrossRef]
- Katewa, S.D.; Katyare, S.S. Antimalarials inhibit human erythrocyte membrane acetylcholinesterase. Drug Chem. Toxicol. 2005, 28, 467–482. [Google Scholar] [CrossRef]
- Dawson, L.J.; Caulfield, V.L.; Stanbury, J.B.; Field, A.E.; Christmas, S.E.; Smith, P.M. Hydroxychloroquine therapy in patients with primary Sjögren’s syndrome may improve salivary gland hypofunction by inhibition of glandular cholinesterase. Rheumatology 2005, 44, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Bian, Y.; Sun, Y.; Kang, C.; Yu, S.; Fu, T.; Li, W.; Pei, Y.; Sun, H. Identification of 4-aminoquinoline core for the design of new cholinesterase inhibitors. PeerJ 2016, 4, e2140. [Google Scholar] [CrossRef] [Green Version]
- Bosak, A.; Opsenica, D.M.; Šinko, G.; Zlatar, M.; Kovarik, Z. Structural aspects of 4-aminoquinolines as reversible inhibitors of human acetylcholinesterase and butyrylcholinesterase. Chem. Biol. Interact. 2019, 308, 101–109. [Google Scholar] [CrossRef]
- Zanon, V.S.; Lima, J.A.; Cuya, T.S.; Lima, F.R.S.; da Fonseca, A.C.C.; Gomez, J.G.; Ribeiro, R.R.; França, T.C.C.; Vargas, M.D. In-vitro evaluation studies of 7-chloro-4-aminoquinoline Schiff bases and their copper complexes as cholinesterase inhibitors. J. Inorg. Biochem. 2019, 191, 183–193. [Google Scholar] [CrossRef]
- Cai, R.; Wang, L.-N.; Fan, J.-J.; Geng, S.-Q.; Liu, Y.-M. New 4-N-phenylaminoquinoline derivatives as antioxidant, metal chelating and cholinesterase inhibitors for Alzheimer’s disease. Bioorg. Chem. 2019, 93, 103328. [Google Scholar] [CrossRef]
- Chidan Kumar, C.S.; Kwong, H.C.; Mah, S.H.; Chia, T.S.; Loh, W.-S.; Quah, C.K.; Lim, G.K.; Chandraju, S.; Fun, H.C. Synthesis and crystallographic insight into the structural aspects of some novel adamantane-based ester derivatives. Molecules 2015, 20, 18827–18846. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Sun, J.; Wang, S.; Bu, W.; Yao, M.; Gao, K.; Song, Y.; Zhao, J.; Lu, C.; Zhang, E.; et al. Synthesis, crystal structure, superoxide scavenging activity, anticancer and docking studies of novel adamantyl nitroxide derivatives. J. Mol. Struct. 2016, 1108, 611–617. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Shevtsova, E.F.; Kovaleva, N.V.; Rudakova, E.V.; Neganova, M.E.; Dubova, L.G.; Shevtsov, P.N.; Aksinenko, A.Y.; Sokolov, V.B.; Bachurin, S.O. Aminoadamantane conjugates with carbazole derivatives as potential multitarget agents for the treatment of Alzheimer’s disease. Effect of the spacer structure. Russ. Chem. Bull. 2018, 67, 2121–2126. [Google Scholar] [CrossRef]
- Aleksic, I.; Jeremic, J.; Milivojevic, D.; Ilic-Tomic, T.; Šegan, S.; Zlatović, M.; Opsenica, D.M.; Senerovic, L. N-benzyl derivatives of long-chained 4-amino-7-chloro-quionolines as inhibitors of pyocyanin production in Pseudomonas aeruginosa. ACS Chem. Biol. 2019, 14, 2800–2809. [Google Scholar] [CrossRef]
- Terzić, N.; Konstantinović, J.; Tot, M.; Burojević, J.; Djurković-Djaković, O.; Srbljanović, J.; Štajner, T.; Verbić, T.; Zlatović, M.; Machado, M.; et al. Reinvestigating old pharmacophores: Are 4-aminoquinolines and tetraoxanes potential two-stage antimalarials? J. Med. Chem. 2016, 59, 264–281. [Google Scholar] [CrossRef]
- Konstantinović, J.; Videnović, M.; Orsini, S.; Bogojević, K.; D’Alessandro, S.; Scaccabarozzi, D.; Terzić Jovanović, N.; Gradoni, L.; Basilico, N.; Šolaja, B.A. Novel aminoquinoline deriva-tives significantly reduce parasite load in Leishmania infantum infected mice. ACS Med. Chem. Lett. 2018, 9, 629–634. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. New and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Eyer, P.; Worek, F.; Kiderlen, D.; Sinko, G.; Stuglin, A.; Simeon-Rudolf, V.; Reiner, E. Molar absorption coefficients for the reduced Ellman reagent: Reassessment. Anal. Biochem. 2003, 312, 224–227. [Google Scholar] [CrossRef]
- Simeon-Rudolf, V.; Šinko, G.; Štuglin, A.; Reiner, E. Inhibition of human blood acetylcholinesterase and butyrylcholinesterase by ethopropazine. Croat. Chem. Acta 2001, 74, 173–182. [Google Scholar]
- Chemicalize. Calculation Module. 2018. Available online: https://chemicalize.com/ (accessed on 22 May 2022).
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Xu, Y.; Colletier, J.-P.; Weik, M.; Jiang, H.; Moult, J.; Silman, I.; Sussman, J.L. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: X-ray versus molecular dynamics. Biophys. J. 2008, 95, 2500–2511. [Google Scholar] [CrossRef] [Green Version]
- Šinko, G. Assessment of scoring functions and in silico parameters for AChE-ligand interactions as a tool for predicting inhibition potency. Chem. Biol. Interact. 2019, 308, 216–223. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. Ferric reducing/antioxidant power assay: Direct measure of total antioxidant activity of biological fluids and modified version for simultaneous measurement of total antioxidant power and ascorbic acid concentration. Methods Enzymol. 1999, 299, 15–27. [Google Scholar]
- Jalili-Baleh, L.; Nadri, H.; Forootanfar, H.; Küçükkılınç, T.T.; Ayazgök, B.; Sharifzadeh, M.; Rahimifard, M.; Baeeri, M.; Abdollahi, M.; Foroumadi, A.; et al. Chromone-lipoic acid conjugate: Neuroprotective agent having acceptable butyrylcholinesterase inhibition, antioxidant and copper-chelation activities. Daru 2021, 29, 23–38. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Central nervous system multiparameter optimization desirability. ACS Chem. Neurosci. 2016, 7, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, W.J.; Lauri, G. Prediction of intestinal permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]
- Schrödinger Suite Release; Maestro, Epik, QikProp, Schrödinger, LLC: New York, NY, USA, 2021.
- Kolossváry, I.; Guida, W.C. Low-mode conformational search elucidated: Application to C39H80 and flexible docking of 9-deazaguanine inhibitors into PNP. J. Comput. Chem. 1999, 20, 1671–1684. [Google Scholar] [CrossRef]
- Polak, E.; Ribiere, G. Note sur la convergence de méthodes de directions conjuguées. ESAIM Math. Model. Numer. Anal. 1969, 3, 35–43. [Google Scholar] [CrossRef]
- Porcelli, F.; Delfini, M.; Del Giudice, M.R. The kinetic inhibition of acetylcholinesterase from human erythrocyte by tacrine and some tacrine derivatives. Bioorg. Chem. 1999, 27, 197–205. [Google Scholar] [CrossRef]
- Ahmed, M.; Rocha, J.B.T.; Corrêa, M.; Mazzani, C.M.; Zanin, R.F.; Morsch, A.L.B.; Morsch, V.M.; Schetinger, M.R.C. Inhibition of two different cholinesterases by tacrine. Chem. Biol. Interact. 2006, 162, 165–171. [Google Scholar] [CrossRef]
- Darvesh, S.; Walsh, R.; Kumar, R.; Caines, A.; Roberts, S.; Magee, D.; Rockwood, K.; Martin, E. Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2003, 17, 117–126. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Kelder, J.; Grootenhuis, P.D.; Bayada, D.M.; Delbressine, L.P.; Ploemen, J.P. Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A. Substituent Constants for Correlation Analysis in Chemistry and Biology; John Wiley & Sons: New York, NY, USA, 1979; pp. 18–43. [Google Scholar]
- Dąbrowska, M.; Starek, M.; Chłon-Rzepa, G.; Zagorska, A.; Komsta, Ł.; Jankowska, A.; Slusarczyk, M.; Pawłowski, M. Estimation of the lipophilicity of purine-2,6-dione-based TRPA1 antagonists and PDE4/7 inhibitors with analgesic activity. Bioorg. Med. Chem. Lett. 2021, 49, 128318. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, Methods and Principles in Medicinal Chemistry, 11th ed.; Mannhold, R., Kubinyi, H., Timmerman, H., Eds.; Wiley-Vch: Weinheim, Germany, 2000. [Google Scholar]
- Fernández-Bachiller, M.I.; Pérez, C.; Campillo, N.E.; Páez, J.A.; González-Muñoz, G.C.; Usán, P.; García-Palomero, E.; López, M.G.; Villarroya, M.; García, A.G.; et al. Tacrine-melatonin hybrids as multifunctional agents for Alzheimer’s disease, with cholinergic, antiox-idant, and neuroprotective properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Muñoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Digiacomo, M.; Chen, Z.; Wang, S.; Lapucci, A.; Macchia, M.; Yang, X.; Chu, J.; Han, Y.; Pi, R.; Rapposelli, S. Synthesis and pharmacological evaluation of multifunctional tacrine derivatives against several disease pathways of AD. Bioorg. Med. Chem. Lett. 2015, 25, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Reichert, C.O.; de Freitas, F.A.; Sampaio-Silva, J.; Rokita-Rosa, L.; de Lima Barros, P.; Levy, D.; Bydlowski, S.P. Ferroptosis mechanisms involved in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 8765. [Google Scholar] [CrossRef] [PubMed]
- Šolaja, B.A.; Opsenica, D.; Smith, K.S.; Milhous, W.K.; Terzić, N.; Opsenica, I.; Burnett, J.C.; Nuss, J.; Gussio, R.; Bavari, S. Novel 4-aminoquinolines active against chloroquine-resistant and sensitive P. falciparum strains that also inhibit botulinum serotype A. J. Med. Chem. 2008, 51, 4388–4391. [Google Scholar] [CrossRef]
- Aleksic, I.; Šegan, S.; Andrić, F.; Zlatović, M.; Morić, I.; Opsenica, D.M.; Senerovic, L. Long-chained 4-aminoquinolines as quorum sensing inhibitors in Serratia marcescens and Pseudomonas aeruginosa. ACS Chem. Biol. 2017, 12, 1425–1434. [Google Scholar] [CrossRef] [Green Version]
- Solomon, V.R.; Pundir, S.; Lee, H. Examination of novel 4-aminoquinoline derivatives designed and synthesized by a hybrid pharmacophore approach to enhance their anticancer activities. Sci. Rep. 2019, 9, 6315. [Google Scholar] [CrossRef] [Green Version]
- Valente, S.; Liu, Y.; Schnekenburger, M.; Zwergel, C.; Cosconati, S.; Gros, C.; Tardugno, M.; Labella, D.; Florean, C.; Minden, S.; et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem Cells. J. Med. Chem. 2014, 57, 701–713. [Google Scholar] [CrossRef]
- Simanek, E.E.; Mammen, M.; Gordon, D.M.; Chin, D.; Mathias, J.P.; Seto, C.T.; Whitesides, G.M. Design and synthesis of hydrogen-bonded aggregates: Theory and computation applied to three systems based on the cyanuric acid-melamine lattice. Tetrahedron 1995, 51, 607–619. [Google Scholar] [CrossRef]
- Viswas, R.S.; Pundir, S.; Lee, H. Design and synthesis of 4-piperazinyl quinoline derived urea/thioureas for anti-breast cancer activity by a hybrid pharmacophore approach. J. Enz. Inh. Med. Chem. 2019, 34, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Warhurst, D.C.; Craig, J.C.; Adagu ISMeyer, D.J.; Lee, S.Y. The relationship of physico-chemical properties and structure to the differential antiplasmodial activity of the cinchona alkaloids. Malar J. 2003, 2, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki/µM | IS | |
|---|---|---|---|
| AChE | BChE | ||
| 1 | 1.2 ± 0.1 (c) | 2.1 ± 0.2 (m) | 1.8 |
| 2 * | 0.77 ± 0.09 (m) | 3.2 ± 0.4 (m) | 4.2 |
| 3 | 1.0 ± 0.0 (n) | 0.92 ± 0.04 (m) | 0.9 |
| 4 | 0.67 ± 0.02 (m) | 0.76 ± 0.06 (m) | 1.1 |
| 5 | 0.075 ± 0.06 (m) | 0.091 ± 0.007 (m) | 1.2 |
| 6 | 1.2 ± 0.1 (n) | 3.5 ± 0.3 (m) | 2.9 |
| 7 | 1.1 ± 0.0 (n) | 2.6 ± 0.2 (c) | 2.4 |
| 8 | 1.6 ± 0.1 (n) | 0.92 ±0.07 (m) | 0.6 |
| 9 | 5.2 ± 0.2 (n) | 1.5 ± 0.2 (c) | 0.3 |
| 10 | 2.4 ± 0.2 (n) | 1.0 ± 0.1 (n) | 0.4 |
| 11 | 0.33 ± 0.01 (m) | 0.82 ± 0.04 (c) | 2.5 |
| 12 | 0.56 ± 0.02 (m) | 1.2 ± 0.1 (c) | 2.1 |
| 13 | 0.91 ± 0.05 (m) | 1.8 ± 0.3 (c) | 1.5 |
| 14 | 1.9 ± 0.1 (m) | 0.15 ± 0.01 (m) | 0.1 |
| 15 | 0.74 ± 0.03 (m) C | 0.65 ± 0.04 (m) M | 0.9 |
| 16 | 0.52 ± 0.02 (c) | 0.38 ± 0.03 (m) | 0.7 |
| 17 | 3.3 ± 0.4 (m) M | 2.5 ± 0.5 (m) | 0.8 |
| 18 | 9.0 ± 0.8 (m) | 5.5 ± 0.6 (m) | 0.6 |
| 19 | 3.8± 0.3 (c) | 22 ± 2 (c) | 5.8 |
| 20 | 9.4± 0.4 (c) | 25± 1 (c) | 2.7 |
| 21 | 2.1± 0.4 (m) | 5.6 ± 0.6 (m) | 2.7 |
| 22 | 0.69 ± 0.02 (c) | 3.3 ± 0.2 (c) | 4.8 |
| 23 | 0.44 ± 0.09 (c) | 1.8 ± 0.2 (m) | 4.1 |
| Tacrine | 0.040 ± 0.006 (m) | 0.0063 ± 0.0010 (m) | |
| Compound | pKa1 calc (Quinoline) | pKa2 calc (Terminal Amino-Group) | pKa3 calc/pKa4 calc (Side Chain) |
|---|---|---|---|
| 1 | 7.25 | 9.92 | - |
| 2 * | 7.31 | 10.55 | - |
| 3 | 7.31 | 10.85 | - |
| 4 | 7.31 | 10.86 | - |
| 5 | 7.31 | 10.86 | - |
| 6 | 7.48 | 9.93 | - |
| 7 | 7.53 | 10.55 | - |
| 8 | 7.25 | 9.33 | - |
| 9 | 7.23 | 10.05 | - |
| 10 | 7.31 | 10.98 | - |
| 11 | 7.31 | 10.98 | - |
| 12 | 7.29 | 10.58 | - |
| 13 | 7.51 | 10.58 | - |
| 14 | 7.28 | 10.50 | - |
| 15 | 8.13 | 10.58 | - |
| 16 | 8.13 | 10.78 | - |
| 17 | 6.22 | 6.85 | - |
| 18 | 6.49 | 5.35 | - |
| 19 | 6.45 | 4.71 | - |
| 20 | 6.90 | 9.13 | - |
| 21 | 6.91 | 8.92 | - |
| 22 | 7.12 | 10.50 | 8.07/1.24 |
| 23 | 7.12 | 10.42 | 8.06/1.23 |
| Tacrine | 8.95 | - | - |
| ADMET_BBB Level | |||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |
| Compounds | 4, 5, 9, 14, 16–18, 20, 21 | 1–3, 6–8, 12, 13, 15, 22, 23, Tacrine | - | - | 10, 11, 19 |
| Dependent Variable | Statistical Performance of the Model | Structural Descriptors Included in the QSAR Model ** |
|---|---|---|
| log (Ki/µM, AChE) | RMSEC = 0.222, RMSECV = 0.367, RMSEP = 0.408 R2cal = 0.708, R2CV = 0.333, R2pred = 0.603 PLS1: 62.13% and 55.44% PLS2: 5.16% and 15.39% | MCD (+),2M (−), ACIX3 (+), ACIX5 (+), RSIpw3 (+) |
| log (Ki/µM, BChE) | RMSEC = 0.132, RMSECV = 0.233, RMSEP = 0.333 R2cal = 0.943, R2CV = 0.825, R2pred = 0.777 PLS1: 40.06% and 79.81% PLS2: 17.79% and 8.76% PLS1: 11.44% and 3.62% PLS2: 12.57% and 2.13% | PE-S-OPLS (+),MCD (+), RSIpw3 (+), TCIO6 (+) RCI (−), RFD (−), RP (+), AVCIC5 (−), RF (−), AVCIC4 (−), Nnrs (+), AVCIC3 (−), AVCIC2 (−), CNS (+), PISA (+), AVCIC1 (−), Nnum (+), QPPMDCK (+) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komatović, K.; Matošević, A.; Terzić-Jovanović, N.; Žunec, S.; Šegan, S.; Zlatović, M.; Maraković, N.; Bosak, A.; Opsenica, D.M. 4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease. Pharmaceutics 2022, 14, 1305. https://doi.org/10.3390/pharmaceutics14061305
Komatović K, Matošević A, Terzić-Jovanović N, Žunec S, Šegan S, Zlatović M, Maraković N, Bosak A, Opsenica DM. 4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease. Pharmaceutics. 2022; 14(6):1305. https://doi.org/10.3390/pharmaceutics14061305
Chicago/Turabian StyleKomatović, Katarina, Ana Matošević, Nataša Terzić-Jovanović, Suzana Žunec, Sandra Šegan, Mario Zlatović, Nikola Maraković, Anita Bosak, and Dejan M. Opsenica. 2022. "4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease" Pharmaceutics 14, no. 6: 1305. https://doi.org/10.3390/pharmaceutics14061305
APA StyleKomatović, K., Matošević, A., Terzić-Jovanović, N., Žunec, S., Šegan, S., Zlatović, M., Maraković, N., Bosak, A., & Opsenica, D. M. (2022). 4-Aminoquinoline-Based Adamantanes as Potential Anticholinesterase Agents in Symptomatic Treatment of Alzheimer’s Disease. Pharmaceutics, 14(6), 1305. https://doi.org/10.3390/pharmaceutics14061305