
Design and Evaluation of Autophagy-Inducing Particles for the Treatment of Abnormal Lipid Accumulation


 , , and
, , and
Abstract
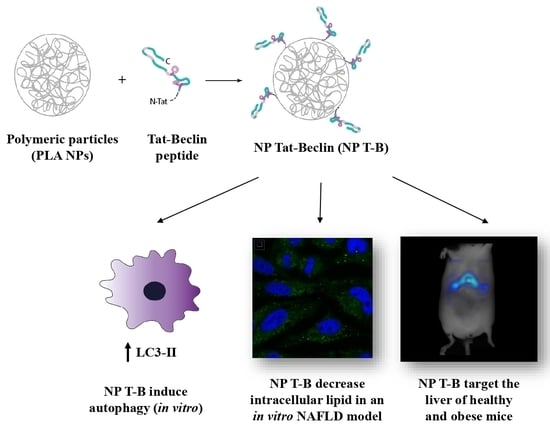
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of Tat-Beclin Polymeric Particles
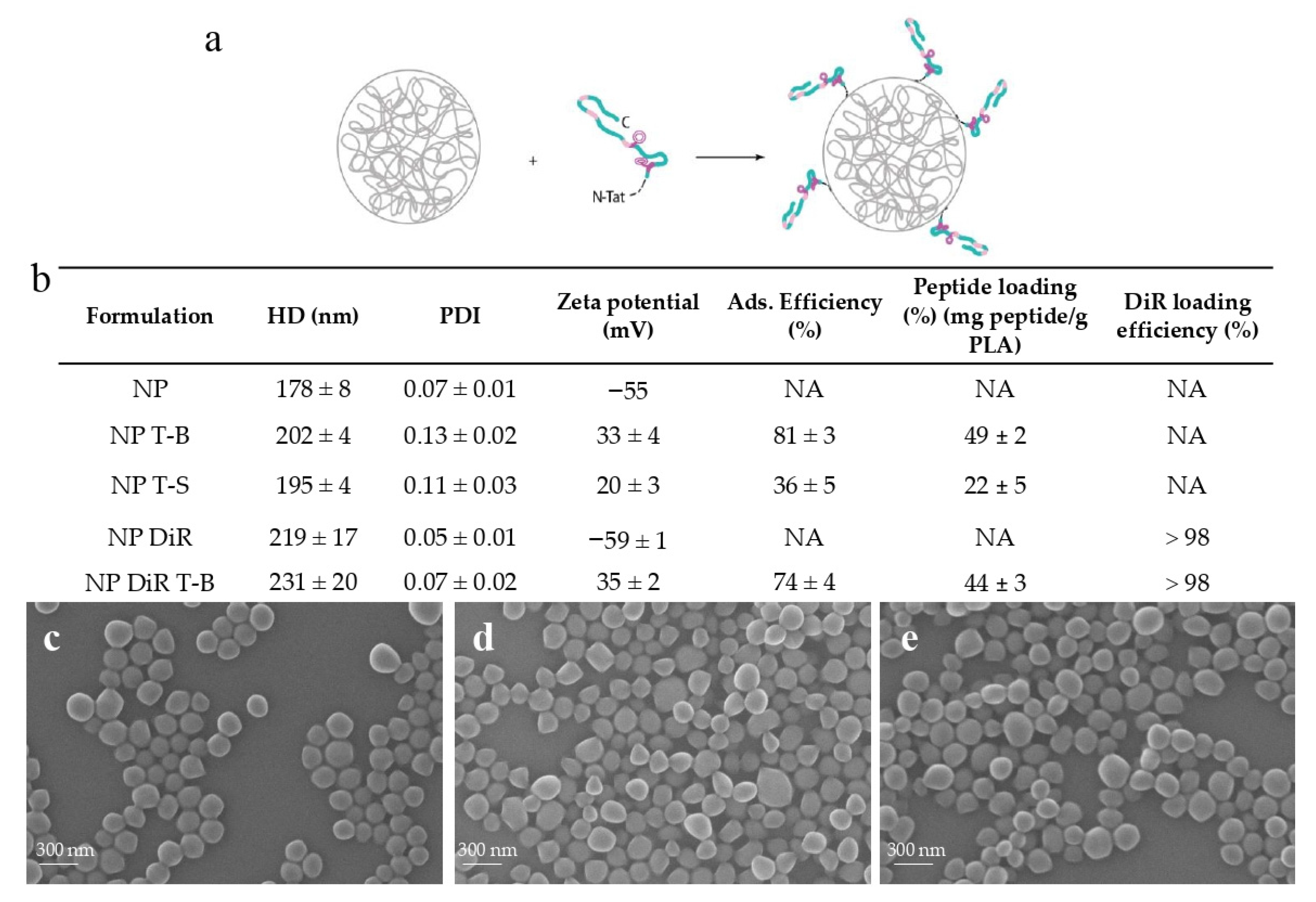
2.2. Physicochemical Characterization of Tat-Beclin NPs
2.3. Cell Culture and Treatments
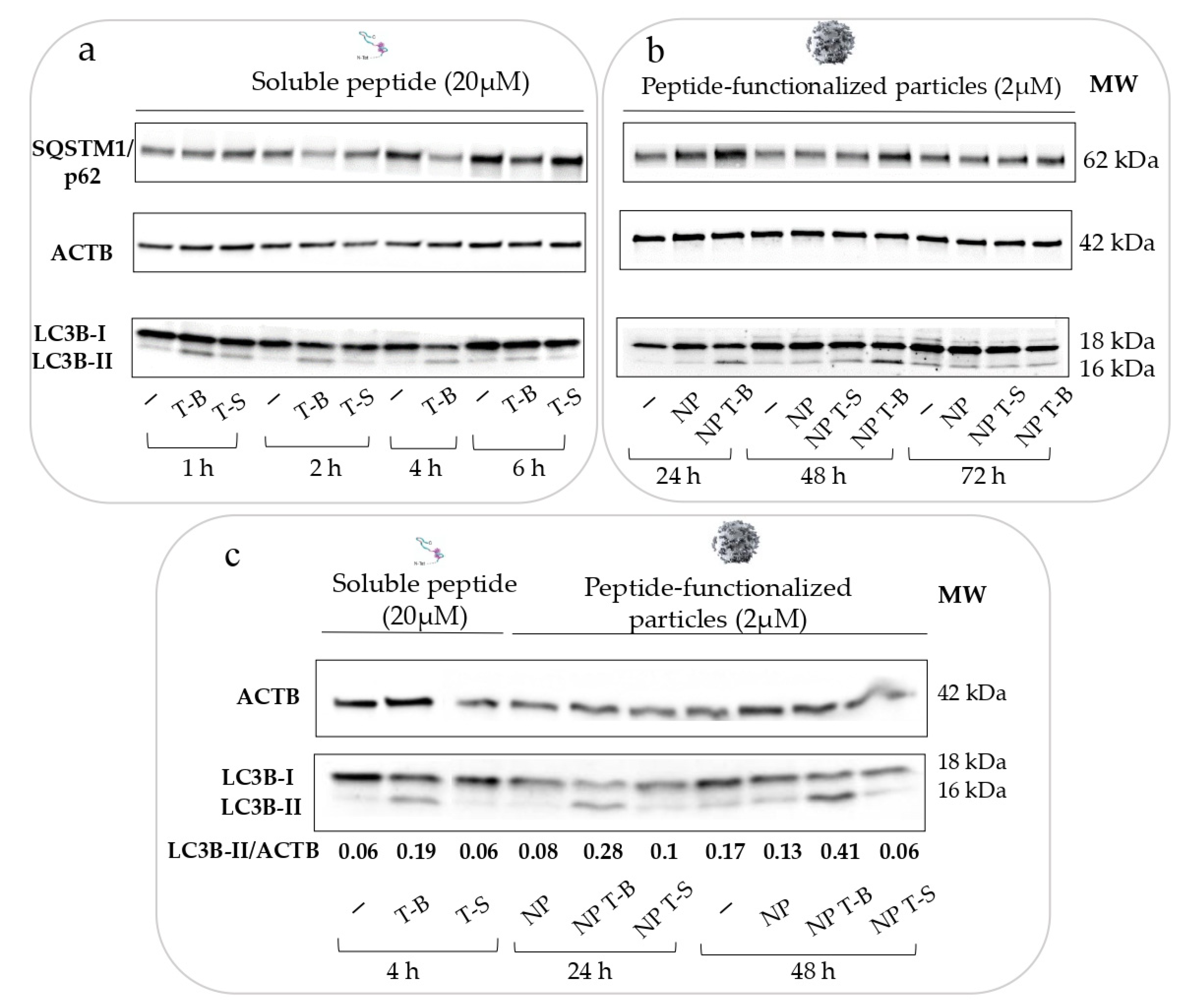
2.4. Evaluation of Autophagy and Autophagic Flux In Vitro
2.5. Immunoblotting and Antibodies
2.6. Cell Viability and Cytotoxicity Evaluation
2.7. Induction of Steatosis in Cells and Quantification of Intracellular Lipid Droplets
2.8. Preparation of Cell Samples for Conventional Electron Microscopy
2.9. Identification of Organelles in Transmission Electron Microscope (TEM) Images
- Autophagosomes have a diameter between 500 nm and 1.5 mm, have a double limiting membrane and enclose cytoplasmic cargo (for example, glycogen, ribosomes, endoplasmic reticulum or mitochondria). The double membrane, however, may not always be visible in EM sections;
- Late autophagic structures, for example autolysosomes, have a single limiting membrane but still contain cytoplasmic cargo. Their cargo appears darker than in autophagosomes, and has partially disintegrated morphology;
- Lysosomes have median diameters of 500–800 nm, often have a characteristic fingerprint or onion-like internal membrane structures, are filled with tiny granular matrix and appear dark in TEM images;
- Endosomes have a single limiting membrane and contain endocytosed material (for example nanoparticles), varying numbers of 50–100 nm internal vesicles, but no cytoplasmic cargo;
- Amphisomes (endosomes fused with autophagosomes) have a single limiting membrane and enclose both endocytosed material, 50–100 nm internal vesicles and cytoplasmic cargo.
2.10. Animal Groups and Handling
2.11. In Vivo Imaging by Tomography
3. Results
3.1. Design and Characterization of Tat-Beclin NPs
3.2. Evaluation of Autophagy in HeLa Cells
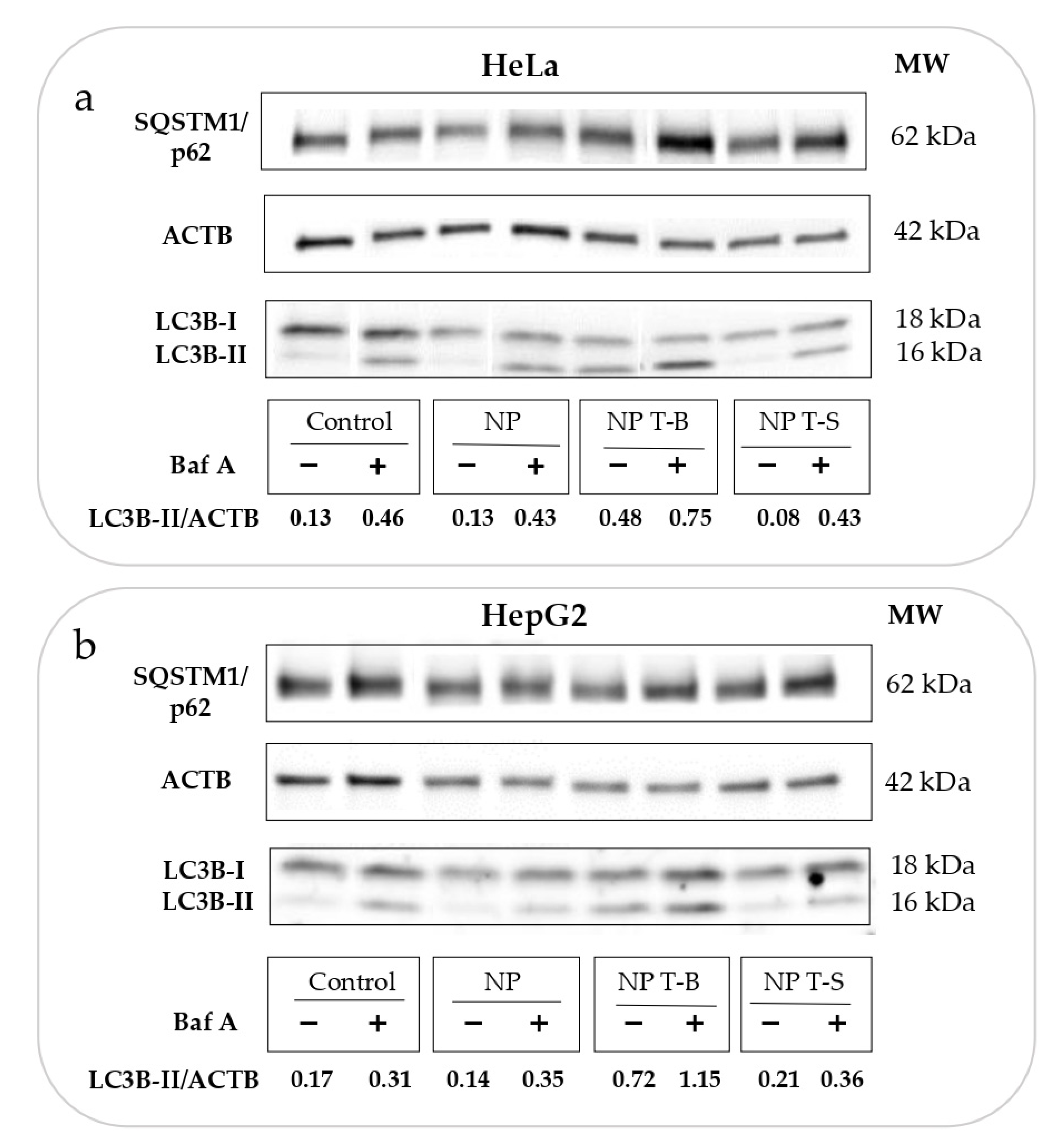
3.3. Evaluation of Autophagic Flux in HeLa and HepG2 Cells
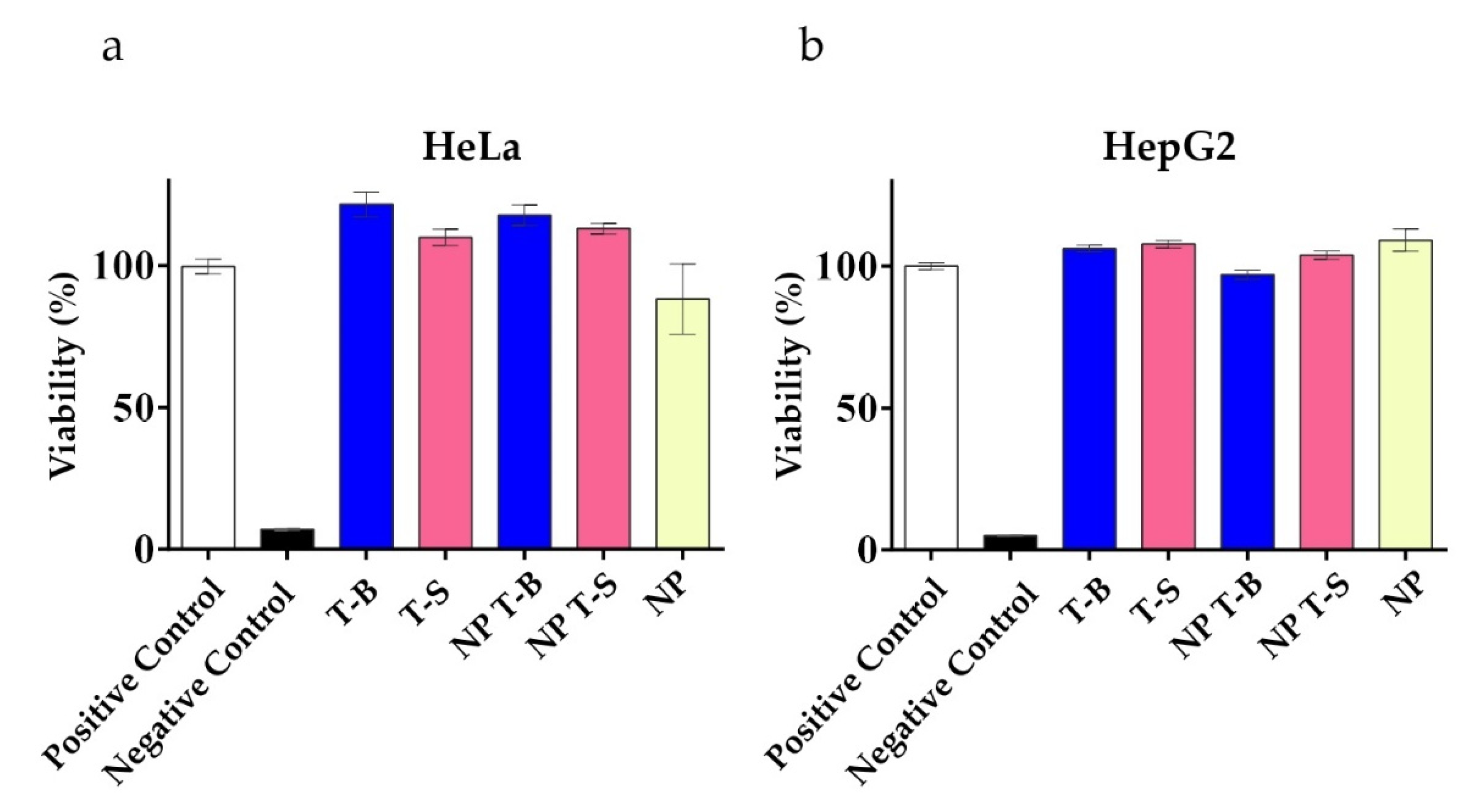
3.4. Evaluation of Viability in HeLa and HepG2 Cells
3.5. Intracellular Fate of Autophagy-Inducing Particles
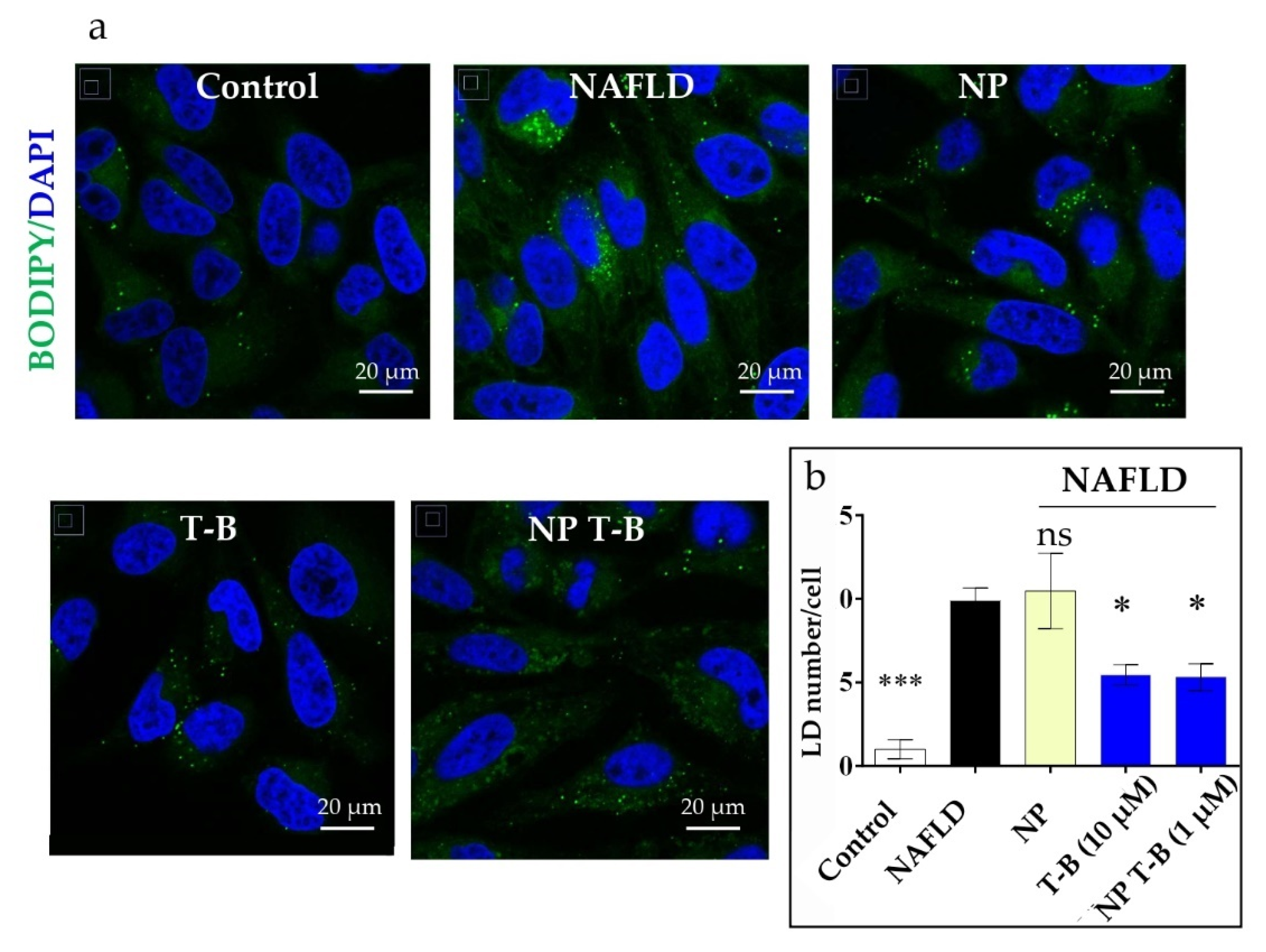
3.6. Evaluation of Intracellular Lipid Droplet Content in an NAFLD Cell Model
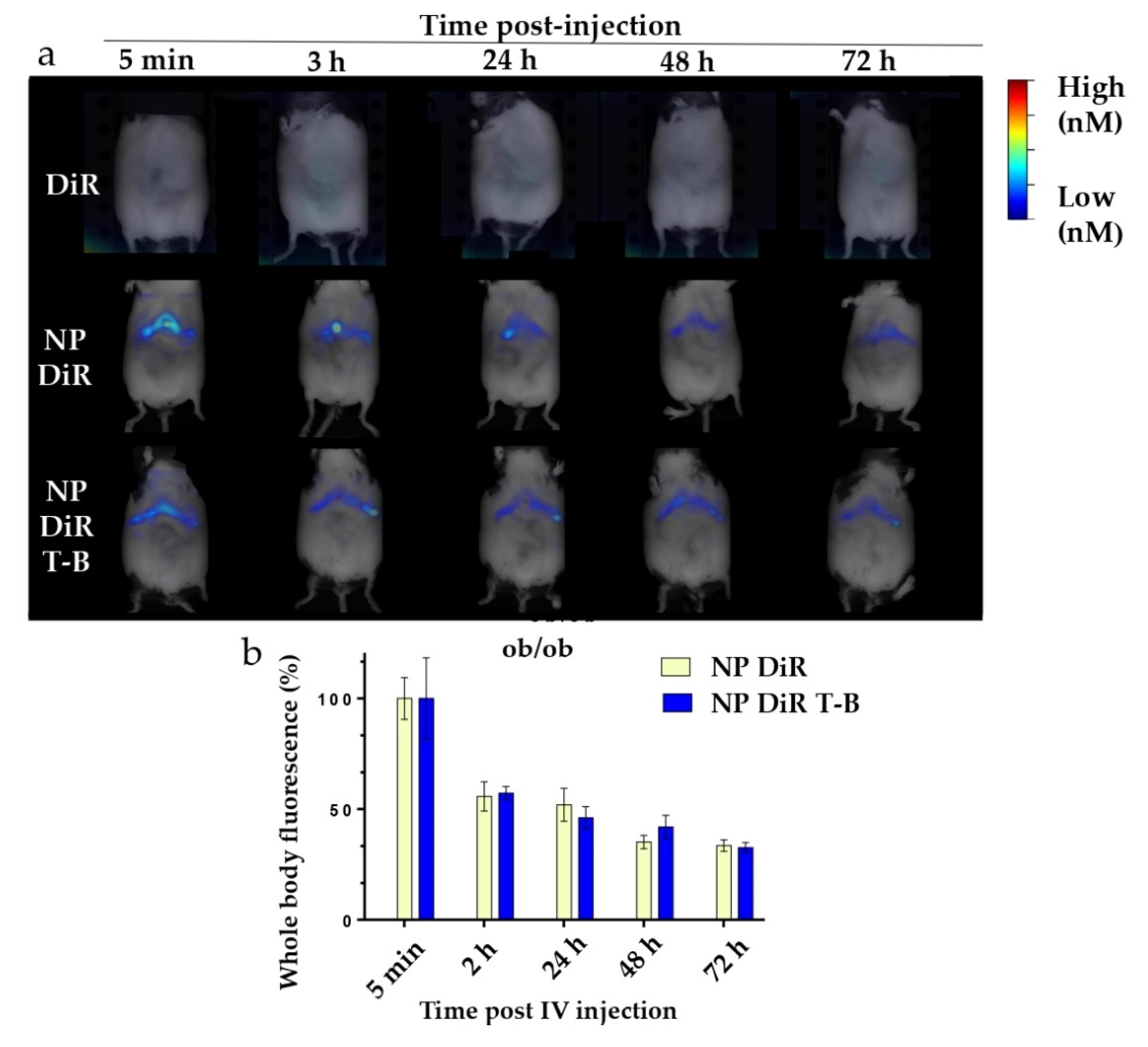
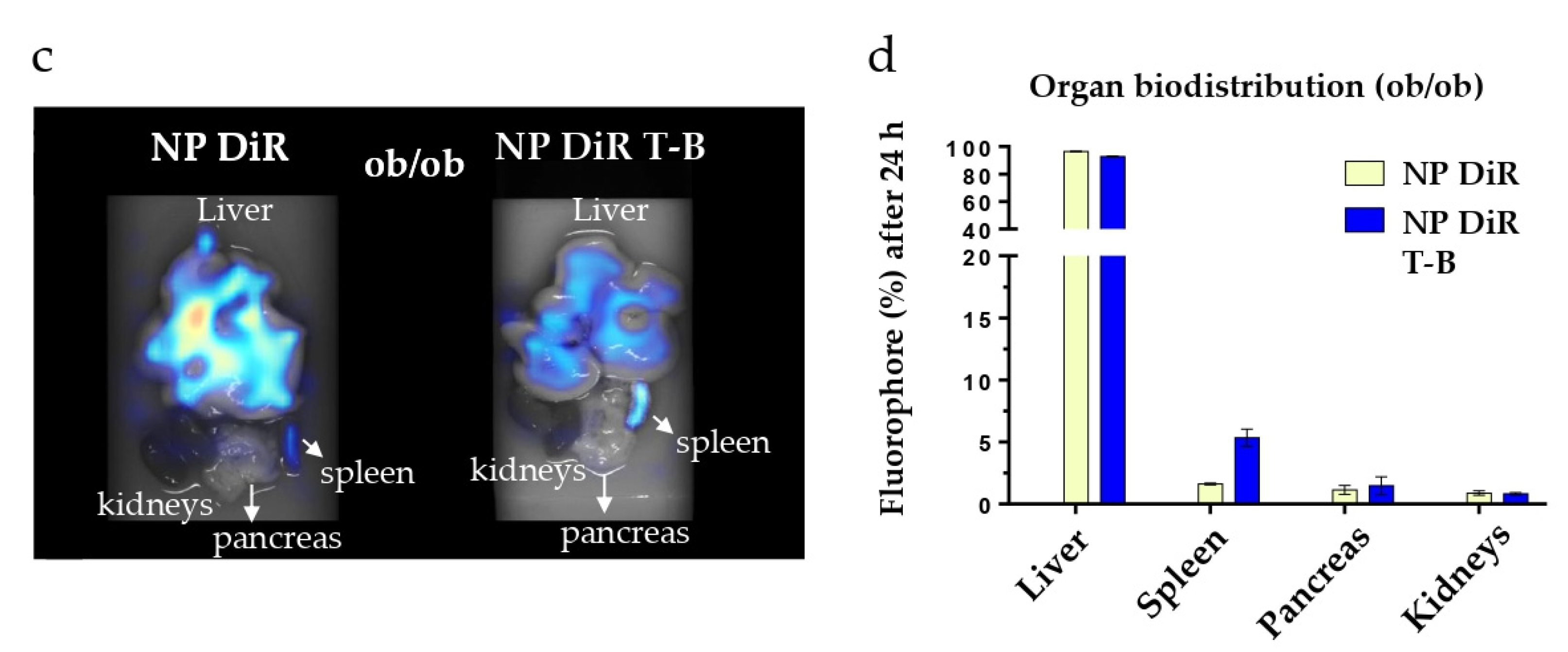
3.7. In Vivo Biodistribution in SKH1 and ob/ob Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular selfdigestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Levine, B. Eating Oneself and Uninvited Guests: Autophagy-Related Pathways in Cellular Defense. Cell 2005, 120, 159–162. [Google Scholar]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2019, 432, 206–230. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Leoni, S.; Tovoli, F.; Napoli, L.; Serio, I.; Ferri, S.; Bolondi, L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J. Gastroenterol. 2018, 24, 3361–3373. [Google Scholar] [CrossRef]
- Filali-Mouncef, Y.; Hunter, C.; Roccio, F.; Zagkou, S.; Dupont, N.; Primard, C.; Proikas-Cezanne, T.; Reggiori, F. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy 2021, 18, 50–72. [Google Scholar] [CrossRef]
- Gual, P.; Gilgenkrantz, H.; Lotersztajn, S. Autophagy in chronic liver diseases: The two faces of Janus. Am. J. Physiol. Physiol. 2017, 312, C263–C273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef]
- González-Rodríguez, Á.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [Green Version]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Chang, C.; Young, L.N.; Morris, K.L.; von Bülow, S.; Schöneberg, J.; Yamamoto-Imoto, H.; Oe, Y.; Yamamoto, K.; Nakamura, S.; Stjepanovic, G.; et al. Bidirectional Control of Autophagy by BECN1 BARA Domain Dynamics. Mol. Cell 2018, 73, 339–353.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Luk, B.T.; Hamidy, M.; Zhang, L.; Spector, S.A. Induction of a Na+/K+-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy 2018, 14, 1359–1375. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Morel, E.; Codogno, P.; Oury, F. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline Article Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Yao, X.; Zhang, Q.-J.; Zhu, M.; Liu, Z.-P.; Ci, B.; Xie, Y.; Carlson, D.; Rothermel, B.A.; Sun, Y.; et al. Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 2018, 138, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.-P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload–Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Moncalvo, F.; Espinoza, M.I.M.; Cellesi, F. Nanosized Delivery Systems for Therapeutic Proteins: Clinically Validated Technologies and Advanced Development Strategies. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef]
- van Witteloostuijn, S.B.; Pedersen, S.L.; Jensen, K.J. Half-Life Extension of Biopharmaceuticals using Chemical Methods: Alternatives to PEGylation. ChemMedChem 2016, 11, 2474–2495. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Koning, G.A.; Ten Hagen, T.L.M.; Fens, M.H.A.M.; Schraa, A.J.; Janssen, A.P.C.A.; Kok, R.J.; Molema, G.; Storm, G. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. J. Control. Release 2003, 91, 115–122. [Google Scholar] [CrossRef]
- Sonju, J.J.; Dahal, A.; Singh, S.S.; Jois, S.D. Peptide-functionalized liposomes as therapeutic and diagnostic tools for cancer treatment. J. Control. Release 2020, 329, 624–644. [Google Scholar] [CrossRef] [PubMed]
- James, R.; Manoukian, O.S.; Kumbar, S.G. Poly(lactic acid) for delivery of bioactive macromolecules. Adv. Drug Deliv. Rev. 2016, 107, 277–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigmes, D.; Trimaille, T. Advances in amphiphilic polylactide/vinyl polymer based nano-assemblies for drug delivery. Adv. Colloid Interface Sci. 2021, 294, 102483. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Warzecha, K.T.; Tacke, F. Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg. Nutr. 2014, 3, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Böttger, R.; Pauli, G.; Chao, P.H.; ALFayez, N.; Hohenwarter, L.; Li, S.D. Lipid-based nanoparticle technologies for liver targeting. Adv. Drug Deliv. Rev. 2020, 154–155, 79–101. [Google Scholar] [CrossRef]
- Gu, L.; Zhang, F.; Wu, J.; Zhuge, Y. Nanotechnology in Drug Delivery for Liver Fibrosis. Front. Mol. Biosci. 2022, 8. [Google Scholar] [CrossRef]
- Zhao, R.; Zhu, M.; Zhou, S.; Feng, W.; Chen, H. Rapamycin-Loaded mPEG-PLGA Nanoparticles Ameliorate Hepatic Steatosis and Liver Injury in Non-alcoholic Fatty Liver Disease. Front Chem. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Wu, Q.; Tian, A.-L.; Pan, H.; Kepp, O.; Kroemer, G. Autophagic flux assessment by immunoblot. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and measuring autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L.; Kovács, A.L. Double membranes vs. lipid bilayers, and their significance for correct identification of macroautophagic structures. Autophagy 2011, 7, 931–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, E.-L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Eskelinen, E.-L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes… Wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef] [Green Version]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E. Chapter 10 Monitoring Autophagy by Electron Microscopy in Mammalian Cells. In Methods in Enzymology; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 452. [Google Scholar] [CrossRef]
- Valenti, S.; Diaz, A.; Romanini, M.; del Valle, L.J.; Puiggalí, J.; Tamarit, J.L. Amorphous binary dispersions of chloramphenicol in enantiomeric pure and racemic poly-lactic acid: Morphology, molecular relaxations, and controlled drug release. Int. J. Pharm. 2019, 568, 118565. [Google Scholar] [CrossRef] [PubMed]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [CrossRef] [Green Version]
- Reifarth, M.; Hoeppener, S.; Schubert, U.S. Uptake and Intracellular Fate of Engineered Nanoparticles in Mammalian Cells: Capabilities and Limitations of Transmission Electron Microscopy-Polymer-Based Nanoparticles. Adv. Mater. 2018, 30. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Lim, Y.-M.; Kim, K.H.; Jeon, Y.-E.; Park, K.; Kim, J.; Hwang, H.-Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C.W. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef]
- Leskinen, T.; Witos, J.; Valle-Delgado, J.J.; Lintinen, K.; Kostiainen, M.; Wiedmer, S.K.; Österberg, M.; Mattinen, M.-L. Adsorption of Proteins on Colloidal Lignin Particles for Advanced Biomaterials. Biomacromolecules 2017, 18, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Keloglu, N.; Verrier, B.; Trimaille, T.; Sohier, J. Controlled association and delivery of nanoparticles from jet-sprayed hybrid microfibrillar matrices. Colloids Surf. B Biointerfaces 2016, 140, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, D.; Exbrayat-Héritier, C.; Rambaud, B.; Megy, S.; Terreux, R.; Verrier, B.; Primard, C. Surface charge modulation of rifampicin-loaded PLA nanoparticles to improve antibiotic delivery in Staphylococcus aureus biofilms. J. Nanobiotechnol. 2021, 19, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the Endosomal Escape of Cell-Penetrating Peptides and Their Cargos: Strategies and Challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef] [PubMed]
- Fretz, M.M.; Koning, G.A.; Mastrobattista, E.; Jiskoot, W.; Storm, G. OVCAR-3 cells internalize TAT-peptide modified liposomes by endocytosis. Biochim. Biophys. Acta BBA Biomembr. 2004, 1665, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P.; Rammohan, R.; Weissig, V.; Levchenko, T.S. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 8786–8791. [Google Scholar] [CrossRef] [Green Version]
- Stern, S.T.; Adiseshaiah, P.P.; Crist, R.M. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part. Fibre Toxicol. 2012, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.-H.; Tsai, J.-C.; Chen, C.-W.; Yan, S.-J.; Wang, Y.-J. Mechanisms of silver nanoparticle-induced toxicity and important role of autophagy. Nanotoxicology 2016, 10, 1021–1040. [Google Scholar] [CrossRef]
- Eidi, H.; Joubert, O.; Némos, C.; Grandemange, S.; Mograbi, B.; Foliguet, B.; Tournebize, J.; Maincent, P.; Le Faou, A.; Aboukhamis, I.; et al. Drug delivery by polymeric nanoparticles induces autophagy in macrophages. Int. J. Pharm. 2012, 422, 495–503. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Moosavi, M.A.; Tavakol, S.; Vardar, D.Ö.; Hosseini, A.; Rahmati, M.; Dini, L.; Hussain, S.; Mandegary, A.; Klionsky, D.J. Necrotic, apoptotic and autophagic cell fates triggered by nanoparticles. Autophagy 2019, 15, 4–33. [Google Scholar] [CrossRef] [Green Version]
- De Mazière, A.; van der Beek, J.; van Dijk, S.; de Heus, C.; Reggiori, F.; Koike, M.; Klumperman, J. An optimized protocol for immuno-electron microscopy of endogenous LC3. Autophagy 2022, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hadadi-Bechor, S.; Haim, Y.; Pecht, T.; Gat, R.; Tarnovscki, T.; Gericke, M.; Rudich, A. Autophagy differentially regulates macrophage lipid handling depending on the lipid substrate (oleic acid vs. acetylated-LDL) and inflammatory activation state. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2019, 1864, 158527. [Google Scholar] [CrossRef]
- Jindal, A.; Bruzzì, S.; Sutti, S.; Locatelli, I.; Bozzola, C.; Paternostro, C.; Parola, M.; Albano, E. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp. Mol. Pathol. 2015, 99, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the liver in obese and diabetic ob/ob and db/db mice fed a standard or high-calorie diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H.; Ong, Z.Y.; Xu, K.; Ee, P.L.R.; Zheng, S.; Hedrick, J.L.; Yang, Y.-Y. Polymer- and lipid-based nanoparticle therapeutics for the treatment of liver diseases. Nano Today 2010, 5, 296–312. [Google Scholar] [CrossRef]
- Egusquiaguirre, S.P.; Beziere, N.; Pedraz, J.L.; Hernández, R.M.; Ntziachristos, V.; Igartua, M. Optoacoustic imaging enabled biodistribution study of cationic polymeric biodegradable nanoparticles. Contrast Media Mol. Imaging 2015, 10, 421–427. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Relevance of Autophagy in Parenchymal and Non-Parenchymal Liver Cells for Health and Disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Baratta, J.L.; Ngo, A.; Lopez, B.; Kasabwalla, N.; Longmuir, K.J.; Robertson, R.T. Cellular organization of normal mouse liver: A histological, quantitative immunocytochemical, and fine structural analysis. Histochem. Cell Biol. 2009, 131, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Dalzon, B.; Lebas, C.; Jimenez, G.; Gutjahr, A.; Terrat, C.; Exposito, J.; Verrier, B.; Lethias, C. Poly(lactic acid) nanoparticles targeting α5β1 integrin as vaccine delivery vehicle, a prospective study. PLoS ONE 2016, 11, e0167663. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagkou, S.; Marais, V.; Zeghoudi, N.; Guillou, E.L.; Eskelinen, E.-L.; Panasyuk, G.; Verrier, B.; Primard, C. Design and Evaluation of Autophagy-Inducing Particles for the Treatment of Abnormal Lipid Accumulation. Pharmaceutics 2022, 14, 1379. https://doi.org/10.3390/pharmaceutics14071379
Zagkou S, Marais V, Zeghoudi N, Guillou EL, Eskelinen E-L, Panasyuk G, Verrier B, Primard C. Design and Evaluation of Autophagy-Inducing Particles for the Treatment of Abnormal Lipid Accumulation. Pharmaceutics. 2022; 14(7):1379. https://doi.org/10.3390/pharmaceutics14071379
Chicago/Turabian StyleZagkou, Stavroula, Valentine Marais, Narimane Zeghoudi, Edouard Le Guillou, Eeva-Liisa Eskelinen, Ganna Panasyuk, Bernard Verrier, and Charlotte Primard. 2022. "Design and Evaluation of Autophagy-Inducing Particles for the Treatment of Abnormal Lipid Accumulation" Pharmaceutics 14, no. 7: 1379. https://doi.org/10.3390/pharmaceutics14071379
APA StyleZagkou, S., Marais, V., Zeghoudi, N., Guillou, E. L., Eskelinen, E. -L., Panasyuk, G., Verrier, B., & Primard, C. (2022). Design and Evaluation of Autophagy-Inducing Particles for the Treatment of Abnormal Lipid Accumulation. Pharmaceutics, 14(7), 1379. https://doi.org/10.3390/pharmaceutics14071379