
Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs




Abstract
:1. Introduction
2. Oligonucleotide Therapeutics as New Targeted Anti-Cancer Drugs for Challenging or Undruggable Proteins
2.1. Oligonucleotide Therapeutics Targeting the MYC Gene Family
2.1.1. Oligonucleotide Therapeutics Targeting MYC
2.1.2. Oligonucleotide Therapeutics Targeting MYCN
2.2. Oligonucleotide Therapeutics Targeting the RAS Gene Family
Oligonucleotide Therapeutics Targeting KRAS
2.3. Oligonucleotide Therapeutics Targeting STAT3
2.4. Oligonucleotide Therapeutics Targeting BCL-2
3. Oligonucleotide Therapeutics as New Targeted Anti-Cancer Drugs for Non-Coding RNAs
3.1. microRNA Therapeutics
3.1.1. Cobomarsen
3.1.2. TargomiRs (a miR-16 Mimic and a MIR16-Based miRNA Mimetic)
3.1.3. MRX34 (MiR-34a Mimic)
4. Challenges and Solutions for the Clinical Success of Oligonucleotide Therapeutics
5. Challenges and Solutions Related to the Oligonucleotides for Cancer Therapy
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic Heterogeneity in Cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Padma, V.V. An Overview of Targeted Cancer Therapy. BioMedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Dalmartello, M.; La Vecchia, C.; Bertuccio, P.; Boffetta, P.; Levi, F.; Negri, E.; Malvezzi, M. European Cancer Mortality Predictions for the Year 2022 with Focus on Ovarian Cancer. Ann. Oncol. 2022, 33, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Printezi, M.I.; Kilgallen, A.B.; Bond, M.J.G.; Štibler, U.; Putker, M.; Teske, A.J.; Cramer, M.J.; Punt, C.J.A.; Sluijter, J.P.G.; Huitema, A.D.R.; et al. Toxicity and Efficacy of Chronomodulated Chemotherapy: A Systematic Review. Lancet Oncol. 2022, 23, e129–e143. [Google Scholar] [CrossRef]
- Lowenthal, R.M.; Eaton, K. Toxicity of Chemotherapy. Hematol. Oncol. Clin. N. Am. 1996, 10, 967–990. [Google Scholar] [CrossRef]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Bakan, A.; Nevins, N.; Lakdawala, A.S.; Bahar, I. Druggability Assessment of Allosteric Proteins by Dynamics Simulations in the Presence of Probe Molecules. J. Chem. Theory Comput. 2012, 8, 2435–2447. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Barril, X. Understanding and Predicting Druggability. A High-Throughput Method for Detection of Drug Binding Sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Winkler, J.D.; Crews, C.M. Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 2017, 174, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.T.; Tsanov, K.M.; Pearson, D.S.; Roels, F.; Spina, C.S.; Ebright, R.; Seligson, M.; de Soysa, Y.; Cahan, P.; Theißen, J.; et al. Multiple Mechanisms Disrupt the Let-7 MicroRNA Family in Neuroblastoma. Nature 2016, 535, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Perini, G.; Milazzo, G.; Narayan, N.; Ekert, P.G. Letting the Breaks off MYCN. Cell Death Differ. 2016, 23, 1904–1905. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-Coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Dai, H.; Abdullah, R.; Wu, X.; Li, F.; Ma, Y.; Lu, A.; Zhang, G. Pancreatic Cancer: Nucleic Acid Drug Discovery and Targeted Therapy. Front. Cell Dev. Biol. 2022, 10, 855474. [Google Scholar] [CrossRef]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense Oligonucleotides Targeting Angiogenic Factors as Potential Cancer Therapeutics. Mol. Ther.-Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Tian, Z.; Liang, G.; Cui, K.; Liang, Y.; Wang, Q.; Lv, S.; Cheng, X.; Zhang, L. Insight into the Prospects for RNAi Therapy of Cancer. Front. Pharmacol. 2021, 12, 644718. [Google Scholar] [CrossRef]
- Igarashi, J.; Niwa, Y.; Sugiyama, D. Research and Development of Oligonucleotide Therapeutics in Japan for Rare Diseases. Future Rare Dis. 2022, 2, FRD19. [Google Scholar] [CrossRef]
- Wang, C.; Fang, H.; Zhang, J.; Gu, Y. Targeting “Undruggable” c-Myc Protein by Synthetic Lethality. Front. Med. 2021, 15, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS Mutation: From Undruggable to Druggable in Cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Duffy, M.J.; O’Grady, S.; Tang, M.; Crown, J. MYC as a Target for Cancer Treatment. Cancer Treat. Rev. 2021, 94, 102154. [Google Scholar] [CrossRef]
- Moumné, L.; Marie, A.-C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14, 260. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous Sarcoma Viral RNA Translation by a Specific Oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [Green Version]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M. The Powerful World of Antisense Oligonucleotides: From Bench to Bedside. WIREs RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. 2012, 19, 937–954. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a Bidentate Ribonuclease in the Initiation Step of RNA Interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Thompson, J.D. Clinical Development of Synthetic SiRNA Therapeutics. Drug Discov. Today Ther. Strateg. 2013, 10, e133–e138. [Google Scholar] [CrossRef]
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic MicroRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132. [Google Scholar] [CrossRef] [Green Version]
- Ryan, K.M.; Birnie, G.D. Myc Oncogenes: The Enigmatic Family. Biochem. J. 1996, 314, 713–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammak, S.; Hamdani, N.; Gorrec, F.; Allen, M.D.; Freund, S.M.V.; Bycroft, M.; Zinzalla, G. Crystal Structures and Nuclear Magnetic Resonance Studies of the Apo Form of the C-MYC:MAX BHLHZip Complex Reveal a Helical Basic Region in the Absence of DNA. Biochemistry 2019, 58, 3144–3154. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, M.-E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Knies-Bamforth, U.E.; Fox, S.B.; Poulsom, R.; Evan, G.I.; Harris, A.L. C-Myc Interacts with Hypoxia to Induce Angiogenesis In Vivo by a Vascular Endothelial Growth Factor-Dependent Mechanism. Cancer Res. 2004, 64, 6563–6570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. MiR-9, a MYC/MYCN-Activated MicroRNA, Regulates E-Cadherin and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Doré-Savard, L.; Roussy, G.; Dansereau, M.-A.; Collingwood, M.A.; Lennox, K.A.; Rose, S.D.; Beaudet, N.; Behlke, M.A.; Sarret, P. Central Delivery of Dicer-Substrate SiRNA: A Direct Application for Pain Research. Mol. Ther. 2008, 16, 1331–1339. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D.; et al. Safety and Activity of DCR-MYC, a First-in-Class Dicer-Substrate Small Interfering RNA (DsiRNA) Targeting MYC, in a Phase I Study in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-Myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N-Myc Oncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef]
- Dang, C.V.; Kim, J.; Gao, P.; Yustein, J. The Interplay between MYC and HIF in Cancer. Nat. Rev. Cancer 2008, 8, 51–56. [Google Scholar] [CrossRef]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E.; et al. Differential Expression of Myc Family Genes during Murine Development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too Many Targets, Not Enough Patients: Rethinking Neuroblastoma Clinical Trials. Nat. Rev. Cancer 2018, 18, 389–400. [Google Scholar] [CrossRef]
- Janowski, B.A.; Kaihatsu, K.; Huffman, K.E.; Schwartz, J.C.; Ram, R.; Hardy, D.; Mendelson, C.R.; Corey, D.R. Inhibiting Transcription of Chromosomal DNA with Antigene Peptide Nucleic Acids. Nat. Chem. Biol. 2005, 1, 210–215. [Google Scholar] [CrossRef]
- Tonelli, R.; Purgato, S.; Camerin, C.; Fronza, R.; Bologna, F.; Alboresi, S.; Franzoni, M.; Corradini, R.; Sforza, S.; Faccini, A.; et al. Anti-Gene Peptide Nucleic Acid Specifically Inhibits MYCN Expression in Human Neuroblastoma Cells Leading to Cell Growth Inhibition and Apoptosis. Mol. Cancer Ther. 2005, 4, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, R.; McIntyre, A.; Camerin, C.; Walters, Z.S.; Di Leo, K.; Selfe, J.; Purgato, S.; Missiaglia, E.; Tortori, A.; Renshaw, J.; et al. Antitumor Activity of Sustained N-Myc Reduction in Rhabdomyosarcomas and Transcriptional Block by Antigene Therapy. Clin. Cancer Res. 2012, 18, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; Van Ryn, C.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association of MYCN Copy Number with Clinical Features, Tumor Biology, and Outcomes in Neuroblastoma: A Report from the Children’s Oncology Group: MYCN Copy Number in Neuroblastoma. Cancer 2017, 123, 4224–4235. [Google Scholar] [CrossRef] [Green Version]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M.; et al. ATF4 Regulates MYC-Mediated Neuroblastoma Cell Death upon Glutamine Deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Montemurro, L.; Raieli, S.; Angelucci, S.; Bartolucci, D.; Amadesi, C.; Lampis, S.; Scardovi, A.L.; Venturelli, L.; Nieddu, G.; Cerisoli, L.; et al. A Novel MYCN-Specific Antigene Oligonucleotide Deregulates Mitochondria and Inhibits Tumor Growth in MYCN-Amplified Neuroblastoma. Cancer Res. 2019, 79, 6166–6177. [Google Scholar] [CrossRef] [Green Version]
- Raieli, S.; Di Renzo, D.; Lampis, S.; Amadesi, C.; Montemurro, L.; Pession, A.; Hrelia, P.; Fischer, M.; Tonelli, R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021, 11, 625207. [Google Scholar] [CrossRef] [PubMed]
- Lampis, S.; Raieli, S.; Montemurro, L.; Bartolucci, D.; Amadesi, C.; Bortolotti, S.; Angelucci, S.; Scardovi, A.L.; Nieddu, G.; Cerisoli, L.; et al. The MYCN Inhibitor BGA002 Restores the Retinoic Acid Response Leading to Differentiation or Apoptosis by the MTOR Block in MYCN-Amplified Neuroblastoma. J. Exp. Clin. Cancer Res. 2022, 41, 160. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Mo, S.P.; Coulson, J.M.; Prior, I.A. RAS Variant Signalling. Biochem. Soc. Trans. 2018, 46, 1325–1332. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS Isoforms and Mutations in Cancer at a Glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Fang, G.; Rudolph, J. Ras Inhibition via Direct Ras Binding—Is There a Path Forward? Bioorg. Med. Chem. Lett. 2012, 22, 5766–5776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.-K. K- and N-Ras Are Geranylgeranylated in Cells Treated with Farnesyl Protein Transferase Inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorde Khvalevsky, E.; Gabai, R.; Rachmut, I.H.; Horwitz, E.; Brunschwig, Z.; Orbach, A.; Shemi, A.; Golan, T.; Domb, A.J.; Yavin, E.; et al. Mutant KRAS Is a Druggable Target for Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20723–20728. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Khvalevsky, E.Z.; Hubert, A.; Gabai, R.M.; Hen, N.; Segal, A.; Domb, A.; Harari, G.; David, E.B.; Raskin, S.; et al. RNAi Therapy Targeting KRAS in Combination with Chemotherapy for Locally Advanced Pancreatic Cancer Patients. Oncotarget 2015, 6, 24560–24570. [Google Scholar] [CrossRef] [Green Version]
- Ramot, Y.; Rotkopf, S.; Gabai, R.M.; Zorde Khvalevsky, E.; Muravnik, S.; Marzoli, G.A.; Domb, A.J.; Shemi, A.; Nyska, A. Preclinical Safety Evaluation in Rats of a Polymeric Matrix Containing an SiRNA Drug Used as a Local and Prolonged Delivery System for Pancreatic Cancer Therapy. Toxicol. Pathol. 2016, 44, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Wancewicz, E.V.; Witchell, D.; Swayze, E.E. Short Antisense Oligonucleotides with Novel 2′–4′ Conformationaly Restricted Nucleoside Analogues Show Improved Potency without Increased Toxicity in Animals. J. Med. Chem. 2009, 52, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-Dependent Tumors with AZD4785, a High-Affinity Therapeutic Antisense Oligonucleotide Inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Wen, Z.; Darnell, J.E. Stat3: A STAT Family Member Activated by Tyrosine Phosphorylation in Response to Epidermal Growth Factor and Interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef]
- Liu, Y.; Li, P.-K.; Li, C.; Lin, J. Inhibition of STAT3 Signaling Blocks the Anti-Apoptotic Activity of IL-6 in Human Liver Cancer Cells. J. Biol. Chem. 2010, 285, 27429–27439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively Activated Stat3 Induces Tumorigenesis and Enhances Cell Motility of Prostate Epithelial Cells through Integrin Β6. Mol. Cell. Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Suzui, M.; Yasumatu, R.; Nakashima, T.; Kuratomi, Y.; Azuma, K.; Tomita, K.; Komiyama, S.; Weinstein, I.B. Constitutive Activation of Signal Transducers and Activators of Transcription 3 Correlates with Cyclin D1 Overexpression and May Provide a Novel Prognostic Marker in Head and Neck Squamous Cell Carcinoma. Cancer Res. 2002, 62, 3351–3355. [Google Scholar] [PubMed]
- Danoch, H.; Kalechman, Y.; Albeck, M.; Longo, D.L.; Sredni, B. Sensitizing B- and T- Cell Lymphoma Cells to Paclitaxel/Abraxane—Induced Death by AS101 via Inhibition of the VLA-4—IL10—Survivin Axis. Mol. Cancer Res. 2015, 13, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, K.; Susa, M.; Choy, E.; Yang, C.; Hornicek, F.J.; Mankin, H.J.; Duan, Z. Oleanane Triterpenoid CDDO-Me Induces Apoptosis in Multidrug Resistant Osteosarcoma Cells through Inhibition of Stat3 Pathway. BMC Cancer 2010, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.-K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The Novel Curcumin Analog FLLL32 Decreases STAT3 DNA Binding Activity and Expression, and Induces Apoptosis in Osteosarcoma Cell Lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Reilley, M.J.; McCoon, P.; Cook, C.; Lyne, P.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; et al. STAT3 Antisense Oligonucleotide AZD9150 in a Subset of Patients with Heavily Pretreated Lymphoma: Results of a Phase 1b Trial. J. Immunother. Cancer 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In Vivo Delivery of SiRNA to Immune Cells by Conjugation to a TLR9 Agonist Enhances Antitumor Immune Responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic Targeting of Innate Immunity with Toll-like Receptor Agonists and Antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef]
- Barchet, W.; Wimmenauer, V.; Schlee, M.; Hartmann, G. Accessing the Therapeutic Potential of Immunostimulatory Nucleic Acids. Curr. Opin. Immunol. 2008, 20, 389–395. [Google Scholar] [CrossRef]
- Krieg, A.M. Toll-like Receptor 9 (TLR9) Agonists in the Treatment of Cancer. Oncogene 2008, 27, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinman, D.M.; Currie, D.; Gursel, I.; Verthelyi, D. Use of CpG Oligodeoxynucleotides as Immune Adjuvants. Immunol. Rev. 2004, 199, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the Chromosome Breakpoint of Neoplastic B Cells with the t(14;18) Chromosome Translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 Inhibition to Overcome Resistance to Chemo- and Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. [Google Scholar] [CrossRef] [Green Version]
- Cotter, F.E.; Johnson, P.; Hall, P.; Pocock, C.; al Mahdi, N.; Cowell, J.K.; Morgan, G. Antisense Oligonucleotides Suppress B-Cell Lymphoma Growth in a SCID-Hu Mouse Model. Oncogene 1994, 9, 3049–3055. [Google Scholar]
- Raynaud, F.I.; Orr, R.M.; Goddard, P.M.; Lacey, H.A.; Lancashire, H.; Judson, I.R.; Beck, T.; Bryan, B.; Cotter, F.E. Pharmacokinetics of G3139, a Phosphorothioate Oligodeoxynucleotide Antisense to Bcl-2, after Intravenous Administration or Continuous Subcutaneous Infusion to Mice. J. Pharmacol. Exp. Ther. 1997, 281, 420–427. [Google Scholar]
- Gagliardi, M.; Ashizawa, A.T. Making Sense of Antisense Oligonucleotide Therapeutics Targeting Bcl-2. Pharmaceutics 2022, 14, 97. [Google Scholar] [CrossRef]
- Ebrahim, A.S.; Kandouz, M.; Liddane, A.; Sabbagh, H.; Hou, Y.; Li, C.; Al-Katib, A. PNT2258, a Novel Deoxyribonucleic Acid Inhibitor, Induces Cell Cycle Arrest and Apoptosis via a Distinct Mechanism of Action: A New Class of Drug for Non-Hodgkin’s Lymphoma. Oncotarget 2016, 7, 42374–42384. [Google Scholar] [CrossRef] [Green Version]
- Qadir, M.I.; Bukhat, S.; Rasul, S.; Manzoor, H.; Manzoor, M. RNA Therapeutics: Identification of Novel Targets Leading to Drug Discovery. J. Cell. Biochem. 2020, 121, 898–929. [Google Scholar] [CrossRef]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small Non-Coding RNA and Cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT) A Promoter-Level Mammalian Expression Atlas. Nature 2014, 507, 462–470. [CrossRef] [PubMed] [Green Version]
- The ENCODE Project Consortium an Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [CrossRef] [PubMed]
- Ning, B.; Yu, D.; Yu, A.-M. Advances and Challenges in Studying Noncoding RNA Regulation of Drug Metabolism and Development of RNA Therapeutics. Biochem. Pharmacol. 2019, 169, 113638. [Google Scholar] [CrossRef] [PubMed]
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. In Non-Coding RNAs in Colorectal Cancer; Slaby, O., Calin, G.A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; Volume 937, pp. 3–17. ISBN 978-3-319-42057-8. [Google Scholar]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long Non-Coding RNA: Classification, Biogenesis and Functions in Blood Cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef]
- Srijyothi, L.; Ponne, S.; Prathama, T.; Ashok, C.; Baluchamy, S. Roles of Non-Coding RNAs in Transcriptional Regulation. In Transcriptional and Post-Transcriptional Regulation; Ghedira, K., Ed.; InTech: London, UK, 2018; ISBN 978-1-78923-791-7. [Google Scholar]
- Kwok, Z.H.; Ni, K.; Jin, Y. Extracellular Vesicle Associated Non-Coding RNAs in Lung Infections and Injury. Cells 2021, 10, 965. [Google Scholar] [CrossRef]
- Slaby, O.; Laga, R.; Sedlacek, O. Therapeutic Targeting of Non-Coding RNAs in Cancer. Biochem. J. 2017, 474, 4219–4251. [Google Scholar] [CrossRef]
- Dozmorov, M.G.; Giles, C.B.; Koelsch, K.A.; Wren, J.D. Systematic Classification of Non-Coding RNAs by Epigenomic Similarity. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef] [Green Version]
- Rasool, M.; Malik, A.; Zahid, S.; Basit Ashraf, M.A.; Qazi, M.H.; Asif, M.; Zaheer, A.; Arshad, M.; Raza, A.; Jamal, M.S. Non-Coding RNAs in Cancer Diagnosis and Therapy. Non-Coding RNA Res. 2016, 1, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin Signature Reveals over a Thousand Highly Conserved Large Non-Coding RNAs in Mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Geisler, S.; Coller, J. RNA in Unexpected Places: Long Non-Coding RNA Functions in Diverse Cellular Contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Maruyama, R.; Yamamoto, E.; Niinuma, T.; Kai, M. Relationship Between Noncoding RNA Dysregulation and Epigenetic Mechanisms in Cancer. Adv. Exp. Med. Biol. 2016, 927, 109–135. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to MiRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Harries, L.W. RNA Biology Provides New Therapeutic Targets for Human Disease. Front. Genet. 2019, 10, 205. [Google Scholar] [CrossRef]
- Wang, T.; Shigdar, S.; Shamaileh, H.A.; Gantier, M.P.; Yin, W.; Xiang, D.; Wang, L.; Zhou, S.-F.; Hou, Y.; Wang, P.; et al. Challenges and Opportunities for SiRNA-Based Cancer Treatment. Cancer Lett. 2017, 387, 77–83. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-Coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and Activity of MicroRNA-Loaded Minicells in Patients with Recurrent Malignant Pleural Mesothelioma: A First-in-Man, Phase 1, Open-Label, Dose-Escalation Study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Foss, F.M.; Querfeld, C.; Porcu, P.; Kim, Y.H.; Pacheco, T.; Halwani, A.S.; DeSimone, J.; William, B.M.; Seto, A.G.; Ruckman, J.; et al. Phase 1 Trial Evaluating MRG-106, a Synthetic Inhibitor of MicroRNA-155, in Patients with Cutaneous t-Cell Lymphoma (CTCL). J. Clin. Oncol. 2017, 35, 7564. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How Close Are MiRNAs from Clinical Practice? A Perspective on the Diagnostic and Therapeutic Market. EJIFCC 2019, 30, 114–127. [Google Scholar]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an Oligonucleotide Inhibitor of MiR-155, Co-Ordinately Regulates Multiple Survival Pathways to Reduce Cellular Proliferation and Survival in Cutaneous T-Cell Lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadou, E.; Seto, A.G.; Beatty, X.; Hermreck, M.; Gilles, M.-E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D.; et al. Cobomarsen, an Oligonucleotide Inhibitor of MiR-155, Slows DLBCL Tumor Cell Growth In Vitro and In Vivo. Clin. Cancer Res. 2021, 27, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Witten, L.; Slack, F.J. MiR-155 as a Novel Clinical Target for Hematological Malignancies. Carcinogenesis 2020, 41, 2–7. [Google Scholar] [CrossRef]
- Fuertes, T.; Ramiro, A.R.; de Yebenes, V.G. MiRNA-Based Therapies in B Cell Non-Hodgkin Lymphoma. Trends Immunol. 2020, 41, 932–947. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA Therapeutics—Challenges and Potential Solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for MicroRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring Expression of MiR-16: A Novel Approach to Therapy for Malignant Pleural Mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef]
- Bader, A.G. MiR-34—A MicroRNA Replacement Therapy Is Headed to the Clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [Green Version]
- Bouchie, A. First MicroRNA Mimic Enters Clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
- Daige, C.L.; Wiggins, J.F.; Priddy, L.; Nelligan-Davis, T.; Zhao, J.; Brown, D. Systemic Delivery of a MiR34a Mimic as a Potential Therapeutic for Liver Cancer. Mol. Cancer Ther. 2014, 13, 2352–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 Study of MRX34, a Liposomal MiR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, Biodistribution and Cell Uptake of Antisense Oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Herkt, M.; Thum, T. Pharmacokinetics and Proceedings in Clinical Application of Nucleic Acid Therapeutics. Mol. Ther. 2021, 29, 521–539. [Google Scholar] [CrossRef]
- Boursereau, R.; Donadieu, A.; Dabertrand, F.; Dubayle, D.; Morel, J.-L. Blood Brain Barrier Precludes the Cerebral Arteries to Intravenously-Injected Antisense Oligonucleotide. Eur. J. Pharmacol. 2015, 747, 141–149. [Google Scholar] [CrossRef]
- Mendonça, M.C.P.; Kont, A.; Aburto, M.R.; Cryan, J.F.; O’Driscoll, C.M. Advances in the Design of (Nano)Formulations for Delivery of Antisense Oligonucleotides and Small Interfering RNA: Focus on the Central Nervous System. Mol. Pharm. 2021, 18, 1491–1506. [Google Scholar] [CrossRef]
- Klabenkova, K.; Fokina, A.; Stetsenko, D. Chemistry of Peptide-Oligonucleotide Conjugates: A Review. Molecules 2021, 26, 5420. [Google Scholar] [CrossRef]
- Quemener, A.M.; Centomo, M.L.; Sax, S.L.; Panella, R. Small Drugs, Huge Impact: The Extraordinary Impact of Antisense Oligonucleotides in Research and Drug Development. Molecules 2022, 27, 536. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.-H. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef]
- Bost, J.P.; Barriga, H.; Holme, M.N.; Gallud, A.; Maugeri, M.; Gupta, D.; Lehto, T.; Valadi, H.; Esbjörner, E.K.; Stevens, M.M.; et al. Delivery of Oligonucleotide Therapeutics: Chemical Modifications, Lipid Nanoparticles, and Extracellular Vesicles. ACS Nano 2021, 15, 13993–14021. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Moyle, P.M.; Toth, I. Endosome Escape Strategies for Improving the Efficacy of Oligonucleotide Delivery Systems. Curr. Med. Chem. 2015, 22, 3326–3346. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, S.; Roscigno, G.; Affinito, A.; Ingenito, F.; Quintavalle, C.; Condorelli, G. Potential and Challenges of Aptamers as Specific Carriers of Therapeutic Oligonucleotides for Precision Medicine in Cancer. Cancers 2019, 11, 1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of Antisense Oligonucleotide and SiRNA-Based Therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and Avoiding SiRNA Off-Target Effects for Target Identification and Therapeutic Application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Andersson, P. Preclinical Safety Assessment of Therapeutic Oligonucleotides. Methods Mol. Biol. 2022, 2434, 355–370. [Google Scholar] [CrossRef]
- Koziolkiewicz, M.; Gendaszewska, E.; Maszewska, M.; Stein, C.A.; Stec, W.J. The Mononucleotide-Dependent, Nonantisense Mechanism of Action of Phosphodiester and Phosphorothioate Oligonucleotides Depends upon the Activity of an Ecto-5′-Nucleotidase. Blood 2001, 98, 995–1002. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Sproat, B.S.; Lamond, A.I.; Beijer, B.; Neuner, P.; Ryder, U. Highly Efficient Chemical Synthesis of 2′-O-Methyloligoribonucleotides and Tetrabiotinylated Derivatives; Novel Probes That Are Resistant to Degradation by RNA or DNA Specific Nucleases. Nucleic Acids Res. 1989, 17, 3373–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Moulton, J.D.; Yan, Y. Using Morpholinos to Control Gene Expression. Curr. Protoc. Mol. Biol. 2008, 83, 26.8.1–26.8.29. [Google Scholar] [CrossRef]
- Larsen, H.J.; Bentin, T.; Nielsen, P.E. Antisense Properties of Peptide Nucleic Acid. Biochim. Biophys. Acta 1999, 1489, 159–166. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.A. A Review of Issues in the Pharmacokinetics and Toxicology of Phosphorothioate Antisense Oligonucleotides. Biochim. Biophys. Acta-Gene Struct. Expr. 1999, 1489, 69–84. [Google Scholar] [CrossRef]
- Lindow, M.; Vornlocher, H.-P.; Riley, D.; Kornbrust, D.J.; Burchard, J.; Whiteley, L.O.; Kamens, J.; Thompson, J.D.; Nochur, S.; Younis, H.; et al. Assessing Unintended Hybridization-Induced Biological Effects of Oligonucleotides. Nat. Biotechnol. 2012, 30, 920–923. [Google Scholar] [CrossRef]
- Hagedorn, P.H.; Yakimov, V.; Ottosen, S.; Kammler, S.; Nielsen, N.F.; Høg, A.M.; Hedtjärn, M.; Meldgaard, M.; Møller, M.R.; Ørum, H.; et al. Hepatotoxic Potential of Therapeutic Oligonucleotides Can Be Predicted from Their Sequence and Modification Pattern. Nucleic Acid Ther. 2013, 23, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Lange, M.J.; Burke, D.H.; Chaput, J.C. Activation of Innate Immune Responses by a CpG Oligonucleotide Sequence Composed Entirely of Threose Nucleic Acid. Nucleic Acid Ther. 2019, 29, 51–59. [Google Scholar] [CrossRef]
- Ouattara, D.A.; Remolue, L.; Becker, J.; Perret, M.; Bunescu, A.; Hennig, K.; Biliaut, E.; Badin, A.; Giacomini, C.; Reynier, F.; et al. An Integrated Transcriptomics and Metabolomics Study of the Immune Response of Newly Hatched Chicks to the Cytosine-Phosphate-Guanine Oligonucleotide Stimulation. Poult. Sci. 2020, 99, 4360–4372. [Google Scholar] [CrossRef]
- Schlee, M.; Hornung, V.; Hartmann, G. SiRNA and IsRNA: Two Edges of One Sword. Mol. Ther. 2006, 14, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.-J.; Jang, H. Anticancer Drug Resistance: An Update and Perspective. Drug Resist. Updates 2021, 59, 100796. [Google Scholar] [CrossRef] [PubMed]
- Feizabadi, M.S. Modeling Multi-Mutation and Drug Resistance: Analysis of Some Case Studies. Theor. Biol. Med. Model. 2017, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]



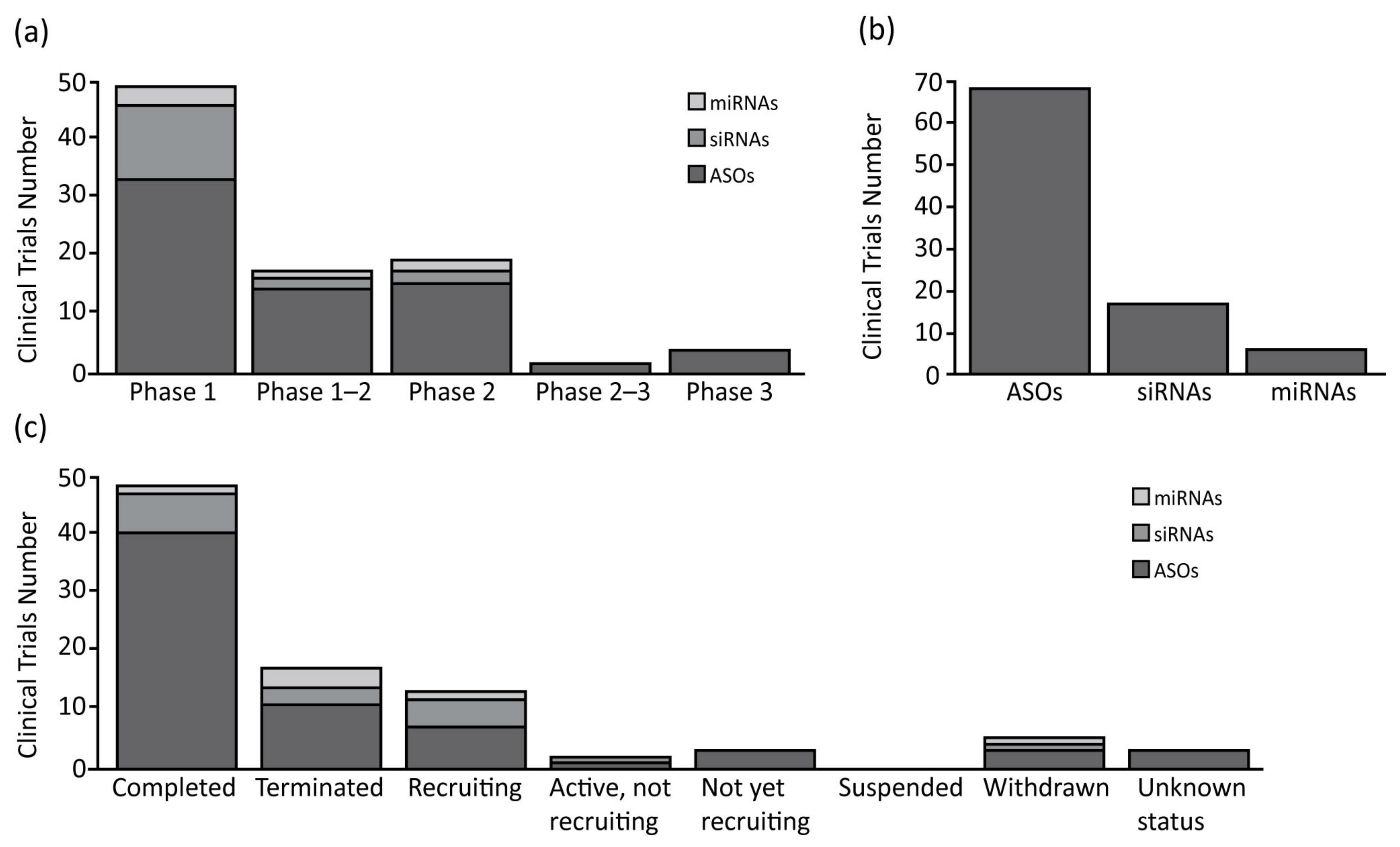{kind=link}
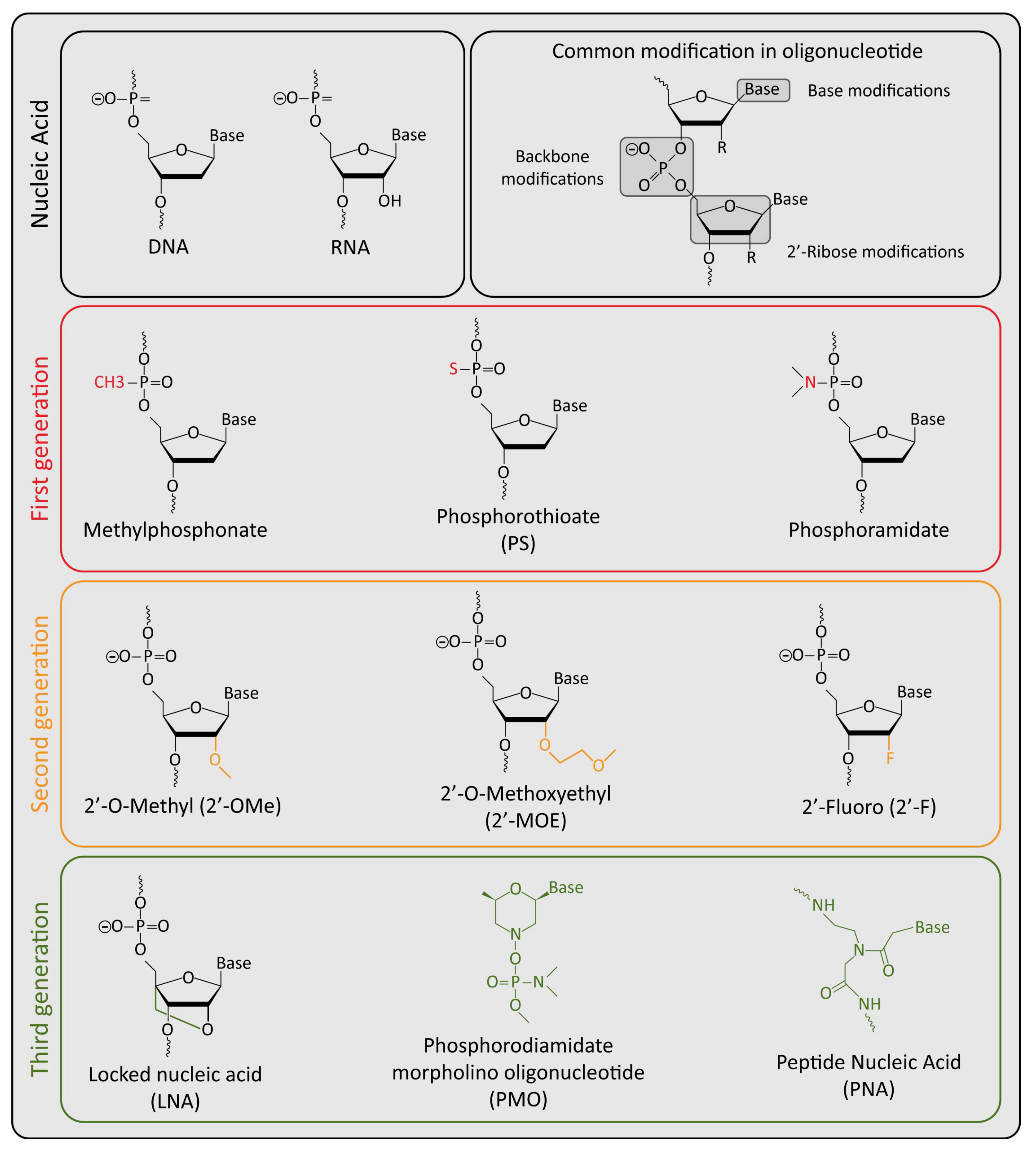{kind=link}
| Oligonucleotide | Target | Drug Type | Cancer Type | Clinical Phase | Clinical Trials ID |
|---|---|---|---|---|---|
| WGI-0301 | AKT1 | ASO | Advanced Solid Tumors | Phase 1 | NCT05267899 |
| AZD5312 | AR | ASO | Advanced Solid Tumors with Androgen Receptor Pathway as a Potential Factor | Phase 1 | NCT02144051 |
| BP1002 (L-Bcl-2 antisense oligonucleotide) | BCL-2 | ASO | Mantle Cell Lymphoma|Peripheral T-cell Lymphoma (PTCL)|Cutaneous T-cell Lymphoma (CTCL)|Chronic Lymphocytic Leukemia (CLL)|Small Lymphocytic Lymphoma (SLL)|Follicular Lymphoma|Marginal Zone Lymphoma|Hodgkin Lymphoma|Waldenstrom Macroglobulinemia|DLBCL | Phase 1 | NCT04072458 |
| PNT2258 | BCL-2 | ASO | Cancer|Lymphoma|Prostate Cancer|Melanoma | Phase 1 | NCT01191775 |
| LErafAON | CRAF | ASO | Neoplasms | Phase 1 | NCT00024648|NCT00024661|NCT00100672 |
| AZD8701 | FOXP3 | ASO | Clear Cell Renal Cell Cancer|Non-Small-Cell Lung Cancer|Triple Negative Breast Neoplasms|Squamous Cell Cancer of Head and Neck|Small-Cell Lung Cancer|Gastroesophageal Cancer|Melanoma|Cervical Cancer|Advanced Solid Tumors | Phase 1 | NCT04504669 |
| BP1001-A (Liposomal Grb2 Antisense Oligonucleotide) | GRB-2 | ASO | Solid Tumor, Adult|Carcinoma, Ovarian Epithelial|Fallopian Tube Neoplasms|Endometrial Cancer|Peritoneal Cancer|Solid Tumor | Phase 1 | NCT04196257 |
| EZN-2968 | HIF-1α | ASO | Neoplasms|Liver Metastases|Carcinoma|Lymphoma | Phase 1 | NCT01120288|NCT00466583 |
| AZD4785 | KRAS | ASO | Non-Small-Cell Lung Cancer|Advanced Solid Tumors | Phase 1 | NCT03101839 |
| TASO-001 | TGF-β2 | ASO | Solid Tumor | Phase 1 | NCT04862767 |
| SD-101 (CpG Oligonucleotide) | TLR9 | ASO | Advanced Malignant Solid Neoplasm|Extracranial Solid Neoplasm|Metastatic Malignant Solid Neoplasm | Phase 1 | NCT03831295 |
| CpG7909 (PF3512676) | TLR9 | ASO | Intraocular Melanoma|Malignant Conjunctival Neoplasm|Melanoma (Skin) | Phase 1 | NCT00471471 |
| ISS 1018 (CpG ODN) | TLR9 | ASO | Colorectal Neoplasms | Phase 1 | NCT00403052 |
| IONIS-STAT3Rx (AZD9150) | STAT3 | ASO | Hepatocellular Carcinoma|Ovarian Cancer|Ascites|Gastrointestinal Cancer|Advanced Cancers|DLBCL|Lymphoma | Phase 1|2 | NCT01839604|NCT02417753|NCT01563302|NCT02549651 |
| ISIS 183750 | eIF4E | ASO | Colorectal Neoplasms|Colorectal Carcinoma|Colorectal Tumors | Phase 1|Phase 2 | NCT01675128 |
| Apatorsen (OGX-427) | HSP-27 | ASO | Squamous Cell Lung Cancer|Bladder Cancer|Urothelial Carcinoma|Prostate Cancer | Phase 1|Phase 2 | NCT02423590|NCT00959868|NCT00487786|NCT01780545|NCT01120470 |
| GTI-2040 | R2 subunit of RNR | ASO | Carcinoma, Renal Cell|Metastases, Neoplasm | Phase 1|Phase 2 | NCT00056173 |
| Aezea (Cenersen) | TP53 | ASO | Myelodysplastic Syndromes|Acute Myelogenous Leukemia | Phase 1|Phase 2 | NCT02243124|NCT00967512 |
| VEGF-Antisense Oligonucleotide | VEGF | ASO | Mesothelioma | Phase 1|Phase 2 | NCT00668499 |
| AEG35156 | XIAP | ASO | Human Mammary Carcinoma|Carcinoma|Pancreas|Non-Small-Cell Lung | Phase 1|Phase 2 | NCT00385775|NCT00558545|NCT00557596|NCT00558922 |
| XIAP Antisense | XIAP | ASO | Leukemia, Myelomonocytic, Acute | Phase 1|Phase 2 | NCT00363974 |
| Oblimersen (G3139) | BCL-2 | ASO | Lymphoma|Prostate Cancer|Lung Cancer|Melanoma (Skin)|Colorectal Cancer|Breast Cancer | Phase 1|Phase 2|Phase 3 | NCT00070083|NCT00080847|NCT00017251|NCT00070343|NCT00016263|NCT00017602|NCT00085228|NCT00030641|NCT00063934|NCT00004870|NCT00005032|NCT00054548|NCT00543231|NCT00543205|NCT00636545|NCT00078234|NCT00021749|NCT00024440|NCT00059813 |
| G4460 (c-myb antisense oligonucleotide) | C-MYB | ASO | Hematologic Malignancies | Phase 2 | NCT00780052|NCT00002592 |
| ISIS 5132 | CRAF | ASO | Breast Cancer | Phase 2 | NCT00003236 |
| BP1001 (Liposomal Grb2 Antisense Oligonucleotide) | GRB-2 | ASO | Recurrent Adult Acute Myeloid Leukemia|Acute Lymphoblastic Leukemia|Myelodysplastic Syndrome|Ph1 Positive CML | Phase 2 | NCT02923986|NCT02781883|NCT01159028 |
| IGV-001 Cell Immunotherapy | IGF type 1 receptor | ASO | Glioblastoma Multiforme|Glioblastoma | Phase 2 | NCT04485949 |
| ISIS 3521 | PKCα | ASO | Breast Cancer | Phase 2 | NCT00003236 |
| STP705 | TGF-β1 and COX-2 | ASO | Squamous Cell Carcinoma in Situ | Phase 2 | NCT04844983 |
| CpG-ODN | TLR9 | ASO | Glioblastoma|Lung Cancer|Hepatocellular Carcinoma|Solid Tumor | Phase 2 | NCT00190424|NCT04952272 |
| Custirsen (OGX-011) | clusterin | ASO | Prostate Cancer|Bladder Cancer|Breast Cancer|Kidney Cancer|Lung Cancer|Ovarian Cancer|Unspecified Adult Solid Tumor | Phase 2|Phase 3 | NCT00054106|NCT00258375|NCT00471432|NCT01083615 |
| INT-1B3 | JNK1 | miRNA | Solid Tumor | Phase 1 | NCT04675996 |
| TargomiRs | Multiple oncogenes, including BCL2, MCL1, CCND1, and WNT3A | miRNA | Malignant Pleural Mesothelioma|Non-Small-Cell Lung Cancer | Phase 1 | NCT02369198 |
| MRX34 | 30 unique oncogenes, including but not limited to MET, MYC, PDGFR-a, CDK4/6 and BCL2 | miRNA | Melanoma | Phase 1|Phase 2 | NCT01829971|NCT02862145 |
| Cobomarsen (MRG-106) | mir-155 | miRNA | Cutaneous T-Cell Lymphoma/Mycosis Fungoides | Phase 2 | NCT03837457|NCT03713320 |
| siRNA-transfected peripheral blood mononuclear cells APN401 | CBLB | siRNA | Metastatic Malignant Neoplasm in the Brain|Metastatic Solid Neoplasm|Recurrent Colorectal Carcinoma|Recurrent Melanoma|Recurrent Pancreatic Cancer|Recurrent Renal Cell Cancer | Phase 1 | NCT03087591|NCT02166255 |
| EphA2-targeting DOPC-encapsulated siRNA | EPHA2 | siRNA | Advanced Malignant Solid Neoplasm | Phase 1 | NCT01591356 |
| NBF-006 | GSTP | siRNA | Non-Small-Cell Lung Cancer|Pancreatic Cancer|Colorectal Cancer | Phase 1 | NCT03819387 |
| Mesenchymal Stromal Cells-derived Exosomes with KRAS G12D siRNA | KRASG12D | siRNA | Metastatic Pancreatic Adenocarcinoma|Pancreatic Ductal Adenocarcinoma | Phase 1 | NCT03608631 |
| Proteasome siRNA and tumor antigen RNA-transfected dendritic cells | LMP2, LMP7, MECL1 | siRNA | Metastatic Melanoma|Absence of CNS Metastases | Phase 1 | NCT00672542 |
| CALAA-01 | M2 subunit of ribonucleotide reductase (R2) | siRNA | Cancer|Solid Tumor | Phase 1 | NCT00689065 |
| TKM-080301 | PLK1 | siRNA | Colorectal Cancer with Hepatic Metastases|Pancreas Cancer with Hepatic Metastase|Gastric Cancer With Hepatic Metastases|Breast Cancer With Hepatic | Phase 1 | NCT01437007 |
| SLN124 | TMPRSS6 | siRNA | Non-transfusion-dependent Thalassemia|Low Risk Myelodysplastic Syndrome | Phase 1 | NCT04176653 |
| DCR-MYC | MYC | siRNA | Solid Tumors|Multiple Myeloma|Non-Hodgkins Lymphoma|Pancreatic Neuroendocrine Tumors|PNET|NHL| Hepatocellular Carcinoma | Phase 1|Phase 2 | NCT02110563|NCT02314052 |
| Atu027 | PNK3 | siRNA | Advanced Solid Tumors|Carcinoma, Pancreatic Ductal | Phase 1|Phase 2 | NCT00938574|NCT01808638 |
| siG12D LODER | KRASG12D | siRNA | Pancreatic Ductal Adenocarcinoma|Pancreatic Cancer | Phase 2 | NCT01188785|NCT01676259 |
| STP705 | TGF-β1, COX-2 mRNA | siRNA | Squamous Cell Carcinoma in Situ | Phase 2 | NCT04844983 |
| CpG-STAT3 siRNA CAS3/SS3 | TLR9 and STAT3 | siRNA | B-Cell Non-Hodgkin Lymphoma|Diffuse Large B-Cell Lymphoma|Follicular Lymphoma|Mantle Cell Lymphoma|Marginal Zone Lymphoma|Small Lymphocytic Lymphoma | Phase 1 | NCT04995536 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartolucci, D.; Pession, A.; Hrelia, P.; Tonelli, R. Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs. Pharmaceutics 2022, 14, 1453. https://doi.org/10.3390/pharmaceutics14071453
Bartolucci D, Pession A, Hrelia P, Tonelli R. Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs. Pharmaceutics. 2022; 14(7):1453. https://doi.org/10.3390/pharmaceutics14071453
Chicago/Turabian StyleBartolucci, Damiano, Andrea Pession, Patrizia Hrelia, and Roberto Tonelli. 2022. "Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs" Pharmaceutics 14, no. 7: 1453. https://doi.org/10.3390/pharmaceutics14071453
APA StyleBartolucci, D., Pession, A., Hrelia, P., & Tonelli, R. (2022). Precision Anti-Cancer Medicines by Oligonucleotide Therapeutics in Clinical Research Targeting Undruggable Proteins and Non-Coding RNAs. Pharmaceutics, 14(7), 1453. https://doi.org/10.3390/pharmaceutics14071453