
The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates

 , , ,
, , ,
Abstract
:1. Introduction
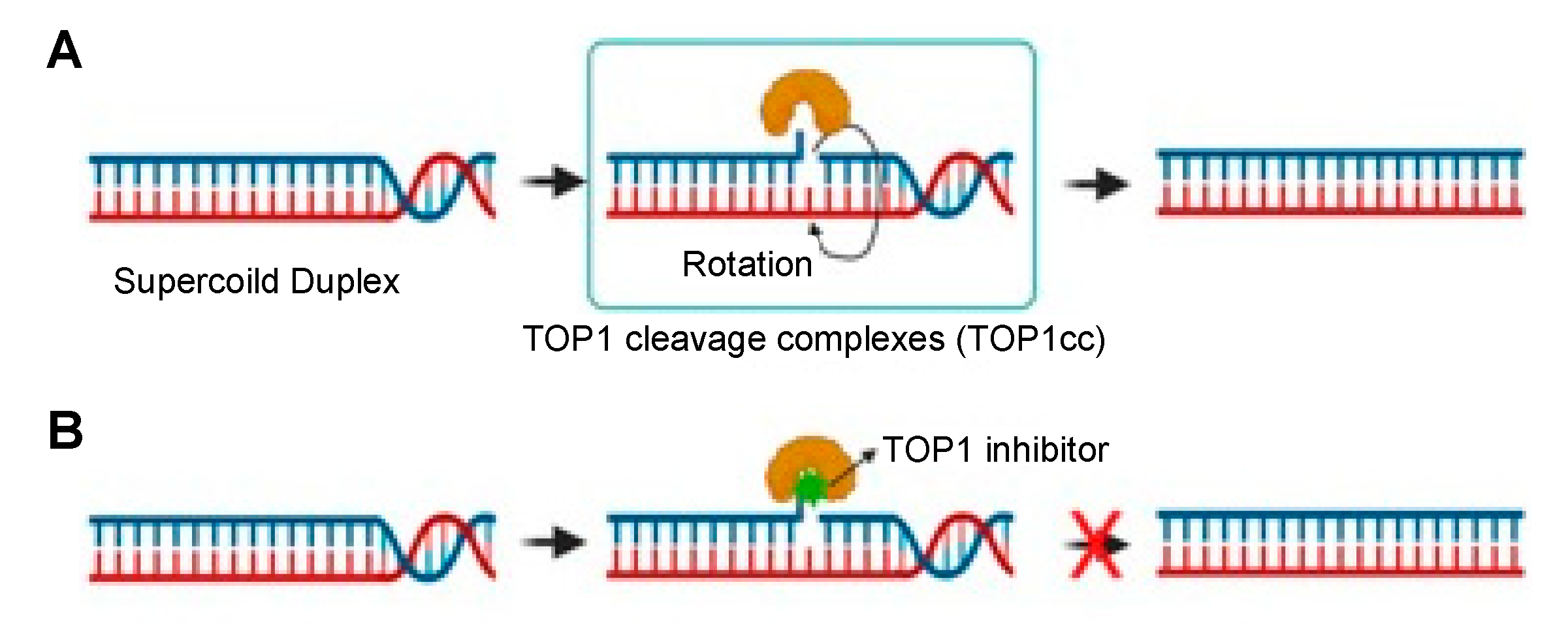
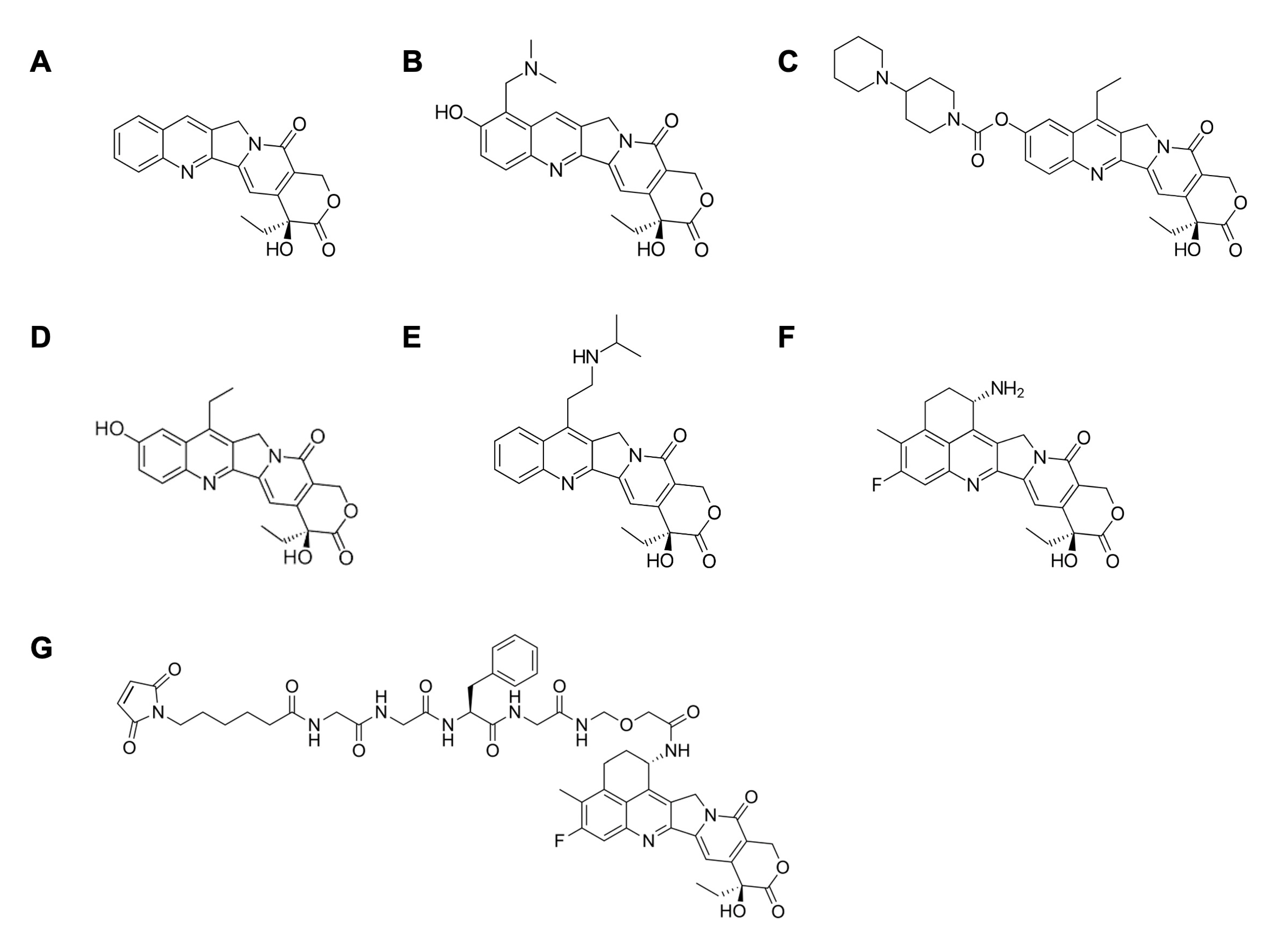
2. TOP1 Inhibitors in Cancer Therapy
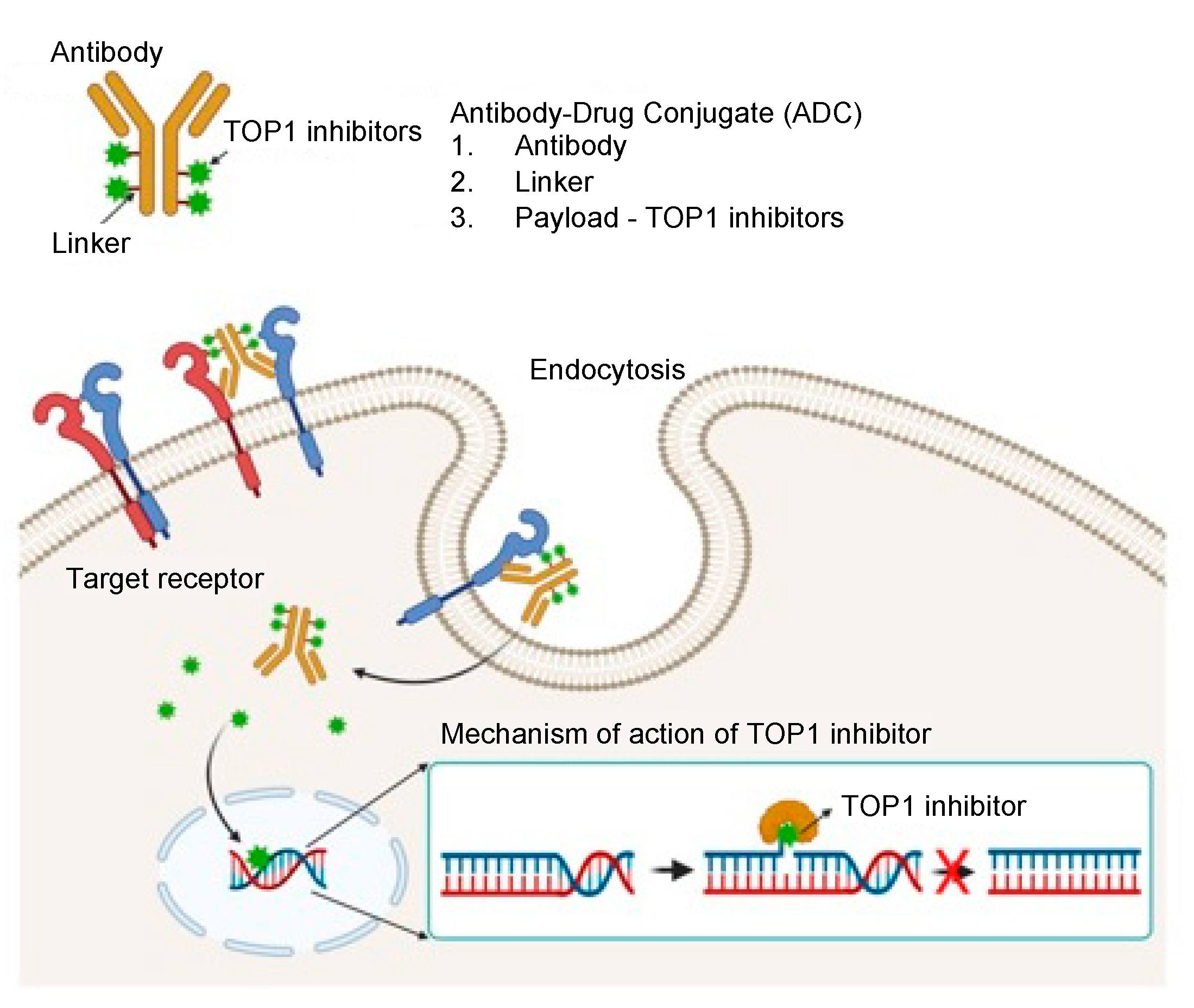
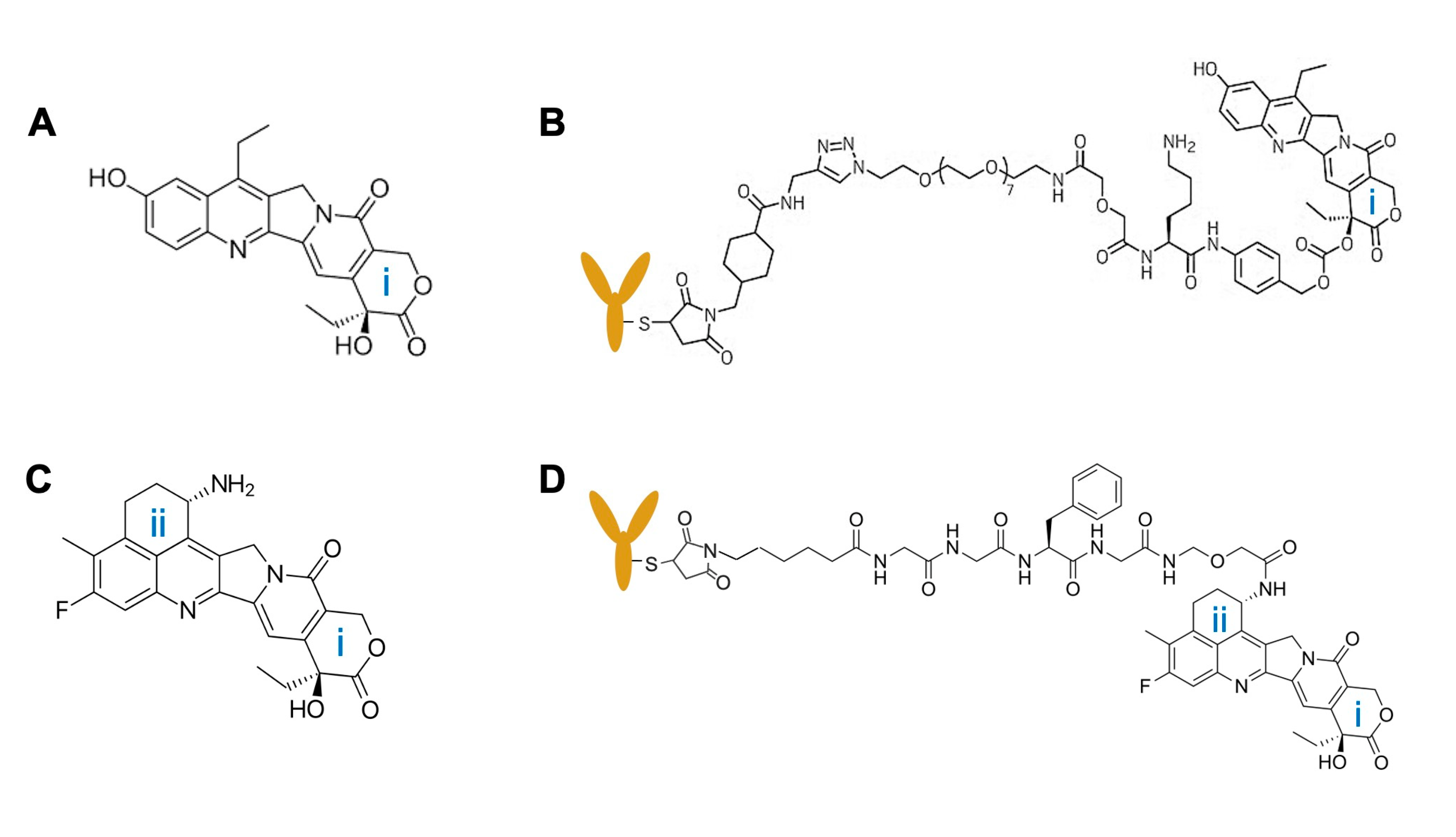
3. Importance of Targeted Delivery of TOP1 Inhibitors: TOP1-ADC
4. Optimization Strategies for TOP1-ADC
4.1. Selection of Antibody Type and Target Antigen
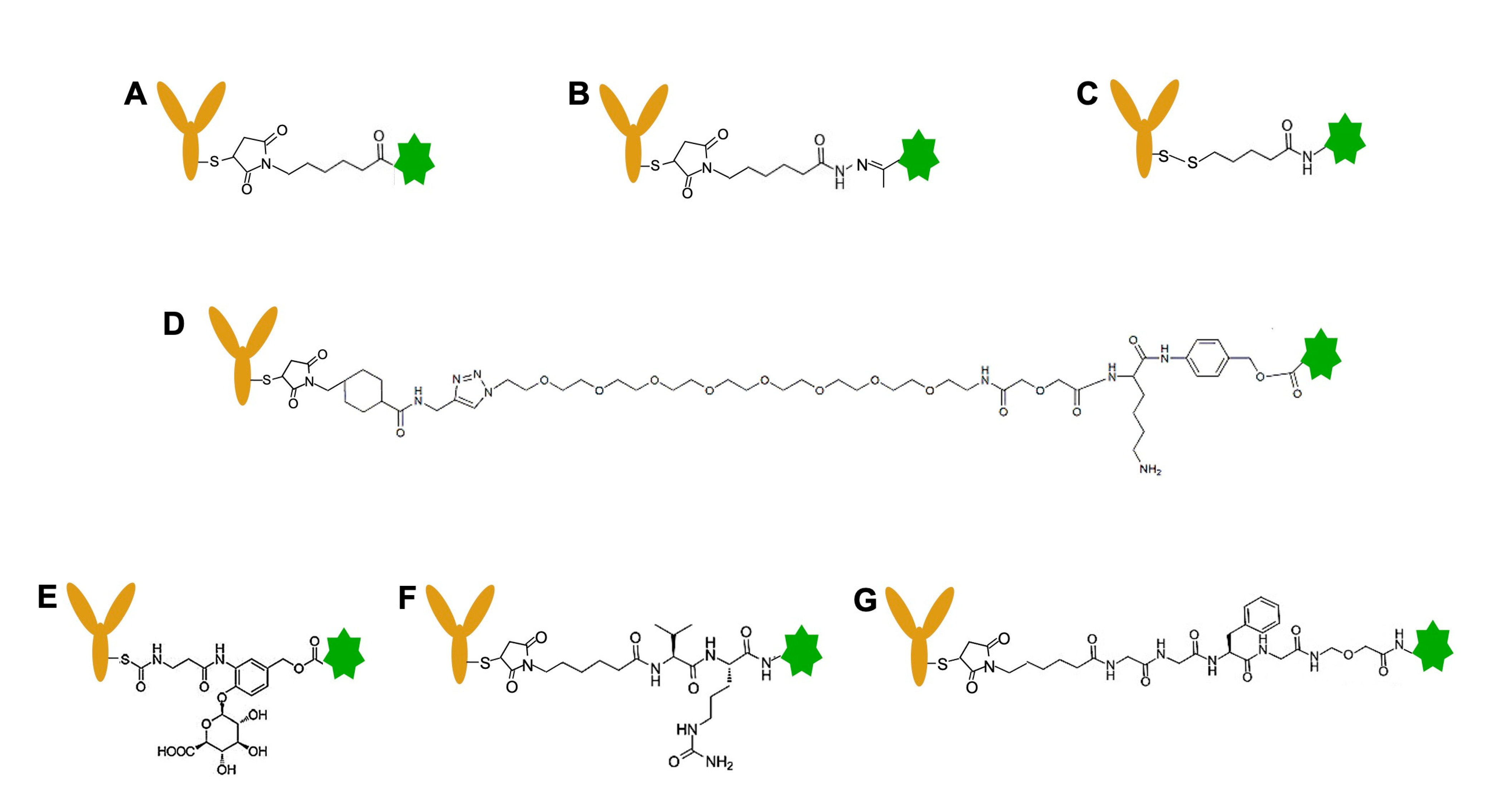
4.2. Selection of Linkers
4.3. Optimization of Drug-to-Antibody Ratio (DAR)
4.4. Linkage Strategy to Generate TOP1-ADC
5. TOP1-ADC in Clinical Trial/FDA Approval
6. Suggestions for Improving Therapy with TOP1-ADC
6.1. DNA Damage Response Modulators
6.2. Immunotherapy
6.3. Establish Assays for Monitoring Topoisomerase Activity
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takahashi, D.T.; Gadelle, D.; Agama, K.; Kiselev, E.; Zhang, H.; Yab, E.; Petrella, S.; Forterre, P.; Pommier, Y.; Mayer, C. Topoisomerase I (TOP1) dynamics: Conformational transition from open to closed states. Nat. Commun. 2022, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Bjornsti, M.A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Research 2019, 8, 1704. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef]
- Ma, J.; Wang, M.D. DNA supercoiling during transcription. Biophys. Rev. 2016, 8, 75–87. [Google Scholar] [CrossRef]
- Manzo, S.G.; Hartono, S.R.; Sanz, L.A.; Marinello, J.; De Biasi, S.; Cossarizza, A.; Capranico, G.; Chedin, F. DNA Topoisomerase I differentially modulates R-loops across the human genome. Genome Biol. 2018, 19, 100. [Google Scholar] [CrossRef]
- Tesauro, C.; Simonsen, A.K.; Andersen, M.B.; Petersen, K.W.; Kristoffersen, E.L.; Algreen, L.; Hansen, N.Y.; Andersen, A.B.; Jakobsen, A.K.; Stougaard, M.; et al. Topoisomerase I activity and sensitivity to camptothecin in breast cancer-derived cells: A comparative study. BMC Cancer 2019, 19, 1158. [Google Scholar] [CrossRef]
- Riemsma, R.; Simons, J.P.; Bashir, Z.; Gooch, C.L.; Kleijnen, J. Systematic Review of topotecan (Hycamtin) in relapsed small cell lung cancer. BMC Cancer 2010, 10, 436. [Google Scholar] [CrossRef]
- De Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin. Pharm. 2018, 57, 1229–1254. [Google Scholar] [CrossRef]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Fu, Y.; Ho, M. DNA damaging agent-based antibody-drug conjugates for cancer therapy. Antib. Ther. 2018, 1, 33–43. [Google Scholar] [CrossRef]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef]
- Pommier, Y.; Kiselev, E.; Marchand, C. Interfacial inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3961–3965. [Google Scholar] [CrossRef]
- Lee, D.H.; Kim, S.-W.; Suh, C.; Lee, J.-S.; Lee, J.H.; Lee, S.-J.; Ryoo, B.Y.; Park, K.; Kim, J.S.; Heo, D.S.; et al. Belotecan, new camptothecin analogue, is active in patients with small-cell lung cancer: Results of a multicenter early phase II study. Ann. Oncol. 2008, 19, 123–127. [Google Scholar] [CrossRef]
- Hevener, K.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef]
- Topoisomerase inhibitors. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson, J.K. (Ed.) Elsevier: Oxford, UK, 2016; pp. 72–85. [Google Scholar]
- Kumazawa, E.; Jimbo, T.; Ochi, Y.; Tohgo, A. Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice. Cancer Chemother. Pharmacol. 1998, 42, 210–220. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, F. Combating P-glycoprotein-mediated multidrug resistance using therapeutic nanoparticles. Curr. Pharm. Des. 2013, 19, 6655–6666. [Google Scholar] [CrossRef]
- Li, W.; Veale, K.H.; Qiu, Q.; Sinkevicius, K.W.; Maloney, E.K.; Costoplus, J.A.; Lau, J.; Evans, H.L.; Setiady, Y.; Ab, O.; et al. Synthesis and Evaluation of Camptothecin Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2019, 10, 1386–1392. [Google Scholar] [CrossRef]
- Ogitani, Y.; Abe, Y.; Iguchi, T.; Yamaguchi, J.; Terauchi, T.; Kitamura, M.; Goto, K.; Goto, M.; Oitate, M.; Yukinaga, H.; et al. Wide application of a novel topoisomerase I inhibitor-based drug conjugation technology. Bioorg. Med. Chem. Lett. 2016, 26, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- FDA Grants Regular Approval to Fam-Trastuzumab Deruxtecan-Nxki for Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-fam-trastuzumab-deruxtecan-nxki-breast-cancer (accessed on 11 May 2022).
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Ducry, L.; Stump, B. Antibody-drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug. Chem. 2010, 21, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, C.L.; Ferrara, B.; Occhipinti, S.; Boggio, E.; Barrera, G.; Pizzimenti, S.; Giovarelli, M.; Fantozzi, R.; Chiocchetti, A.; Argenziano, M.; et al. Enhanced cytotoxic effect of camptothecin nanosponges in anaplastic thyroid cancer cells in vitro and in vivo on orthotopic xenograft tumors. Drug Deliv. 2017, 24, 670–680. [Google Scholar] [CrossRef]
- Gigliotti, C.L.; Minelli, R.; Cavalli, R.; Occhipinti, S.; Barrera, G.; Pizzimenti, S.; Cappellano, G.; Boggio, E.; Conti, L.; Fantozzi, R.; et al. In Vitro and In Vivo Therapeutic Evaluation of Camptothecin-Encapsulated β-Cyclodextrin Nanosponges in Prostate Cancer. J. Biomed. Nanotechnol. 2016, 12, 114–127. [Google Scholar] [CrossRef]
- Chae, S.; Kim, D.; Lee, K.-J.; Lee, D.; Kim, Y.-O.; Jung, Y.C.; Rhee, S.D.; Kim, K.R.; Lee, J.-O.; Ahn, S.; et al. Encapsulation and Enhanced Delivery of Topoisomerase I Inhibitors in Functionalized Carbon Nanotubes. ACS Omega 2018, 3, 5938–5945. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, W.; He, R.; Ismail, M.; Ling, L.; Yao, C.; Fu, Z.; Li, X. Dual 7-ethyl-10-hydroxycamptothecin conjugated phospholipid prodrug assembled liposomes with in vitro anticancer effects. Bioorg. Med. Chem. 2017, 25, 3247–3258. [Google Scholar] [CrossRef]
- Falchook, G.S.; Bendell, J.C.; Ulahannan, S.V.; Sen, S.; Vilimas, R.; Kriksciukaite, K.; Mei, L.; Jerkovj, G.; Sarapa, N.; Bilodeau, M.T.; et al. Pen-866, a miniature drug conjugate of a heat shock protein 90 (HSP90) ligand linked to SN38 for patients with advanced solid malignancies: Phase I and expansion cohort results. J. Clin. Oncol. 2020, 38, 3515. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target 2022, 7, 93. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef]
- Pettinato, M.C. Introduction to Antibody-Drug Conjugates. Antibodies 2021, 10, 42. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct Target Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer. Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Yonesaka, K.; Takegawa, N.; Watanabe, S.; Haratani, K.; Kawakami, H.; Sakai, K.; Chiba, Y.; Maeda, N.; Kagari, T.; Hirotani, K.; et al. An HER3-targeting antibody-drug conjugate incorporating a DNA topoisomerase I inhibitor U3-1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene 2019, 38, 1398–1409. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, E.; Gandullo-Sánchez, L.; Ocaña, A.; Pandiella, A. Novel ADCs and Strategies to Overcome Resistance to Anti-HER2 ADCs. Cancers 2021, 14, 154. [Google Scholar] [CrossRef]
- Lyu, H.; Han, A.; Polsdofer, E.; Liu, S.; Liu, B. Understanding the biology of HER3 receptor as a therapeutic target in human cancer. Acta Pharm. Sin. B 2018, 8, 503–510. [Google Scholar] [CrossRef]
- Ocaña, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Moek, K.L.; de Groot, D.J.A.; de Vries, E.G.E.; Fehrmann, R.S.N. The antibody-drug conjugate target landscape across a broad range of tumour types. Ann. Oncol. 2017, 28, 3083–3091. [Google Scholar] [CrossRef]
- Mathur, R.; Weiner, G.J. Picking the Optimal Target for Antibody-Drug Conjugates. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e103–e107. [Google Scholar] [CrossRef]
- Criscitiello, C.; Morganti, S.; Curigliano, G. Antibody-drug conjugates in solid tumors: A look into novel targets. J. Hematol. Oncol. 2021, 14, 20. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-Drug Conjugates for Cancer Therapy: Chemistry to Clinical Implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Ricart, A.D. Antibody-drug conjugates of calicheamicin derivative: Gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin. Cancer Res. 2011, 17, 6417–6427. [Google Scholar] [CrossRef] [PubMed]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- Rossin, R.; Versteegen, R.M.; Wu, J.; Khasanov, A.; Wessels, H.J.; Steenbergen, E.J.; Hoeve, W.T.; Janssen, H.M.; van Onzen, A.H.A.M.; Hudson, P.J.; et al. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9, 1484. [Google Scholar] [CrossRef]
- Klaus, T.; Deshmukh, S. pH-responsive antibodies for therapeutic applications. J. Biomed. Sci. 2021, 28, 11. [Google Scholar] [CrossRef]
- Nani, R.R.; Gorka, A.P.; Nagaya, T.; Kobayashi, H.; Schnermann, M.J. Near-IR Light-Mediated Cleavage of Antibody-Drug Conjugates Using Cyanine Photocages. Angew. Chem. Int. Ed. Engl. 2015, 54, 13635–13638. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Corso, A.D.; Pignataro, L.; Belvisi, L.; Gennari, C. Innovative Linker Strategies for Tumor-Targeted Drug Conjugates. Chemistry 2019, 25, 14740–14757. [Google Scholar] [CrossRef]
- Zang, C.; Wang, H.; Li, T.; Zhang, Y.; Li, J.; Shang, M.; Du, J.; Xi, Z.; Zhou, C. A light-responsive, self-immolative linker for controlled drug delivery. Chem. Sci. 2019, 10, 8973–8980. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody-drug conjugates: Recent advances in linker chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Conilh, L.; Fournet, G.; Fourmaux, E.; Murcia, A.; Matera, E.-L.; Joseph, B.; Dumontet, C.; Viricel, W. Exatecan Antibody Drug Conjugates Based on a Hydrophilic Polysarcosine Drug-Linker Platform. Pharmaceuticals 2021, 14, 247. [Google Scholar] [CrossRef]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Doronina, S.O.; Setter, J.R.; Bovee, T.D.; Anderson, M.E.; Jonas, M.; Daniho, S.; Kostner, H.; Senter, P.D.; Lyon, R.P. Abstract 4470: Elucidating the role of drug-linker hydrophobicity in the disposition of antibody-drug conjugates. Cancer Res. 2014, 74, 4470. [Google Scholar] [CrossRef]
- Lyon, R.P.; Meyer, D.L.; Setter, J.R.; Senter, P.D. Conjugation of anticancer drugs through endogenous monoclonal antibody cysteine residues. Methods Enzym. 2012, 502, 123–138. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Govindan, S.V.; Zalath, M.B.; Rossi, D.L.; Wang, Y.; Chang, C.H.; Goldenberg, D.M. IMMU-140, a Novel SN-38 Antibody-Drug Conjugate Targeting HLA-DR, Mediates Dual Cytotoxic Effects in Hematologic Cancers and Malignant Melanoma. Mol. Cancer Ther. 2018, 17, 150–160. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan. MAbs 2019, 11, 987–995. [Google Scholar] [CrossRef]
- Das, M. Labetuzumab govitecan in metastatic colorectal cancer. Lancet Oncol. 2017, 18, e563. [Google Scholar] [CrossRef]
- Wata, T.N.; Ishii, C.; Ishida, S.; Ogitani, Y.; Wada, T.; Agatsuma, T. A HER2-Targeting Antibody-Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model. Mol. Cancer Ther. 2018, 17, 1494–1503. [Google Scholar] [CrossRef]
- Koganemaru, S.; Kuboki, Y.; Koga, Y.; Kojima, T.; Yamauchi, M.; Maeda, N.; Kagari, T.; Hirotani, K.; Yasunaga, M.; Matsumura, Y.; et al. U3-1402, a Novel HER3-Targeting Antibody-Drug Conjugate, for the Treatment of Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Takegawa, N.; Nonagase, Y.; Yonesaka, K.; Sakai, K.; Maenishi, O.; Ogitani, Y.; Tamura, T.; Nishio, K.; Nakagawa, K.; Tsurutani, J. DS-8201a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int. J. Cancer 2017, 141, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse polysarcosine-based highly-loaded antibody-drug conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef]
- Yurkovetskiy, A.V.; Bodyak, N.D.; Yin, M.; Thomas, J.D.; Clardy, S.M.; Conlon, P.R.; Stevenson, C.A.; Uttard, A.; Qin, L.; Gumerov, D.R.; et al. Dolaflexin: A Novel Antibody-Drug Conjugate Platform Featuring High Drug Loading and a Controlled Bystander Effect. Mol. Cancer Ther. 2021, 20, 885–895. [Google Scholar] [CrossRef]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef]
- You, J.; Zhang, J.; Wang, J.; Jin, M. Cysteine-Based Coupling: Challenges and Solutions. Bioconjug. Chem. 2021, 32, 1525–1534. [Google Scholar] [CrossRef]
- Wahby, S.; Fashoyin-Aje, L.; Osgood, C.L.; Cheng, J.; Fiero, M.H.; Zhang, L.; Tang, S.; Hamed, S.S.; Song, P.; Charlab, R.; et al. FDA Approval Summary: Accelerated Approval of Sacituzumab Govitecan-hziy for Third-line Treatment of Metastatic Triple-negative Breast Cancer. Clin. Cancer Res. 2021, 27, 1850–1854. [Google Scholar] [CrossRef]
- DeLucia, D.C.; Cardillo, T.M.; Ang, L.; Labrecque, M.P.; Zhang, A.; Hopkins, J.E.; De Sarkar, N.; Coleman, I.; da Costa, R.M.G.; Corey, E.; et al. Regulation of CEACAM5 and Therapeutic Efficacy of an Anti-CEACAM5-SN38 Antibody-drug Conjugate in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2021, 27, 759–774. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody-Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef]
- Li, C.; Ren, C.; Wen, L.; Chen, X.; Chen, B.; Zhang, G.; Wang, Y.; Li, K.; Cao, L.; Jia, M.; et al. Heterogenous and Low Expression of HER2 in Breast Cancer Overcome by DS-8201a in a Heavily Treated Patient: Case Report and Review of the Literature. Clin. Med. Insights Oncol. 2022, 16, 11795549211072880. [Google Scholar] [CrossRef]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients with HER2-Low-Expressing Advanced Breast Cancer: Results from a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef]
- Znojek, P.; Willmore, E.; Curtin, N.J. Preferential potentiation of topoisomerase I poison cytotoxicity by PARP inhibition in S phase. Br. J. Cancer 2014, 111, 1319–1326. [Google Scholar] [CrossRef]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Ditano, J.P.; Donahue, K.L.; Tafe, L.J.; McCleery, C.F.; Eastman, A. Sensitivity of cells to ATR and CHK1 inhibitors requires hyperactivation of CDK2 rather than endogenous replication stress or ATM dysfunction. Sci. Rep. 2021, 11, 7077. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef]
- Ho, A.L.; Bendell, J.C.; Cleary, J.M.; Schwartz, G.K.; Burris, H.A.; Oakes, P.; Agbo, F.; Barker, P.N.; Senderowicz, A.M.; Shapiro, G. Phase I, open-label, dose-escalation study of AZD7762 in combination with irinotecan (irino) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2011, 29, 3033. [Google Scholar] [CrossRef]
- Levengood, M.R.; Zhang, X.; Hunter, J.H.; Emmerton, K.K.; Miyamoto, J.B.; Lewis, T.S.; Senter, P.D. Orthogonal Cysteine Protection Enables Homogeneous Multi-Drug Antibody-Drug Conjugates. Angew. Chem. Int. Ed. Engl. 2017, 56, 733–737. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Berger, G.; Marloye, M.; Lawler, S.E. Pharmacological Modulation of the STING Pathway for Cancer Immunotherapy. Trends Mol. Med. 2019, 25, 412–427. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Alfonsi, R.; Grassi, L.; Signore, M.; Bonci, D. The Double Face of Exosome-Carried MicroRNAs in Cancer Immunomodulation. Int. J. Mol. Sci. 2018, 19, 1183. [Google Scholar] [CrossRef]
- Meldolesi, J. Extracellular vesicles, news about their role in immune cells: Physiology, pathology and diseases. Clin. Exp. Immunol. 2019, 196, 318–327. [Google Scholar] [CrossRef]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef]
- Borghaei, H.; Besse, B.; Bardia, A.; Mazieres, J.; Popat, S.; Augustine, B.; D’amelio, A.M.; Barrios, D.; Rugo, H.S. Trastuzumab deruxtecan (T-DXd; DS-8201) in combination with pembrolizumab in patients with advanced/metastatic breast or non-small cell lung cancer (NSCLC): A phase Ib, multicenter, study. J. Clin. Oncol. 2020, 38, TPS1100. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lim, K.S.; Valencia, G.M.; Jung, M.; Bull, D.A.; Won, Y.-W. One-Step Method for Instant Generation of Advanced Allogeneic NK Cells. Adv. Sci. 2018, 5, 1800447. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-K.; Hsiao, C.-W.; Yang, S.-H.; Yang, H.-P.; Wu, T.-S.; Lee, C.-Y.; Lin, Y.-L.; Pan, J.; Cheng, Z.-F.; Lai, Y.-D.; et al. A Novel off-the-Shelf Trastuzumab-Armed NK Cell Therapy (ACE1702) Using Antibody-Cell-Conjugation Technology. Cancers 2021, 13, 2724. [Google Scholar] [CrossRef]
- Pfister, T.D.; Reinhold, W.C.; Agama, K.; Gupta, S.; Khin, S.A.; Kinders, R.J.; Parchment, R.E.; Tomaszewski, J.E.; Doroshow, J.H.; Pommier, Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 2009, 8, 1878–1884. [Google Scholar] [CrossRef] [PubMed]
- Pfister, T.D.; Hollingshead, M.; Kinders, R.J.; Zhang, Y.; Evrard, Y.A.; Ji, J.; Khin, S.A.; Borgel, S.; Stotler, H.; Carter, J.; et al. Development and validation of an immunoassay for quantification of topoisomerase I in solid tumor tissues. PLoS ONE 2012, 7, e50494. [Google Scholar] [CrossRef]
- Kinders, R.J.; Hollingshead, M.; Lawrence, S.; Ji, J.; Tabb, B.; Bonner, W.M.; Pommier, Y.; Rubinstein, L.; Evrard, Y.A.; Parchment, R.E.; et al. Development of a validated immunofluorescence assay for γH2AX as a pharmacodynamic marker of topoisomerase I inhibitor activity. Clin. Cancer Res. 2010, 16, 5447–5457. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IgG Subclass | IgG1 | IgG2 | IgG3 | IgG4 |
|---|---|---|---|---|
| Serum half-life | 21 days | 21 days | 7–21 days | 21 days |
| Fcγreceptor binding | High | Low | High | Moderate |
| ADC | Status | Antibody Target | Drug | Linkers | DAR | Target Cancer | Clinical Trial Identifier |
|---|---|---|---|---|---|---|---|
| Sacituzumab govitecan (IMMU-132) | FDA-approved | TROP2 | SN38 | CL2A (pH-sensitive cleavage) | 7.6 | Triple-negative breast cancer/urothelial cancer (phase II) | NCT04320693 |
| Labetuzumab govitecan (IMMU-130) | Phase I | CEACAM5 | SN38 | CL2A (pH-sensitive cleavage) | 7.6 | Colorectal cancer | NCT01270698 |
| Trastuzumab deruxtecan (DS-8201a) | FDA-approved | HER2 | Dxd | GGFG (lysosomal enzyme cleavage) | 8 | Metastatic HER2-positive breast cancer | NCT03384940 |
| Patritumab deruxtecan (U3-1402) | Phase I/II | HER3 | Dxd | GGFG (lysosomal enzyme cleavage) | 8 | NSCLC (phase I) breast cancer (phase I/II) | NCT02980341, NCT03260491 |
| DS-1062 | Phase III | TROP2 | Dxd | GGFG (lysosomal enzyme cleavage) | 4 | Breast cancer, NSCLC | NCT05104866, NCT05215340 |
| DS-7300a | Phase I/II | B7-H3 | Dxd | GGFG (lysosomal enzyme cleavage) | 4 | Solid cancer, SCLC | NCT05280470, NCT04145622 |
| Strategy | Effect | |
|---|---|---|
| DNA damage response modulator | PARP inhibitor | Inhibit DNA stabilization and sensitize cancer cells to TOP1 inhibitor |
| ATR/CHK1 inhibitor | Inhibit cellular recovery and increase the sensitivity of cancer cells to TOP1 inhibitor | |
| Immunotherapy | Checkpoint inhibitor | Inhibit checkpoint molecule that is increased by topoisomerase inhibitor and enhance immunosurveillance |
| ADC-mediated immune cell engineering | Dual effect: increase tumor targeting of immune cells and deliver a drug to cancer | |
| Enzyme activity | Monitoring of topoisomerase activity | Increase drug response through selective treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Lim, K.S.; Blackburn, B.J.; Yun, J.; Putnam, C.W.; Bull, D.A.; Won, Y.-W. The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates. Pharmaceutics 2022, 14, 1707. https://doi.org/10.3390/pharmaceutics14081707
Han S, Lim KS, Blackburn BJ, Yun J, Putnam CW, Bull DA, Won Y-W. The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates. Pharmaceutics. 2022; 14(8):1707. https://doi.org/10.3390/pharmaceutics14081707
Chicago/Turabian StyleHan, Seungmin, Kwang Suk Lim, Brody J. Blackburn, Jina Yun, Charles W. Putnam, David A. Bull, and Young-Wook Won. 2022. "The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates" Pharmaceutics 14, no. 8: 1707. https://doi.org/10.3390/pharmaceutics14081707
APA StyleHan, S., Lim, K. S., Blackburn, B. J., Yun, J., Putnam, C. W., Bull, D. A., & Won, Y. -W. (2022). The Potential of Topoisomerase Inhibitor-Based Antibody–Drug Conjugates. Pharmaceutics, 14(8), 1707. https://doi.org/10.3390/pharmaceutics14081707