
Quality Control Dissolution Data Is Biopredictive for a Modified Release Ropinirole Formulation: Virtual Experiment with the Use of Re-Developed and Verified PBPK Model




Abstract
:1. Introduction
2. Materials and Methods
2.1. PBPK Model Development
2.2. Description of Distribution and Elimination
2.3. Description of the Absorption
- : Dissolution rate
- : Transit time in the n-th segment of the small intestine
- : Amount of solid mass trapped in the formulation and not available for dissolution immediately
- : Drug degradation rate constant (in lumen)
- : Absorption rate constant
- : The unit adjustment factor for the drug transported out of the enterocyte
- : Efflux clearance from the enterocyte
- : Metabolic clearance within the enterocyte
- : Fraction of the drug unbound in the enterocyte
- : Drug concentration in the enterocyte
- : The volume of enterocytes in the segment
- : Blood flow to the intestinal segment
2.4. Prolonged-Release Formulation
2.5. Dissolution Data
2.6. Intestinal Permeability
2.7. Fraction of Drug Unbound in the Enterocytes (fugut)
2.8. Intestinal Metabolism
- The release from the formulation is the only rate-limiting factor in the ropinirole’s absorption.
- In vitro dissolution data, obtained by the quality control method, is biopredictive as for ropinirole’s behavior in the human GIT.
- There is no drug degradation in the intestinal lumen.
- The intestinal absorption is based on passive diffusion, and there are no efflux processes taking place at the intestinal wall.
- Ropinirole intestinal metabolism is negligible.
- The formulation provides stable active substance release in the Parkinson’s patients GIT environment.
2.9. Model Verification and Application
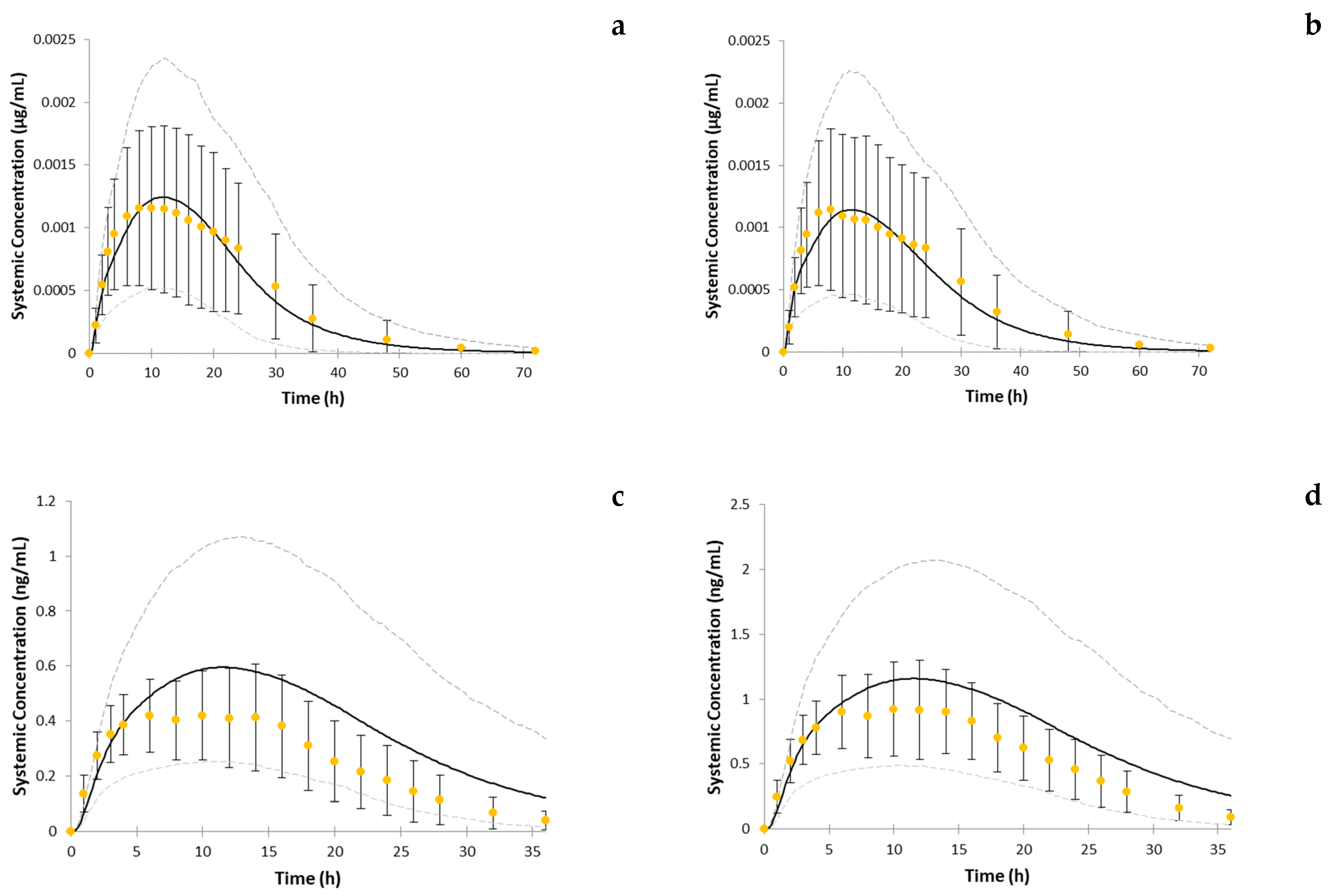
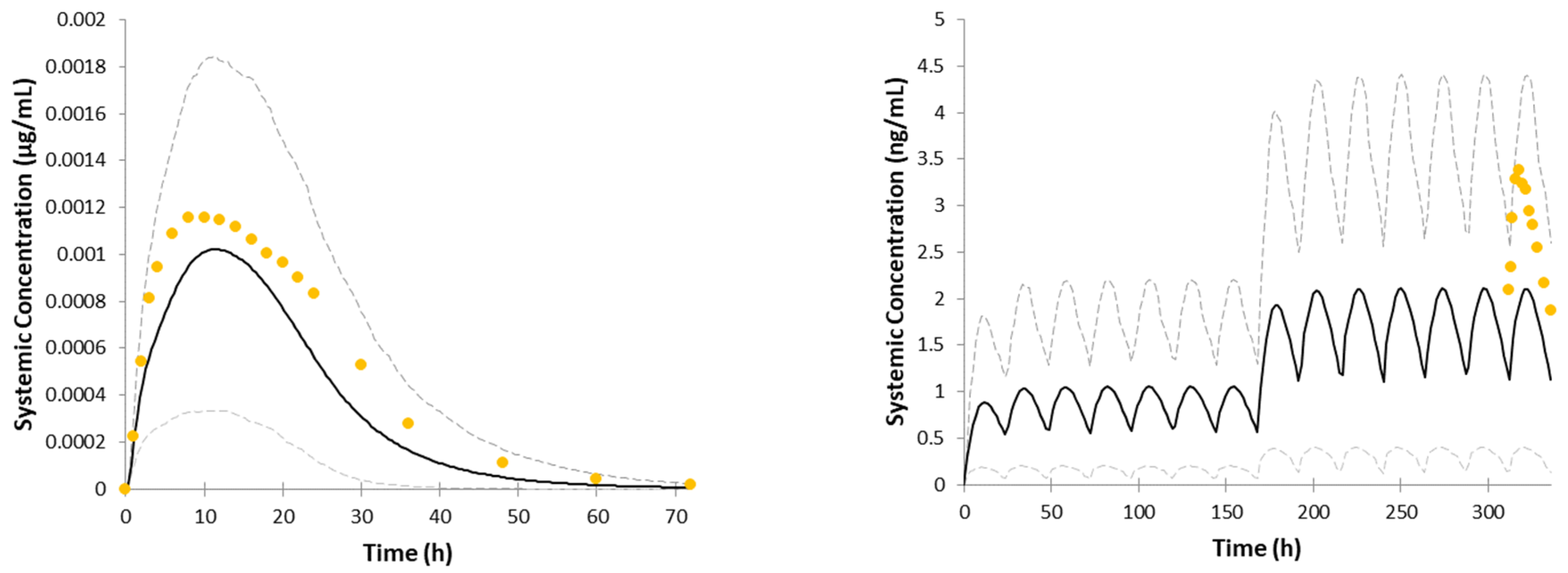
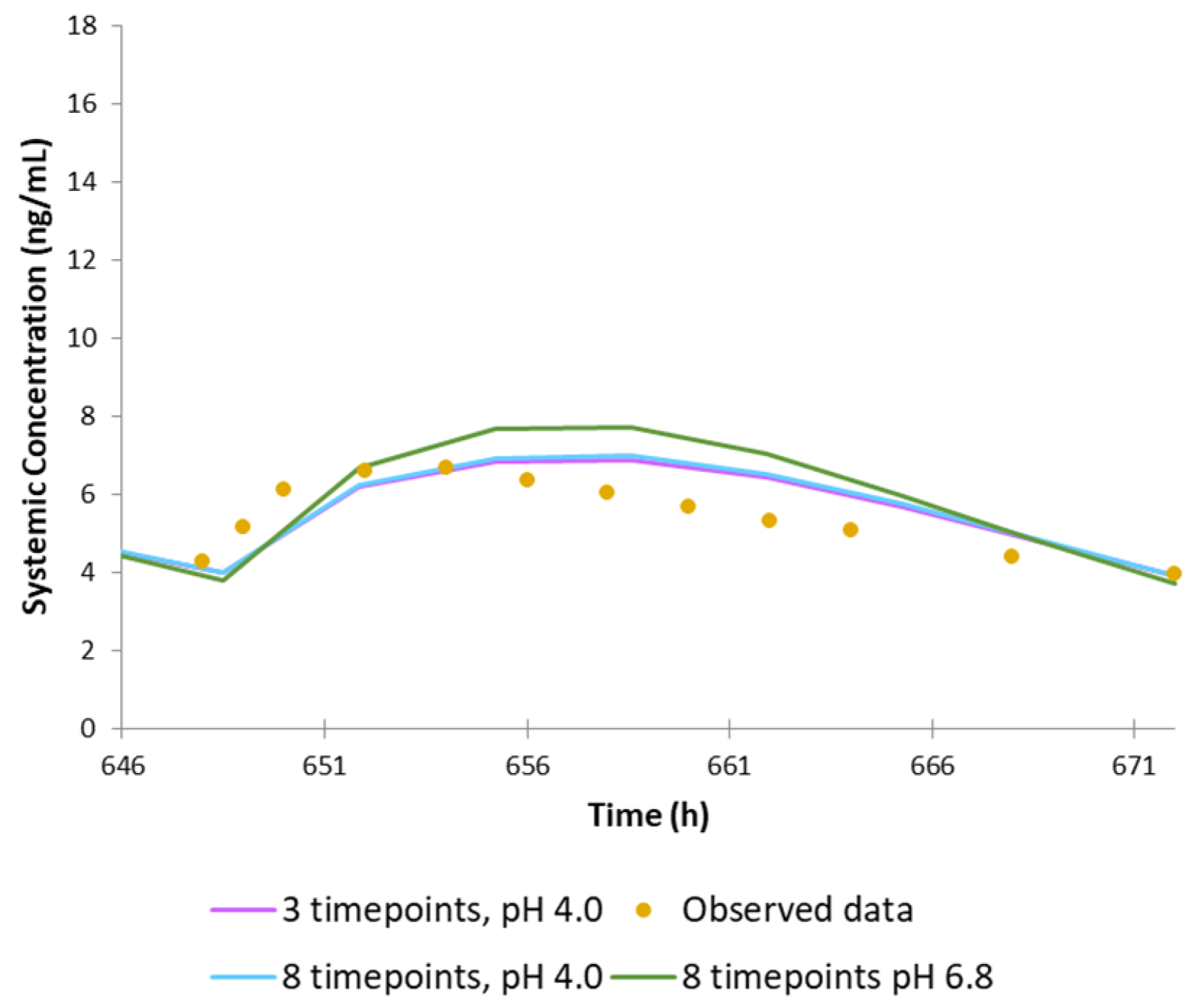
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Nutt, J.G.; Ransom, B.R. Parkinson’s disease. Lancet 2004, 363, 1783–1793. [Google Scholar] [CrossRef] [Green Version]
- Homayoun, H. Parkinson Disease. Ann. Intern. Med. 2018, 169, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Contin, M.; Riva, R.; Albani, F.; Baruzzi, A. Pharmacokinetic optimisation in the treatment of Parkinson’s disease. Clin. Pharm. 1996, 30, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Nyholm, D. Pharmacokinetic optimisation in the treatment of Parkinson’s disease: An update. Clin. Pharm. 2006, 45, 109–136. [Google Scholar] [CrossRef] [PubMed]
- Dobson, A.M.; Cuellar, N. Use of ropinirole in RLS. Nurse. Pract. 2006, 31, 46–48. [Google Scholar] [CrossRef]
- Latt, M.D.; Lewis, S.; Zekry, O.; Fung, V.S.C. Factors to Consider in the Selection of Dopamine Agonists for Older Persons with Parkinson’s Disease. Drugs Aging 2019, 36, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Avoidance of dyskinesia: Preclinical evidence for continuous dopaminergic stimulation. Neurology 2004, 62, S47–S55. [Google Scholar] [CrossRef]
- Drug Approval Package: Requip XL (Ropinirole) NDA #022008. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2008/022008_requip_toc.cfm (accessed on 19 April 2021).
- Stillhart, C.; Vučićević, K.; Augustijns, P.; Basit, A.W.; Batchelor, H.; Flanagan, T.R.; Gesquiere, I.; Greupink, R.; Keszthelyi, D.; Koskinen, M.; et al. Impact of gastrointestinal physiology on drug absorption in special populations—An UNGAP review. Eur. J. Pharm. Sci. 2020, 147, 105280. [Google Scholar] [CrossRef]
- Wollmer, E.; Klein, S. A review of patient-specific gastrointestinal parameters as a platform for developing in vitro models for predicting the in vivo performance of oral dosage forms in patients with Parkinson’s disease. Int. J. Pharm. 2017, 533, 298–314. [Google Scholar] [CrossRef]
- Available online: http://getdata-graph-digitizer.com (accessed on 20 July 2020).
- Shuklinova, O. Development of Physiologically Based Pharmacokinetic Model for the Immediate Release Ropinirole Tablets—Acta Poloniae Pharmaceutica—Drug Research—Czasopisma Naukowe. Available online: https://ptfarm.pl/wydawnictwa/czasopisma/acta-poloniae-pharmaceutica/110/-/29108 (accessed on 12 October 2021).
- Adlard, M.; Okafo, G.; Meenan, E.; Camilleri, P. Rapid estimation of octanol–water partition coefficients using deoxycholate micelles in capillary electrophoresis. J. Chem. Soc. Chem. Commun. 1995, 21, 2241–2243. [Google Scholar] [CrossRef]
- Coufal, P.; Stulík, K.; Claessens, H.A.; Hardy, M.J.; Webb, M. Determination of the dissociation constants of ropinirole and some impurities and their quantification using capillary zone electrophoresis. J. Chromatogr. B Biomed Sci. Appl. 1998, 720, 197–204. [Google Scholar] [CrossRef]
- Swagzdis, J.E.; Wittendorf, R.W.; DeMarinis, R.M.; Mico, B.A. Pharmacokinetics of dopamine-2 agonists in rats and dogs. J. Pharm. Sci. 1986, 75, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Yamamoto, S.; Sano, N.; Tohyama, K.; Kosugi, Y.; Furuta, A.; Hamada, T.; Igari, T.; Fujioka, Y.; Hirabayashi, H.; et al. Direct Drug Delivery of Low-Permeable Compounds to the Central Nervous System Via Intranasal Administration in Rats and Monkeys. Pharm. Res. 2019, 36, 76. [Google Scholar] [CrossRef]
- GSK—A Study Conducted in Healthy Subjects to Demonstrate Bioequivalence between Ropinirole Prolonged Release Tablets Manufactured at Crawley and Aranda. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=112771 (accessed on 6 December 2021).
- GSK—An Open Label, Randomised, Five-Way Crossover Single-Dose Pharmacokinetic Study to Assess Dosage Strength Equivalence of Ropinirole CR in Healthy Male and Female Volunteers. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/219 (accessed on 6 December 2021).
- GSK—An Open-Label, Up-Titration Study to Assess the Dose Proportionality of Ropinirole Controlled Release (CR) and to Demonstrate the Bioequivalence of Ropinirole CR (1 × 8 mg) Compared to the Ropinirole CR (4 × 2 mg) in Parkinson’s Disease Patients Not Receiving Other Dopaminergic Therapies. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/165 (accessed on 6 December 2021).
- GSK—An Open Study to Investigate the Effect of Food on the Pharmacokinetics of Ropinirole from a CR Formulation in Healthy Male Volunteers. [Type ??]. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/164 (accessed on 6 December 2021).
- GSK—Parkinson’s Disease Patient Study on Absorption, Distribution, Metabolism and Excretion of Ropinirole. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=ROP109087 (accessed on 6 December 2021).
- Bloomer, J.C.; Clarke, S.E.; Chenery, R.J. In vitro identification of the P450 enzymes responsible for the metabolism of ropinirole. Drug Metab. Dispos. 1997, 25, 840–844. [Google Scholar] [PubMed]
- GSK—An Open Study to Compare the PK and Tolerability of Ropinirole Administered as 5 Different New Formulations with the Standard, Marketed Formulation in Healthy Volunteers. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/197 (accessed on 19 April 2021).
- GSK—An Open Study to Investigate the Tolerance and Preliminary Pharmacokinetics of Single Intravenous Doses of 100, 200, 400, 600 and 800 mg SK&F 101468 Following Domperidone Pre-Treatment (20 mg t.i.d.) in Healthy Male Volunteers. Available online: https://www.gsk-studyregister.com/en/trial-details/?id=101468/009 (accessed on 19 April 2021).
- Kaye, C.M.; Nicholls, B. Clinical pharmacokinetics of ropinirole. Clin. Pharm. 2000, 39, 243–254. [Google Scholar] [CrossRef]
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Conte, U.; Maggi, L. Modulation of the dissolution profiles from Geomatrix multi-layer matrix tablets containing drugs of different solubility. Biomaterials 1996, 17, 889–896. [Google Scholar] [CrossRef]
- Available online: https://www.gsk-studyregister.com (accessed on 20 July 2020).
- Fujikawa, M.; Ano, R.; Nakao, K.; Shimizu, R.; Akamatsu, M. Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: Application to prediction of Caco-2 cell permeability. Bioorg. Med. Chem. 2005, 13, 4721–4732. [Google Scholar] [CrossRef]
- Sun, D.; Lennernas, H.; Welage, L.S.; Barnett, J.L.; Landowski, C.P.; Foster, D.; Fleisher, D.; Lee, K.-D.; Amidon, G.L. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm. Res. 2002, 19, 1400–1416. [Google Scholar] [CrossRef]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Jamei, M.; Marciniak, S.; Feng, K.; Barnett, A.; Tucker, G.; Rostami-Hodjegan, A. The Simcyp population-based ADME simulator. Expert. Opin. Drug Metab. Toxicol. 2009, 5, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Howgate, E.M.; Rowland Yeo, K.; Proctor, N.J.; Tucker, G.T.; Rostami-Hodjegan, A. Prediction of in vivo drug clearance from in vitro data. I: Impact of inter-individual variability. Xenobiotica 2006, 36, 473–497. [Google Scholar] [CrossRef]
- de Mey, C.; Enterling, D.; Meineke, I.; Yeulet, S. Interactions between domperidone and ropinirole, a novel dopamine D2-receptor agonist. Br. J. Clin. Pharmacol. 1991, 32, 483–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetty, M.; Johnson, T.N.; Polak, S.; Salem, F.; Doki, K.; Rostami-Hodjegan, A. Physiologically based pharmacokinetic modelling to guide drug delivery in older people. Adv. Drug Deliv. Rev. 2018, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Grimstein, M.; Fan, J.; Grillo, J.A.; Huang, S.-M.; Zhu, H.; Wang, Y. Application of PBPK Modeling and Simulation for Regulatory Decision Making and Its Impact on US Prescribing Information: An Update on the 2018-2019 Submissions to the US FDA’s Office of Clinical Pharmacology. J. Clin. Pharmacol. 2020, 60 (Suppl. S1), S160–S178. [Google Scholar] [CrossRef]
- Wagner, C.; Zhao, P.; Pan, Y.; Hsu, V.; Grillo, J.; Huang, S.M.; Sinha, V. Application of Physiologically Based Pharmacokinetic (PBPK) Modeling to Support Dose Selection: Report of an FDA Public Workshop on PBPK. CPT Pharmacomet. Syst. Pharm. 2015, 4, 226–230. [Google Scholar] [CrossRef]
- Wu, F.; Shah, H.; Li, M.; Duan, P.; Zhao, P.; Suarez, S.; Raines, K.; Zhao, Y.; Wang, M.; Lin, H.; et al. Biopharmaceutics Applications of Physiologically Based Pharmacokinetic Absorption Modeling and Simulation in Regulatory Submissions to the U.S. Food and Drug Administration for New Drugs. AAPS J. 2021, 23, 1–14. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Value | Reference |
|---|---|---|---|
| Ropinirole physicochemical properties and blood binding | |||
| MW (g/mol) | Molecular weight | 260.38 | Chemicalize.com |
| LogPo:w | Neutral species octanol: buffer partition coefficient | 2.7 | [14] |
| Type of the compound | Monoprotic base | ||
| pKa | Dissociation constant | 9.79 | [15] |
| B/P | Blood-to-plasma partition ratio | 1.09 | [16] |
| fu | Fraction unbound in plasma | 0.68 | [16] |
| Absorption model | Advanced Dissolution, Absorption, and Metabolism (ADAM) model | ||
| fa | Fraction available from a dosage form | 0.99 | Simcyp® predicted |
| ka (h−1) | First-order absorption rate constant | 2.19 | Simcyp® predicted |
| Papp (PAMPA, 10−6 cm/s) | Apparent permeability in PAMPA | 26.8 | [17] |
| Peff, man (10−4 cm/s) | Effective human jejunum permeability | 5.01 | Simcyp® predicted |
| Weibull fit parameters | alpha | 33.70 | Study 112771, Aranda site [18] |
| beta | 1.33 | ||
| alpha | 28.95 | Study 112771, Crawley site [18] | |
| beta | 1.23 | ||
| alpha | 33.66 | Study 101468/219, 1 mg [19] | |
| beta | 1.34 | ||
| alpha | 30.86 | Study 101468/219, 2 mg [19] | |
| beta | 1.29 | ||
| alpha | 29.57 | Study 101468/219, 3 mg [19] | |
| beta | 1.20 | ||
| alpha | 33.71 | Study 101468/165, 2 mg [20] | |
| beta | 1.35 | ||
| alpha | 24.12 | Study 101468/165, 4 mg [20] | |
| beta | 1.22 | ||
| alpha | 34.02 | Study 101468/165, 8 mg [20] | |
| beta | 1.37 | ||
| alpha | 17.14 | Study101468/164 [21] | |
| beta | 1.08 | ||
| alpha | 38.44 | Study ROP109087, 4 mg [22] | |
| beta | 1.35 | ||
| alpha | 26.55 | Study ROP109087, 8 mg [22] | |
| beta | 1.22 | ||
| alpha | 22.24 | Study ROP109087, 12 mg [22] | |
| beta | 1.19 | ||
| Distribution Model | Full PBPK | ||
| Vss (L/kg) | Volume of distribution at steady state | 3.37 | Simcyp® predicted Method 2 |
| Elimination | |||
| Enzyme kinetic parameters for IVIVE | |||
| N-despropylation | Enzyme | Value | |
| Vmax (nmol/h/mg) | CYP1A2 | 7.83 | [23] |
| Km (µmol) | CYP1A2 | 34.63 | |
| Vmax (nmol/h/mg) | CYP3A4 | 523.33 | |
| Km (µmol) | CYP3A4 | 2700.00 | |
| Hydroxylation | |||
| Vmax (nmol/h/mg) | CYP1A2 | 6.93 | |
| Km (µmol) | CYP1A2 | 45.87 | |
| Vmax (nmol/h/mg) | CYP3A4 | 255.33 | |
| Km (µmol) | CYP3A4 | 3933.33 | |
| fumic | Fraction unbound in an in vitro microsomal preparation | 0.39 | Estimated based on dataset from Study No. 101468/197 [24] |
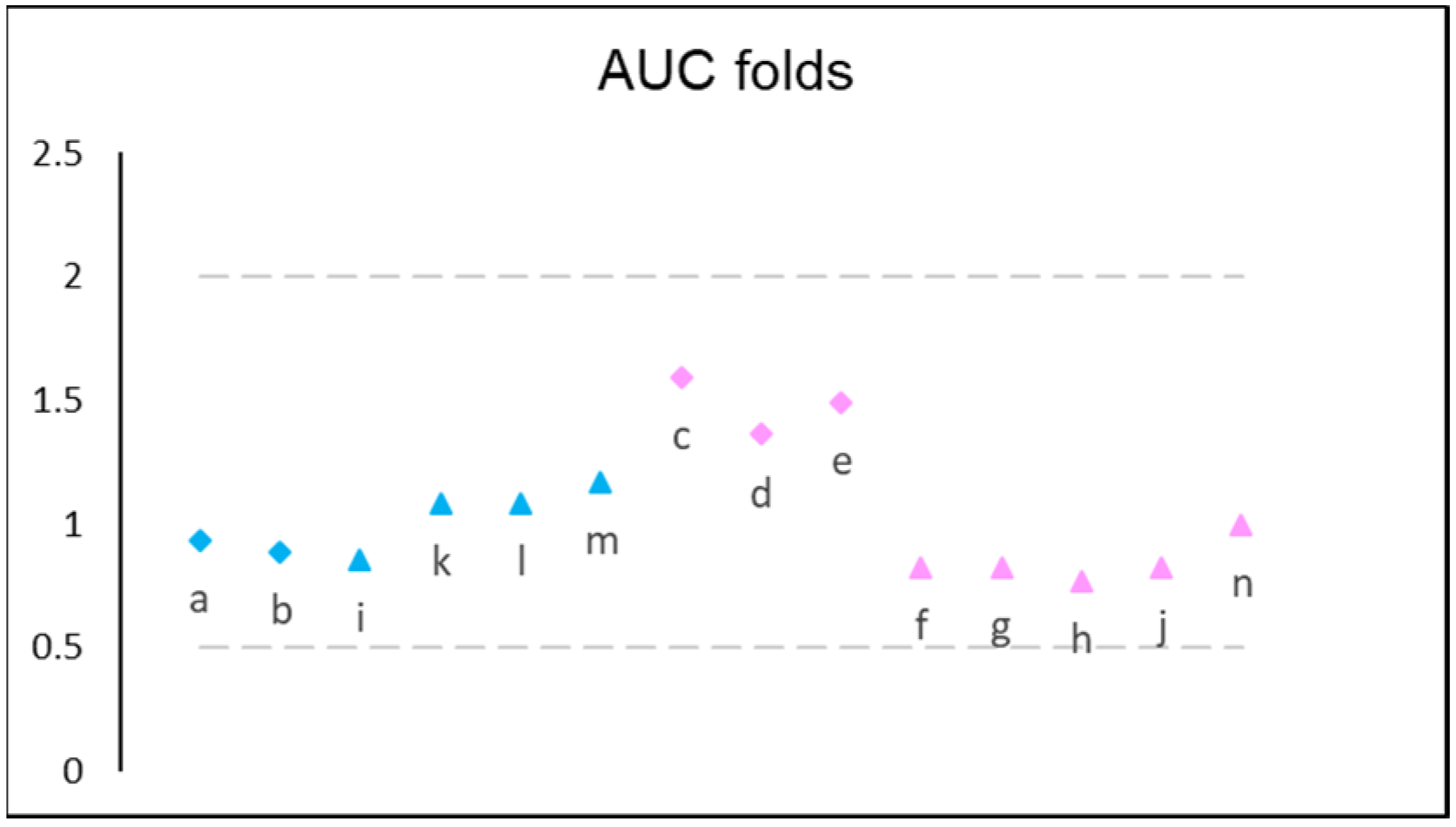
| Study ID * and Reference | Simulation ID | Clinical Study Population | Virtual Population | Subject Age | n of Subjects | PK Assessment Dose, mg | Dosing Regimen | Prandial State |
|---|---|---|---|---|---|---|---|---|
| 112771 [18] | a (Aranda site) b (Crawley site) | HV | Sim-Healthy Volunteers | 18–50 | 50 | 2 | QD | Fasted |
| 101468/219 [19] | c | HV | Sim-Healthy Volunteers | 18–44 | 31–33 | 1 2 3 | QD QD QD | Fed |
| d | ||||||||
| e | ||||||||
| 101468/165 [20] | f | PARKD | Sim-NEurCaucasian | 47–87 | 25 | 2 | QD × 7 days ** | |
| g | 4 | 2 mg QD × 7 days, 4 mg QD × 7 days ** | ||||||
| h | 8 | 2 mg QD × 7 days 4 mg QD × 7 days 6 mg QD × 7 days 8 mg QD × 7 days ** | ||||||
| 101468/164 [21] | i | PARKD | Sim-NEurCaucasian | 34–80 | 21 | 8 | 2 mg QD × 7 days 4 mg QD × 7 days 6 mg QD × 7 days 8 mg QD × 7 days ** | Fasted |
| j | Fed | |||||||
| ROP109087 [22] | k | PARKD | Sim-NEurCaucasian | 47–81 | 27 | 4 | 2 mg QD × 7 days 4 mg QD × 7 days ** | Fasted |
| l | 8 | 2 mg QD × 7 days 4 mg QD × 7 days 6 mg QD × 7 days 8 mg QD × 7 days ** | ||||||
| m | 12 | 2 mg QD × 7 days, 4 mg QD × 7 days 6 mg QD × 7 days 8 mg QD × 7 days 12 mg QD × 7 days ** | Fasted | |||||
| n | Fed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuklinova, O.; Dorożyński, P.; Kulinowski, P.; Polak, S. Quality Control Dissolution Data Is Biopredictive for a Modified Release Ropinirole Formulation: Virtual Experiment with the Use of Re-Developed and Verified PBPK Model. Pharmaceutics 2022, 14, 1514. https://doi.org/10.3390/pharmaceutics14071514
Shuklinova O, Dorożyński P, Kulinowski P, Polak S. Quality Control Dissolution Data Is Biopredictive for a Modified Release Ropinirole Formulation: Virtual Experiment with the Use of Re-Developed and Verified PBPK Model. Pharmaceutics. 2022; 14(7):1514. https://doi.org/10.3390/pharmaceutics14071514
Chicago/Turabian StyleShuklinova, Olha, Przemysław Dorożyński, Piotr Kulinowski, and Sebastian Polak. 2022. "Quality Control Dissolution Data Is Biopredictive for a Modified Release Ropinirole Formulation: Virtual Experiment with the Use of Re-Developed and Verified PBPK Model" Pharmaceutics 14, no. 7: 1514. https://doi.org/10.3390/pharmaceutics14071514
APA StyleShuklinova, O., Dorożyński, P., Kulinowski, P., & Polak, S. (2022). Quality Control Dissolution Data Is Biopredictive for a Modified Release Ropinirole Formulation: Virtual Experiment with the Use of Re-Developed and Verified PBPK Model. Pharmaceutics, 14(7), 1514. https://doi.org/10.3390/pharmaceutics14071514