

Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects

 , ,
, ,
Abstract
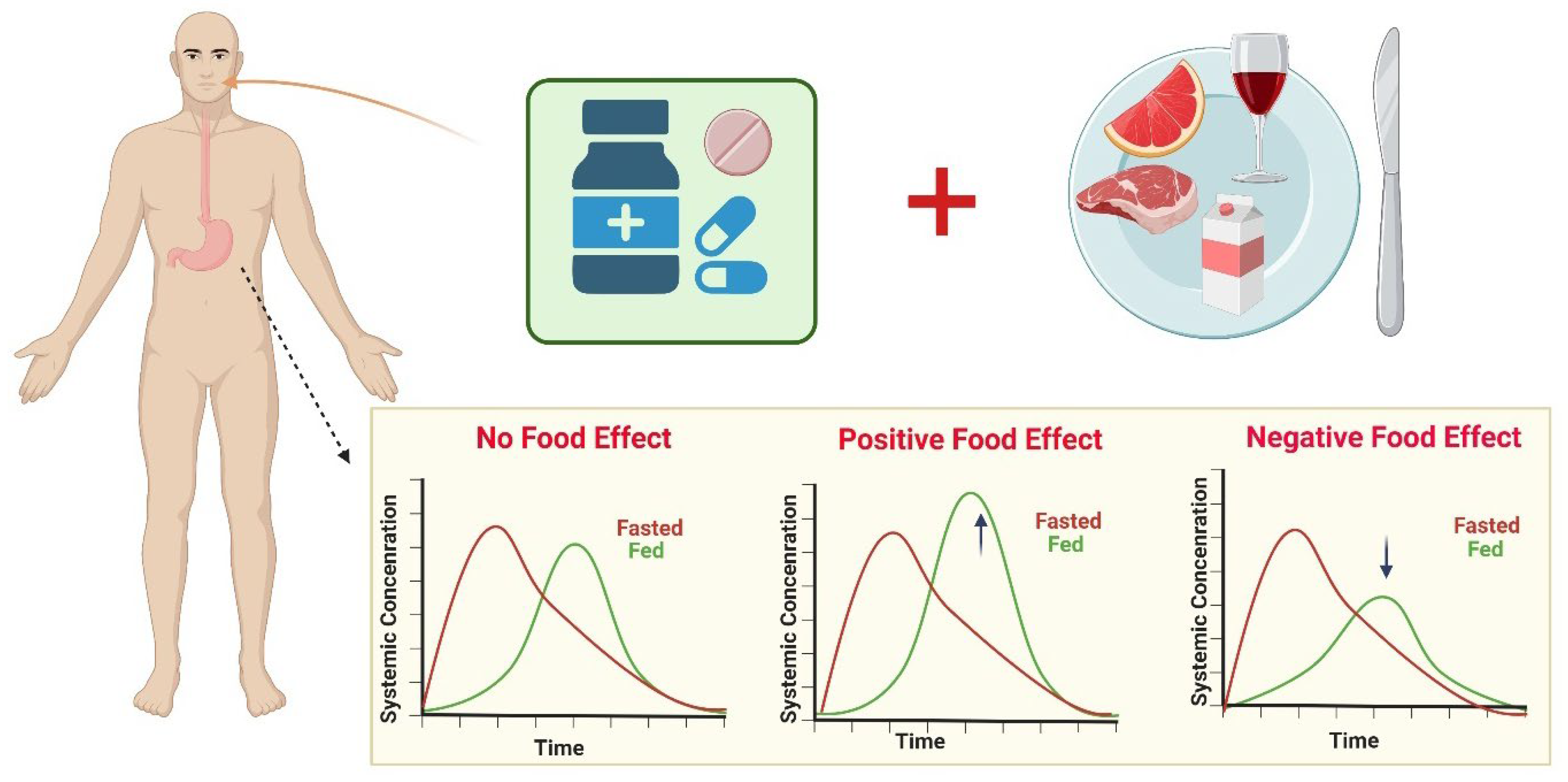
:1. Introduction
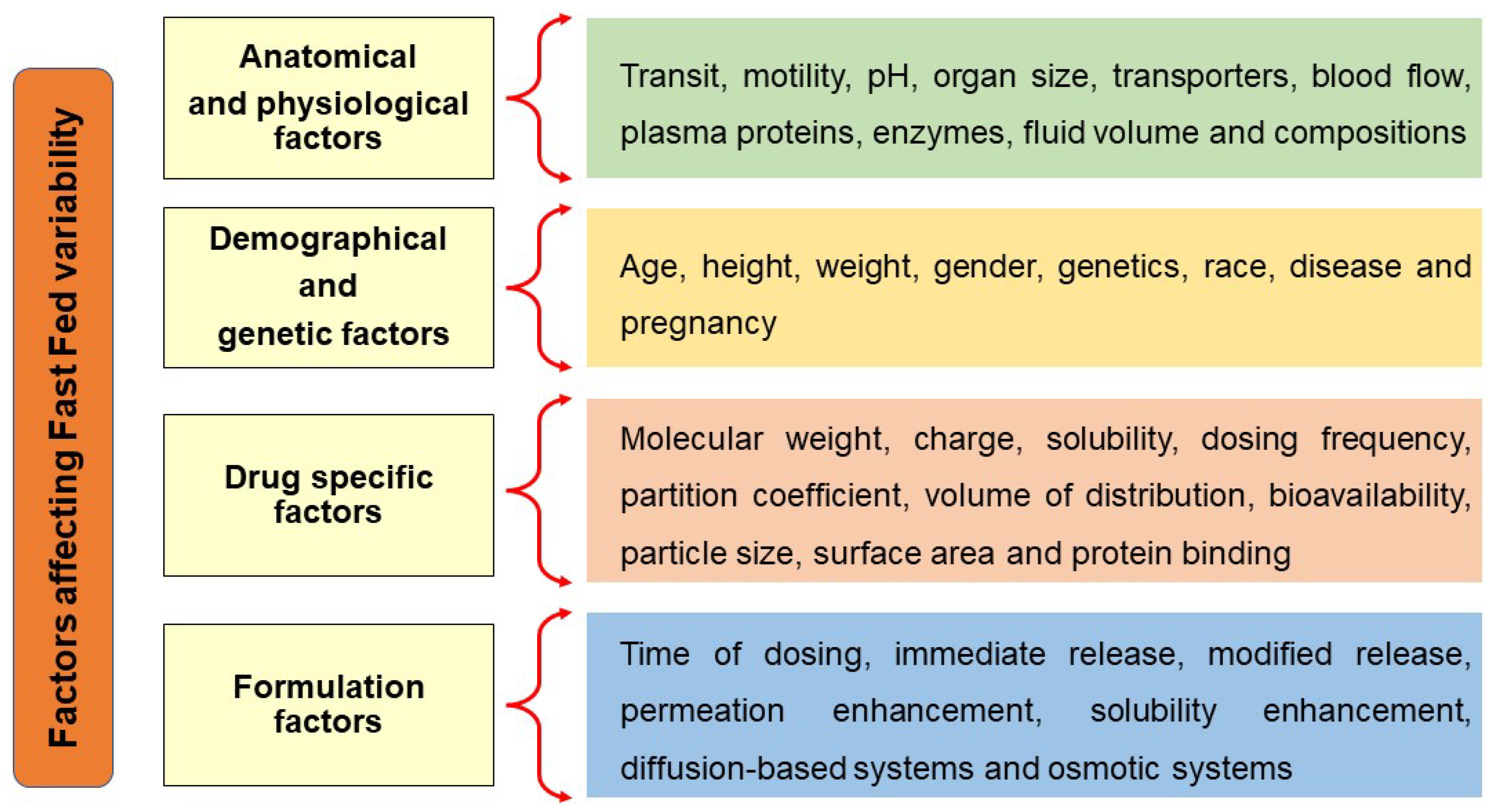
2. Factors Influencing Fast-Fed Variability
2.1. Anatomical and Physiological Factors
2.1.1. Gastrointestinal Transit
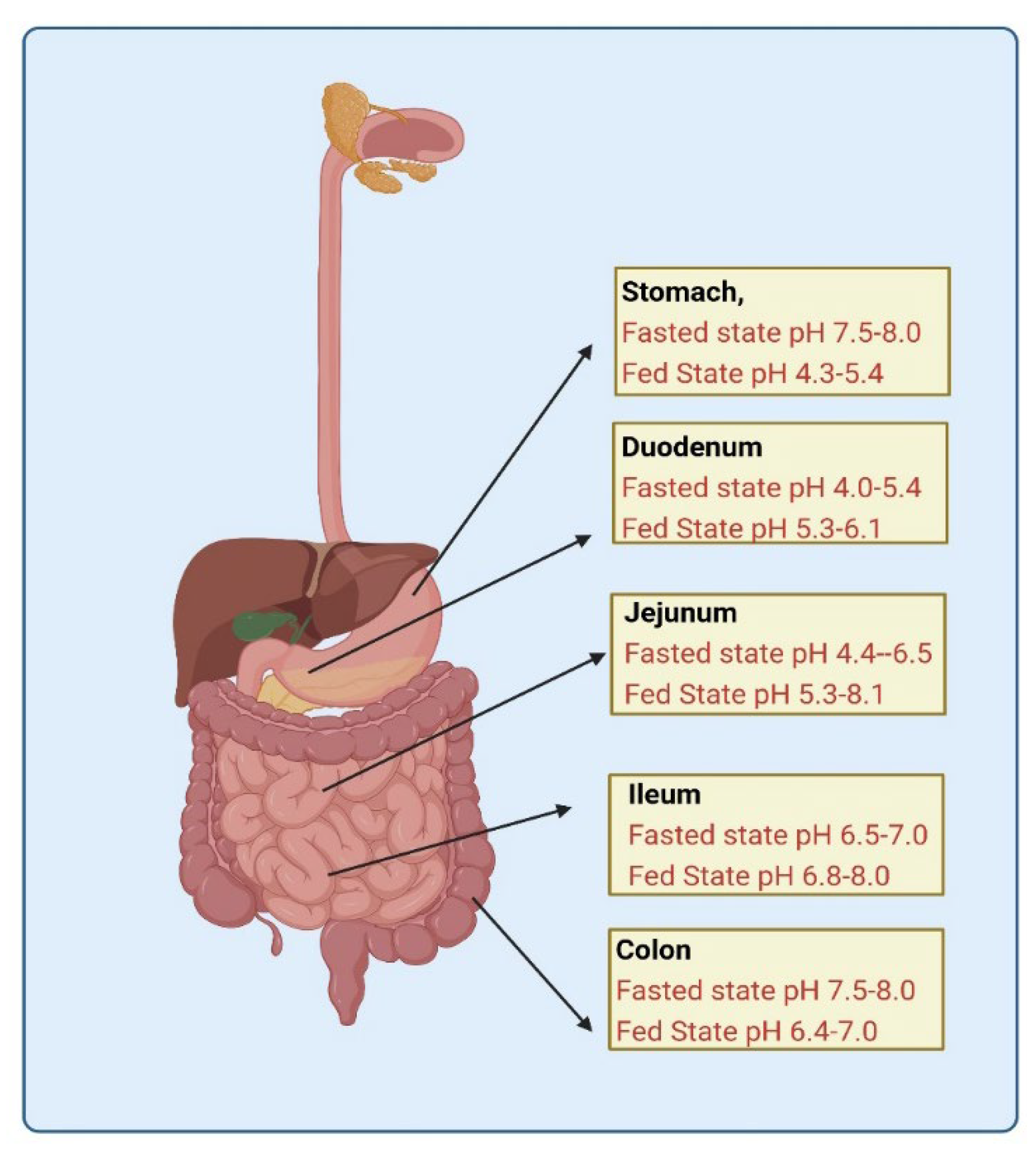
2.1.2. Gastric pH
2.1.3. Enzyme Content and Transporters
2.1.4. Hormonal Changes
2.1.5. Gastric Fluid Volume and Micellar Solubilization of Lipophilic Drugs
2.2. Demographic and Genetic Factors
2.3. Drug-Specific Factors
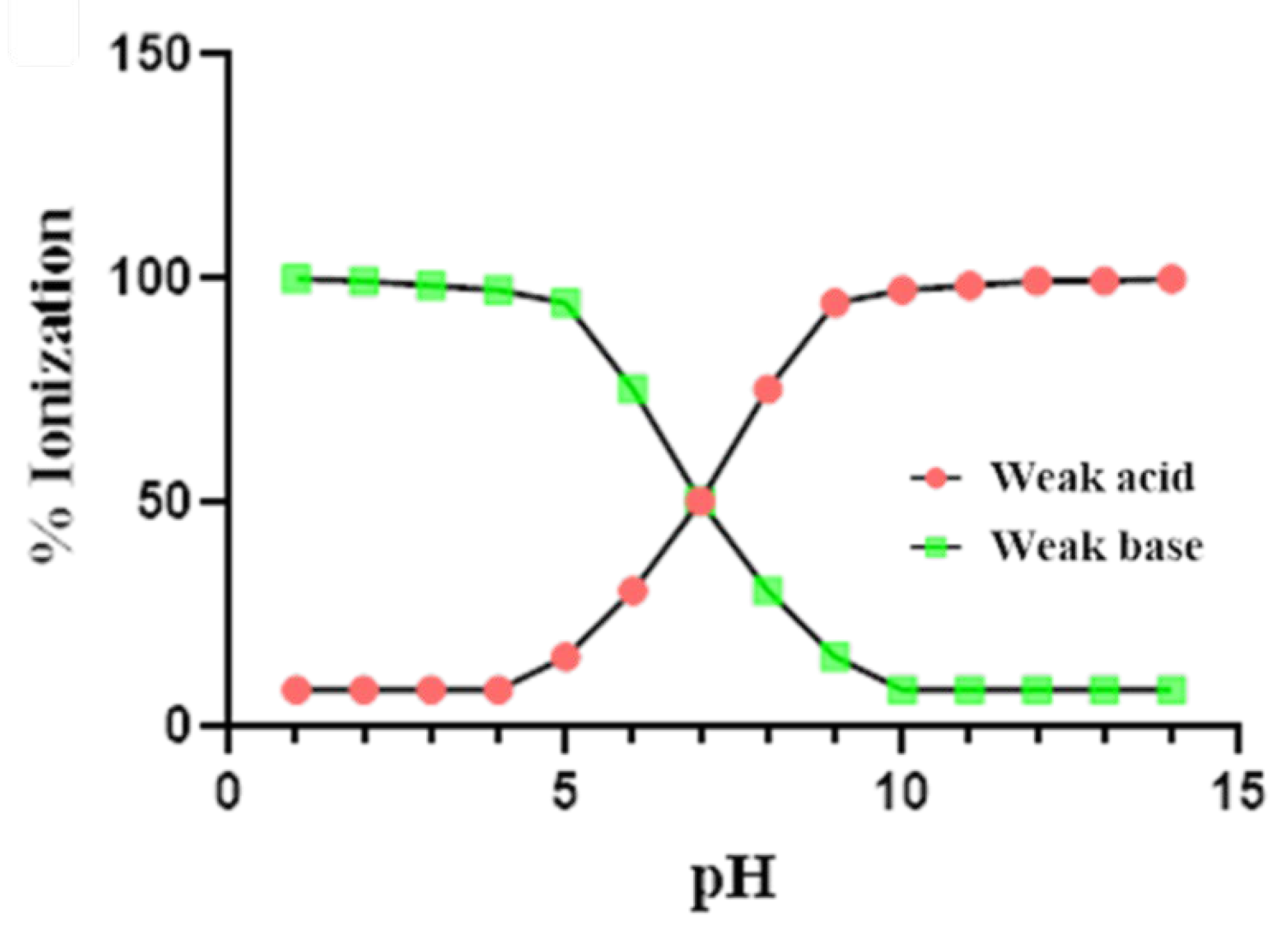
2.3.1. Charge
2.3.2. Partition Coefficient
2.3.3. Molecular Weight
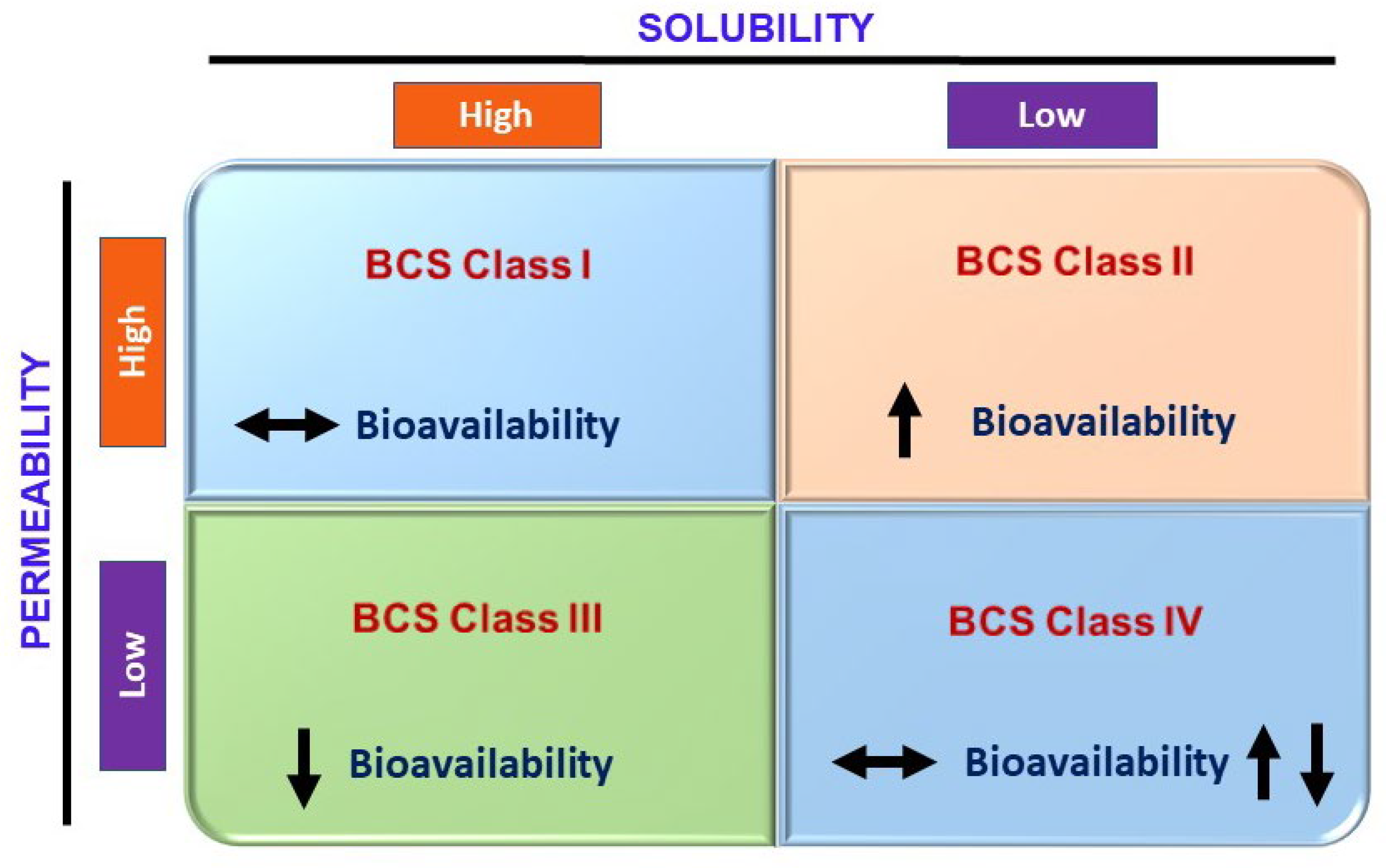
2.3.4. Solubility
2.3.5. Particle Size and Surface Area
2.3.6. Pharmacokinetic Factors
2.4. Formulation-Related Factors
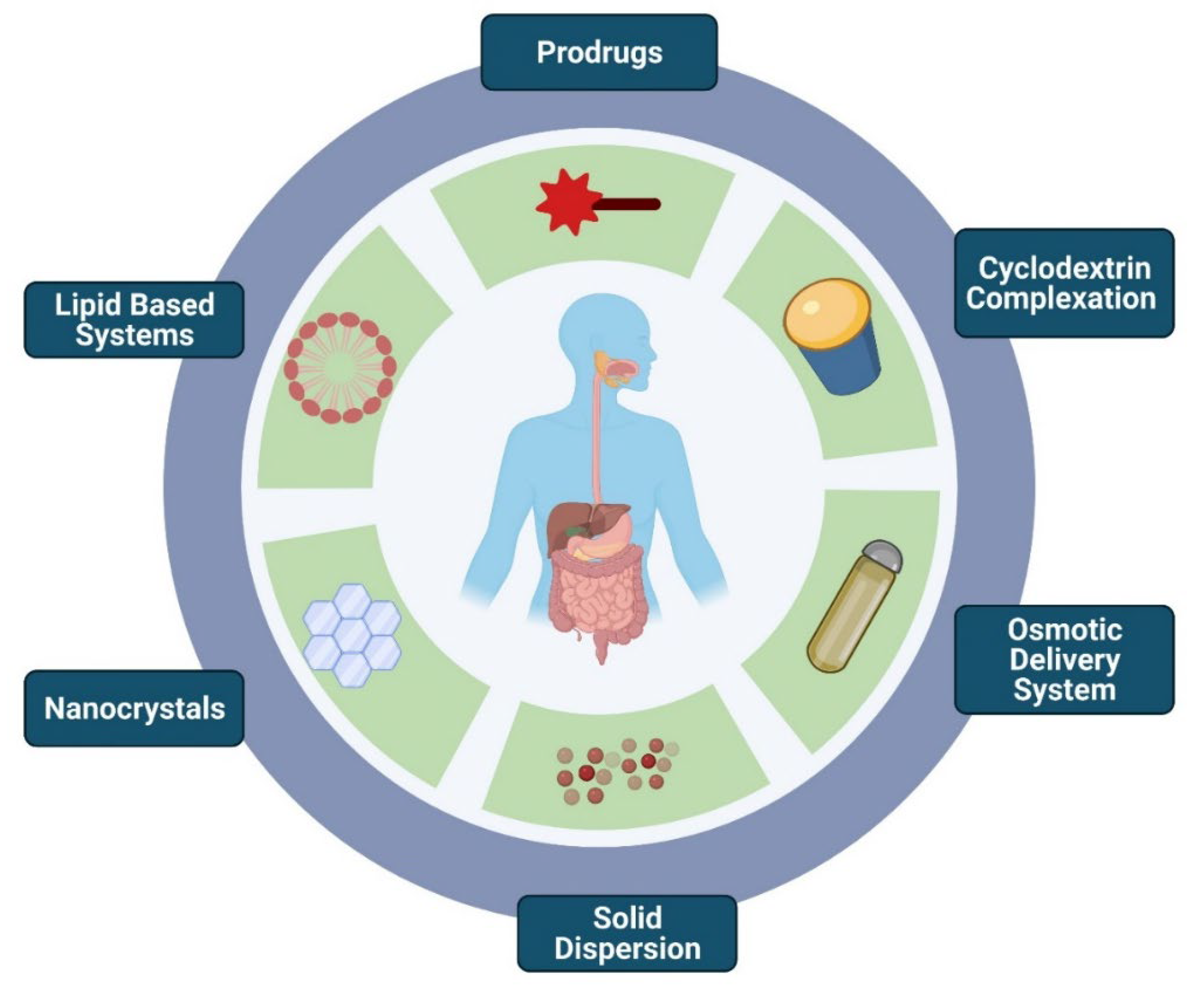
3. Formulation Strategies to Overcome Fast-Fed Variability
3.1. Prodrugs
3.2. Cyclodextrin Complexation
3.3. Osmotic Delivery System
3.4. Amorphous Solid Dispersion
3.5. Nanocrystal Technology
3.6. Lipid-Based Systems
4. Interplay of Different Molecular Properties Contributing to Fast-Fed Variability
5. Regulatory Aspects
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Jamei, M.; Turner, D.; Yang, J.; Neuhoff, S.; Polak, S.; Rostami-Hodjegan, A.; Tucker, G. Population-based mechanistic prediction of oral drug absorption. AAPS J. 2009, 11, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Abuhelwa, A.Y.; Williams, D.B.; Upton, R.N.; Foster, D.J.R. Food, gastrointestinal pH, and models of oral drug absorption. Eur. J. Pharm. Biopharm. 2017, 112, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Herbrink, M.; Nuijen, B.; Schellens, J.H.M.; Beijnen, J.H. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat. Rev. 2015, 41, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Vinarov, Z.; Abrahamsson, B.; Artursson, P.; Batchelor, H.; Berben, P.; Bernkop-Schnürch, A.; Butler, J.; Ceulemans, J.; Davies, N.; Dupont, D.; et al. Current challenges and future perspectives in oral absorption research: An opinion of the UNGAP network. Adv. Drug Deliv. Rev. 2021, 171, 289–331. [Google Scholar] [CrossRef]
- Watts, A.B.; Williams, R.O., III. Formulation and production strategies for enhancing bioavailability of poorly absorbed drugs. In Preclinical Drug Development; CRC Press: Boca Raton, FL, USA, 2020; pp. 173–207. ISBN 9780429142192. [Google Scholar]
- Benedetti, M.S.; Whomsley, R.; Canning, M. Drug metabolism in the paediatric population and in the elderly. Drug Discov. Today 2007, 12, 599–610. [Google Scholar] [CrossRef]
- Breitkreutz, J.; Boos, J. Paediatric and geriatric drug delivery. Expert Opin. Drug Deliv. 2007, 4, 37–45. [Google Scholar] [CrossRef]
- Shono, Y.; Jantratid, E.; Kesisoglou, F.; Reppas, C.; Dressman, J.B. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur. J. Pharm. Biopharm. 2010, 76, 95–104. [Google Scholar] [CrossRef]
- Abbas, R.; Hsyu, P.H. Clinical Pharmacokinetics and Pharmacodynamics of Bosutinib. Clin. Pharmacokinet. 2016, 55, 1191–1204. [Google Scholar] [CrossRef]
- Yu, P.; Lu, S.; Zhang, S.; Zhang, W.; Li, Y.; Liu, J. Enhanced oral bioavailability and diminished food effect of lurasidone hydrochloride nanosuspensions prepared by facile nanoprecipitation based on dilution. Powder Technol. 2017, 312, 11–20. [Google Scholar] [CrossRef]
- Sachar, M.; Park, C.H.; Pesco-Koplowitz, L.; Koplowitz, B.; McGinn, A. Effect of food intake on the pharmacokinetics of rivoceranib in healthy subjects. Fundam. Clin. Pharmacol. 2022, 36, 171–181. [Google Scholar] [CrossRef]
- Thombre, A.G.; Wu, X.Y.; Am Ende, M.T. Controlled release technology and design of oral controlled release dosage forms. Chem. Eng. Pharm. Ind. 2019, 381–407. [Google Scholar] [CrossRef]
- FDA. Food-Effect Bioavailability and Fed Bioequivalence Studies; Guidance for Industry; FDA: Silver Spring, MD, USA, 2002; p. 9.
- Lentz, K.A.; Quitko, M.; Morgan, D.G.; Grace, J.E.; Gleason, C.; Marathe, P.H. Development and validation of a preclinical food effect model. J. Pharm. Sci. 2007, 96, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Koziolek, M.; Alcaro, S.; Augustijns, P.; Basit, A.W.; Grimm, M.; Hens, B.; Hoad, C.L.; Jedamzik, P.; Madla, C.M.; Maliepaard, M.; et al. The mechanisms of pharmacokinetic food-drug interactions—A perspective from the UNGAP group. Eur. J. Pharm. Sci. 2019, 134, 31–59. [Google Scholar] [CrossRef] [PubMed]
- Carusi, A. Validation and variability: Dual challenges on the path from systems biology to systems medicine. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2014, 48, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of gastrointestinal tract variability on oral drug absorption and pharmacokinetics: An UNGAP review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Augustijns, P.; Brandl, M.; Brayden, D.J.; Brouwers, J.; Griffin, B.T.; Holm, R.; Jacobsen, A.C.; Lennernäs, H.; Vinarov, Z.; et al. Best practices in current models mimicking drug permeability in the gastrointestinal tract—An UNGAP review. Eur. J. Pharm. Sci. 2022, 170, 106098. [Google Scholar] [CrossRef]
- Meola, T.R.; Bremmell, K.E.; Williams, D.B.; Schultz, H.B.; Prestidge, C.A. Bio-enabling strategies to mitigate the pharmaceutical food effect: A mini review. Int. J. Pharm. 2022, 619, 121695. [Google Scholar] [CrossRef]
- Al-Saffar, A.; Takemi, S.; Saaed, H.K.; Sakata, I.; Sakai, T. Utility of animal gastrointestinal motility and transit models in functional gastrointestinal disorders. Best Pract. Res. Clin. Gastroenterol. 2019, 40–41, 101633. [Google Scholar] [CrossRef]
- Horowitz, M.; Maddox, A.; Bochner, M.; Wishart, J.; Bratasiuk, R.; Collins, P.; Shearman, D. Relationships between gastric emptying of solid and caloric liquid meals and alcohol absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 1989, 257, G291–G298. [Google Scholar] [CrossRef]
- Hörter, D.; Dressman, J.B. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2001, 46, 75–87. [Google Scholar] [CrossRef]
- Dressman, J.B.; Berardi, R.R.; Elta, G.H.; Gray, T.M.; Montgomery, P.A.; Lau, H.S.; Pelekoudas, K.L.; Szpunar, G.J.; Wagner, J.G. Absorption of Flurbiprofen in the Fed and Fasted States. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1992, 9, 901–907. [Google Scholar] [CrossRef]
- Barbara, G.; Feinle-Bisset, C.; Ghoshal, U.C.; Santos, J.; Vanner, S.J.; Vergnolle, N.; Zoetendal, E.G.; Quigley, E.M. The intestinal microenvironment and functional gastrointestinal disorders. Gastroenterology 2016, 150, 1305–1318.e8. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, P.; Orme, M. The effects of food on drug bioavailability. Br. J. Clin. Pharmacol. 1989, 28, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. Gut reaction: Impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug Discov. Today 2019, 24, 417–427. [Google Scholar] [CrossRef]
- Hirsch, C.H.; Maharaj, S.; Bourgeois, J.A. Pharmacotherapy: Safe Prescribing and Adverse Drug Events. In Geriatric Psychiatry; Springer: Cham, Switzerland, 2018; pp. 109–134. [Google Scholar] [CrossRef]
- Ogata, H.; Aoyagi, N.; Ejima, A. Gastric Emptying Rates of Drug Preparations. I. Effects of Size of Dosage Forms, Food and Species on Gastric Emptying Rates. J. Pharmacobiodyn. 1988, 11, 563–570. [Google Scholar] [CrossRef]
- Clarke, G.M.; Newton, J.M.; Short, M.D. Gastrointestinal transit of pellets of differing size and density. Int. J. Pharm. 1993, 100, 81–92. [Google Scholar] [CrossRef]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of pharmaceutical dosage forms through the small intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef]
- Staniforth, D.H.; Baird, I.M.; Fowler, J.; Lister, R.E. The Effects of Dietary Fibre on Upper and Lower Gastro-Intestinal Transit Times and Faecal Bulking. J. Int. Med. Res. 1991, 19, 228–233. [Google Scholar] [CrossRef]
- Helander, H.F.; Fändriks, L. Surface area of the digestive tract-revisited. Scand. J. Gastroenterol. 2014, 49, 681–689. [Google Scholar] [CrossRef]
- Cheng, L.K.; O’Grady, G.; Du, P.; Egbuji, J.U.; Windsor, J.A.; Pullan, A.J. Gastrointestinal system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 65–79. [Google Scholar] [CrossRef]
- Gao, Y.; Gesenberg, C.; Zheng, W. Oral Formulations for preclinical studies: Principle, design, and development considerations. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice, 2nd ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 455–495. ISBN 9780128024478. [Google Scholar]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, T.J.; Hardcastle, J.D. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Fallingborg, J.; Christensen, L.A.; Ingeman-Nielsen, M.; Jacobsen, B.A.; Abildgaard, K.; Rasmussen, H.H. pH-Profile and regional transit times of the normal gut measured by a radiotelemetry device. Aliment. Pharmacol. Ther. 1989, 3, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Vertzoni, M.; Goumas, K.; Reppas, C. Estimating drug solubility in the gastrointestinal tract. Adv. Drug Deliv. Rev. 2007, 59, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Fallingborg, J.; Christensen, L.A.; Ingeman-Nielsen, M.; Jacobsen, B.A.; Abildgaard, K.; Rasmussen, H.H.; Rasmussen, S.N. Measurement of gastrointestinal pH and regional transit times in normal children. J. Pediatr. Gastroenterol. Nutr. 1990, 11, 211–214. [Google Scholar] [CrossRef]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barnett, J.L.; Jarvenpaa, K.M. Upper Gastrointestinal (GI) pH in Young, Healthy Men and Women. Pharm. Res. An Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 756–761. [Google Scholar] [CrossRef]
- Blum, R.A.; D’Andrea, D.T.; Florentino, B.M.; Wilton, J.H.; Hilligoss, D.M.; Gardner, M.J.; Henry, E.B.; Goldstein, H.; Schentag, J.J. Increased gastric pH and the bioavailability of fluconazole and ketoconazole. Ann. Intern. Med. 1991, 114, 755–757. [Google Scholar] [CrossRef]
- Zhou, R.; Moench, P.; Heran, C.; Lu, X.; Mathias, N.; Faria, T.N.; Wall, D.A.; Hussain, M.A.; Smith, R.L.; Sun, D. pH-Dependent dissolution in Vitro and absorption in Vivo of weakly basic drugs: Development of a canine model. Pharm. Res. 2005, 22, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Jaruratanasirikul, S.; Sriwiriyajan, S. Effect of omeprazole on the pharmacokinetics of itraconazole. Eur. J. Clin. Pharmacol. 1998, 54, 159–161. [Google Scholar] [CrossRef]
- Derendorf, H.; VanderMaelen, C.P.; Brickl, R.S.; MacGregor, T.R.; Eisert, W. Dipyridamole bioavailability in subjects with reduced gastric acidity. J. Clin. Pharmacol. 2005, 45, 845–850. [Google Scholar] [CrossRef]
- Carver, P.L.; Fleisher, D.; Zhou, S.Y.; Kaul, D.; Kazanjian, P.; Li, C. Meal composition effects on the oral bioavailability of indinavir in HIV-infected patients. Pharm. Res. 1999, 16, 718–724. [Google Scholar] [CrossRef]
- Lebsack, M.E.; Nix, D.; Ryerson, B.; Toothaker, R.D.; Welage, L.; Norman, A.M.; Schentag, J.J.; Sedman, A.J. Effect of gastric acidity on enoxacin absorption. Clin. Pharmacol. Ther. 1992, 52, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Aoyagi, N.; Kaniwa, N.; Ejima, A.; Sekine, N.; Kitamura, M.; Inoue, Y. Gastric acidity dependent bioavailability of cinnarizine from two commercial capsules in healthy volunteers. Int. J. Pharm. 1986, 29, 113–120. [Google Scholar] [CrossRef]
- Saathoff, N.; Lode, H.; Neider, K.; Depperman, K.M.; Borner, K.; Koeppe, P. Pharmacokinetics of cefpodoxime proxetil and interactions with an antacid and an H2 receptor antagonist. Antimicrob. Agents Chemother. 1992, 36, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Perelló, P.; Maria, F. In Vitro Digestion Behavior of Complex Formulations for Clinical Nutrition Applications Based on Model Systems. Available online: https://www.semanticscholar.org/paper/In-vitro-digestion-behavior-of-complex-formulations-Perelló-Maria/3d9d6e4d1e04d83aff6074a75384b2de4eb42159 (accessed on 25 July 2022).
- Koziolek, M.; Carrière, F.; Porter, C.J.H. Lipids in the Stomach—Implications for the Evaluation of Food Effects on Oral Drug Absorption. Pharm. Res. 2018, 35, 55. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, S.; Zhang, W.; Zecchinati, F.; Mottino, A.; Vore, M. ABC Transporters in Extrahepatic Tissues: Pharmacological Regulation in Heart and Intestine. Curr. Med. Chem. 2018, 26, 1155–1184. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.J.; Isbister, C.; Bobik, A.; Dudley, F.J. Reduction of first-pass hepatic clearance of propranolol by food. Clin. Pharmacol. Ther. 1981, 30, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Clifford, C.P.; Adams, D.A.; Murray, S.; Taylor, G.W.; Wilkins, M.R.; Boobis, A.R.; Davies, D.S. The cardiac effects of terfenadine after inhibition of its metabolism by grapefruit juice. Eur. J. Clin. Pharmacol. 1997, 52, 311–315. [Google Scholar] [CrossRef]
- Bliss, E.S.; Whiteside, E. The gut-brain axis, the human gut microbiota and their integration in the development of obesity. Front. Physiol. 2018, 9, 900. [Google Scholar] [CrossRef]
- Schiller, C.; Fröhlich, C.P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
- Johnson, I.T.; Gee, J.M. Effect of gel-forming gums on the intestinal unstirred layer and sugar transport in vitro. Gut 1981, 22, 398–403. [Google Scholar] [CrossRef]
- Sandhar, B.K.; Goresky, G.V.; Maltby, J.R.; Shaffer, E.A. Effect of oral liquids and ranitidine on gastric fluid volume and pH in children undergoing outpatient surgery. Anesthesiology 1989, 71, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Mudie, D.M.; Amidon, G.L.; Amidon, G.E. Physiological parameters for oral delivery and in vitro testing. Mol. Pharm. 2010, 7, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Hur, S.J.; Lim, B.O.; Decker, E.A.; McClements, D.J. In vitro human digestion models for food applications. Food Chem. 2011, 125, 1–12. [Google Scholar] [CrossRef]
- Lindahl, A.; Ungell, A.L.; Knutson, L.; Lennernäs, H. Characterization of fluids from the stomach and proximal jejunum in men and women. Pharm. Res. 1997, 14, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Falavigna, M.; Klitgaard, M.; Steene, E.; Flaten, G.E. Mimicking regional and fasted/fed state conditions in the intestine with the mucus-PVPA in vitro model: The impact of pH and simulated intestinal fluids on drug permeability. Eur. J. Pharm. Sci. 2019, 132, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sugano, K. Estimation of effective intestinal membrane permeability considering bile micelle solubilisation. Int. J. Pharm. 2009, 368, 116–122. [Google Scholar] [CrossRef]
- Brogna, A.; Ferrara, R.; Bucceri, A.M.; Lanteri, E.; Catalano, F. Influence of aging on gastrointestinal transit time an ultrasonographic and radiologic study. Investig. Radiol. 1999, 34, 357–359. [Google Scholar] [CrossRef]
- Sun, T.; Li, D.; Hu, S.; Huang, L.; Sun, H.; Yang, S.; Wu, B.; Ji, F.; Zhou, D. Aging-dependent decrease in the numbers of enteric neurons, interstitial cells of Cajal and expression of connexin43 in various regions of gastrointestinal tract. Aging 2018, 10, 3851–3865. [Google Scholar] [CrossRef]
- Chi, C.; Li, D.J.; Jiang, Y.J.; Tong, J.; Fu, H.; Wu, Y.H.; Shen, F.M. Vascular smooth muscle cell senescence and age-related diseases: State of the art. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1810–1821. [Google Scholar] [CrossRef]
- Strolin, M.; Whomsley, R.; Baltes, E.L. Differences in absorption, distribution, metabolism and excretion of xenobiotics between the paediatric and adult populations. Expert Opin. Drug Metab. Toxicol. 2005, 1, 447–471. [Google Scholar] [CrossRef]
- Toutain, P.L.; Ferran, A.; Bousquet-Mélou, A. Species differences in pharmacokinetics and pharmacodynamics. Handb. Exp. Pharmacol. 2010, 199, 19–48. [Google Scholar] [CrossRef]
- Donovan, M.D. Sex and racial differences in pharmacological response: Effect of route of administration and drug delivery system on pharmacokinetics. J. Womens Health 2005, 14, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Javaid, N.; Choi, S. Acetylation- and Methylation-Related Epigenetic Proteins in the Context of Their Targets. Genes 2017, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Zhong, X. Epigenetic regulation of drug metabolism and transport. Acta Pharm. Sin. B 2015, 5, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.W. Racial, ethnic and gender differences in response to medicines. Drug Metabol. Drug Interact. 1995, 12, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R. The effects of diet, aging and disease-states on presystemic elimination and oral drug bioavailability in humans. Adv. Drug Deliv. Rev. 1997, 27, 129–159. [Google Scholar] [CrossRef]
- Nicolò, E.; Trapani, D.; Giachetti, P.P.M.B.; Zagami, P.; Curigliano, G. Fed or fasted state for oral therapies in breast cancer treatment? A comprehensive review of clinical practice recommendations. Cancer Treat. Rev. 2021, 100, 102281. [Google Scholar] [CrossRef]
- Hamed, R.; Awadallah, A.; Sunoqrot, S.; Tarawneh, O.; Nazzal, S.; AlBaraghthi, T.; Al Sayyad, J.; Abbas, A. pH-Dependent Solubility and Dissolution Behavior of Carvedilol—Case Example of a Weakly Basic BCS Class II Drug. AAPS PharmSciTech 2016, 17, 418–426. [Google Scholar] [CrossRef]
- Kumar, S.; Ravulapalli, S.Y.; Tiwari, S.K.; Gupta, S.; Nair, A.B.; Jacob, S. Effect of sex and food on the pharmacokinetics of different classes of BCS drugs in rats after cassette administration. Int. J. Pharm. 2021, 610, 121221. [Google Scholar] [CrossRef]
- Williams, H.D.; Ford, L.; Lim, S.; Han, S.; Baumann, J.; Sullivan, H.; Vodak, D.; Igonin, A.; Benameur, H.; Pouton, C.W.; et al. Transformation of Biopharmaceutical Classification System Class I and III Drugs Into Ionic Liquids and Lipophilic Salts for Enhanced Developability Using Lipid Formulations. J. Pharm. Sci. 2018, 107, 203–216. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Charman, W.N. Uptake of drugs into the intestinal lymphatics after oral administration. Adv. Drug Deliv. Rev. 1997, 25, 71–89. [Google Scholar] [CrossRef]
- Shekhawat, P.B.; Pokharkar, V.B. Understanding peroral absorption: Regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm. Sin. B 2017, 7, 260–280. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Ford, L.; Igonin, A.; Shan, Z.; Botti, P.; Morgen, M.M.; Hu, G.; Pouton, C.W.; Scammells, P.J.; Porter, C.J.H.; et al. Unlocking the full potential of lipid-based formulations using lipophilic salt/ionic liquid forms. Adv. Drug Deliv. Rev. 2019, 142, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Acharya, P.C.; Fernandes, C.; Suares, D.; Shetty, S.; Tekade, R.K. Solubility and Solubilization Approaches in Pharmaceutical Product Development. In Dosage Form Design Considerations: Volume I; Academic Press: Cambridge, MA, USA, 2018; pp. 513–547. ISBN 9780128144244. [Google Scholar]
- Auer, S.; Frenkel, D. Suppression of crystal nucleation in polydisperse colloids due to increase of the surface free energy. Nature 2001, 413, 711–713. [Google Scholar] [CrossRef]
- Hecq, J.; Deleers, M.; Fanara, D.; Vranckx, H.; Amighi, K. Preparation and characterization of nanocrystals for solubility and dissolution rate enhancement of nifedipine. Int. J. Pharm. 2005, 299, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Jinno, J.I.; Kamada, N.; Miyake, M.; Yamada, K.; Mukai, T.; Odomi, M.; Toguchi, H.; Liversidge, G.G.; Higaki, K.; Kimura, T. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J. Control. Release 2006, 111, 56–64. [Google Scholar] [CrossRef]
- Wanat, K. Biological barriers, and the influence of protein binding on the passage of drugs across them. Mol. Biol. Rep. 2020, 47, 3221–3231. [Google Scholar] [CrossRef]
- Silmore, L.H.; Willmer, A.R.; Capparelli, E.V.; Rosania, G.R. Food effects on the formulation, dosing, and administration of cannabidiol (CBD) in humans: A systematic review of clinical studies. Pharmacotherapy 2021, 41, 405–420. [Google Scholar] [CrossRef]
- Benziger, D.P.; Kaiko, R.F.; Miotto, J.B.; Fitzmartin, R.D.; Reder, R.F.; Chasin, M. Differential effects of food on the bioavailability of controlled-release oxycodone tablets and immediate-release oxycodone solution. J. Pharm. Sci. 1996, 85, 407–410. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Benet, L.Z. Predicting Drug Disposition via Application of BCS: Transport/Absorption/ Elimination Interplay and Development of a Biopharmaceutics Drug Disposition Classification System. Pharm. Res. 2005, 22, 11–23. [Google Scholar] [CrossRef]
- Yasuji, T.; Kondo, H.; Sako, K. The effect of food on the oral bioavailability of drugs: A review of current developments and pharmaceutical technologies for pharmacokinetic control. Ther. Deliv. 2012, 3, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wong, H. Food effects on oral drug absorption: Application of physiologically-based pharmacokinetic modeling as a predictive tool. Pharmaceutics 2020, 12, 672. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Mandlekar, S.; Marathe, P. Prodrug Design to Improve Pharmacokinetic and Drug Delivery Properties: Challenges to the Discovery Scientists. Curr. Med. Chem. 2010, 17, 3874–3908. [Google Scholar] [CrossRef]
- Jornada, D.H.; Dos Santos Fernandes, G.F.; Chiba, D.E.; De Melo, T.R.F.; Dos Santos, J.L.; Chung, M.C. The prodrug approach: A successful tool for improving drug solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef]
- Barton, P.; Riley, R.J. A new paradigm for navigating compound property related drug attrition. Drug Discov. Today 2016, 21, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Tack, J.; Augustijns, P. Parallel monitoring of plasma and intraluminal drug concentrations in man after oral administration of fosamprenavir in the fasted and fed state. Pharm. Res. 2007, 24, 1862–1869. [Google Scholar] [CrossRef]
- Kaye, A.D. Pharmacology, An Issue of Anesthesiology Clinics E-Book; Elsevier: Amsterdam, The Netherlands, 2017; ISBN 9780323529983. [Google Scholar]
- Lee, J.; Kim, B.; Kim, T.H.; Lee, S.H.; Park, H.D.; Chung, K.; Lee, S.H.; Paek, S.; Kim, E.E.K.; Yoon, S.K.; et al. A Food Effect Study of an Oral Thrombin Inhibitor and Prodrug Approach to Mitigate It. Mol. Pharm. 2016, 13, 1197–1205. [Google Scholar] [CrossRef]
- Angeli, F.; Verdecchia, P.; Pascucci, C.; Poltronieri, C.; Reboldi, G. Pharmacokinetic evaluation and clinical utility of azilsartan medoxomil for the treatment of hypertension. Expert Opin. Drug Metab. Toxicol. 2013, 9, 379–385. [Google Scholar] [CrossRef]
- Swanson, B.N.; Vlasses, P.H.; Ferguson, R.K.; Bergquist, P.A.; Till, A.E.; Irvin, J.D.; Harris, K. Influence of food on the bioavailability of enalapril. J. Pharm. Sci. 1984, 73, 1655–1657. [Google Scholar] [CrossRef]
- Malhotra, B.; Gandelman, K.; Sachse, R.; Wood, N.; Michel, M. The Design and Development of Fesoterodine as a Prodrug of 5- Hydroxymethyl Tolterodine (5-HMT), the Active Metabolite of Tolterodine. Curr. Med. Chem. 2009, 16, 4481–4489. [Google Scholar] [CrossRef]
- Oscier, D.; Orchard, J.A.; Culligan, D.; Cunningham, D.; Johnson, S.; Parker, A.; Klein, M.; Gieschen, H. The bioavailability of oral fludarabine phosphate is unaffected by food. Hematol. J. 2001, 2, 316–321. [Google Scholar] [CrossRef]
- Loftsson, T. Cyclodextrins and the biopharmaceutics classification system of drugs. J. Incl. Phenom. 2002, 44, 63–67. [Google Scholar] [CrossRef]
- Riis, T.; Bauer-Brandl, A.; Wagner, T.; Kranz, H. pH-independent drug release of an extremely poorly soluble weakly acidic drug from multiparticulate extended release formulations. Eur. J. Pharm. Biopharm. 2007, 65, 78–84. [Google Scholar] [CrossRef]
- Rauseo, A.M.; Mazi, P.; Lewis, P.; Burnett, B.; Mudge, S.; Spec, A. Bioavailability of single-dose SUBA-itraconazole compared to conventional itraconazole under fasted and fed conditions. Antimicrob. Agents Chemother. 2021, 65, e00134-21. [Google Scholar] [CrossRef]
- Van De Velde, V.J.S.; Van Peer, A.P.; Heykants, J.J.P.; Woestenborghs, R.J.H.; Van Rooy, P.; De Beule, K.L.; Cauwenbergh, G.F.M.J. Effect of food on the pharmacokinetics of a new hydroxypropyl-β-cyclodextrin formulation of itraconazole. Pharmacotherapy 1996, 16, 424–428. [Google Scholar]
- Thombre, A.G.; Shah, J.C.; Sagawa, K.; Caldwell, W.B. In vitro and in vivo characterization of amorphous, nanocrystalline, and crystalline ziprasidone formulations. Int. J. Pharm. 2012, 428, 8–17. [Google Scholar] [CrossRef]
- Wang, D.; Chen, G.; Ren, L. Preparation and Characterization of the Sulfobutylether-β-Cyclodextrin Inclusion Complex of Amiodarone Hydrochloride with Enhanced Oral Bioavailability in Fasted State. AAPS PharmSciTech 2017, 18, 1526–1535. [Google Scholar] [CrossRef]
- Wang, J.; Huang, B.; Dai, J.; Chen, G.; Ren, L. Inclusion complex of lurasidone hydrochloride with Sulfobutylether-β-cyclodextrin has enhanced oral bioavailability and no food effect. Am. J. Transl. Res. 2022, 14, 1495. [Google Scholar]
- Schapperer, E.; Daumann, H.; Lamouche, S.; Thyroff-Friesinger, U.; Viel, F.; Weitschies, W. Bioequivalence of Sandoz methylphenidate osmotic-controlled release tablet with Concerta® (Janssen-Cilag). Pharmacol. Res. Perspect. 2015, 3, e00072. [Google Scholar] [CrossRef]
- Modi, N.B.; Wang, B.; Hu, W.T.; Gupta, S.K. Effect of food on the pharmacokinetics of osmotic controlled-release methylphenidate HCl in healthy subjects. Biopharm. Drug Dispos. 2000, 21, 23–31. [Google Scholar] [CrossRef]
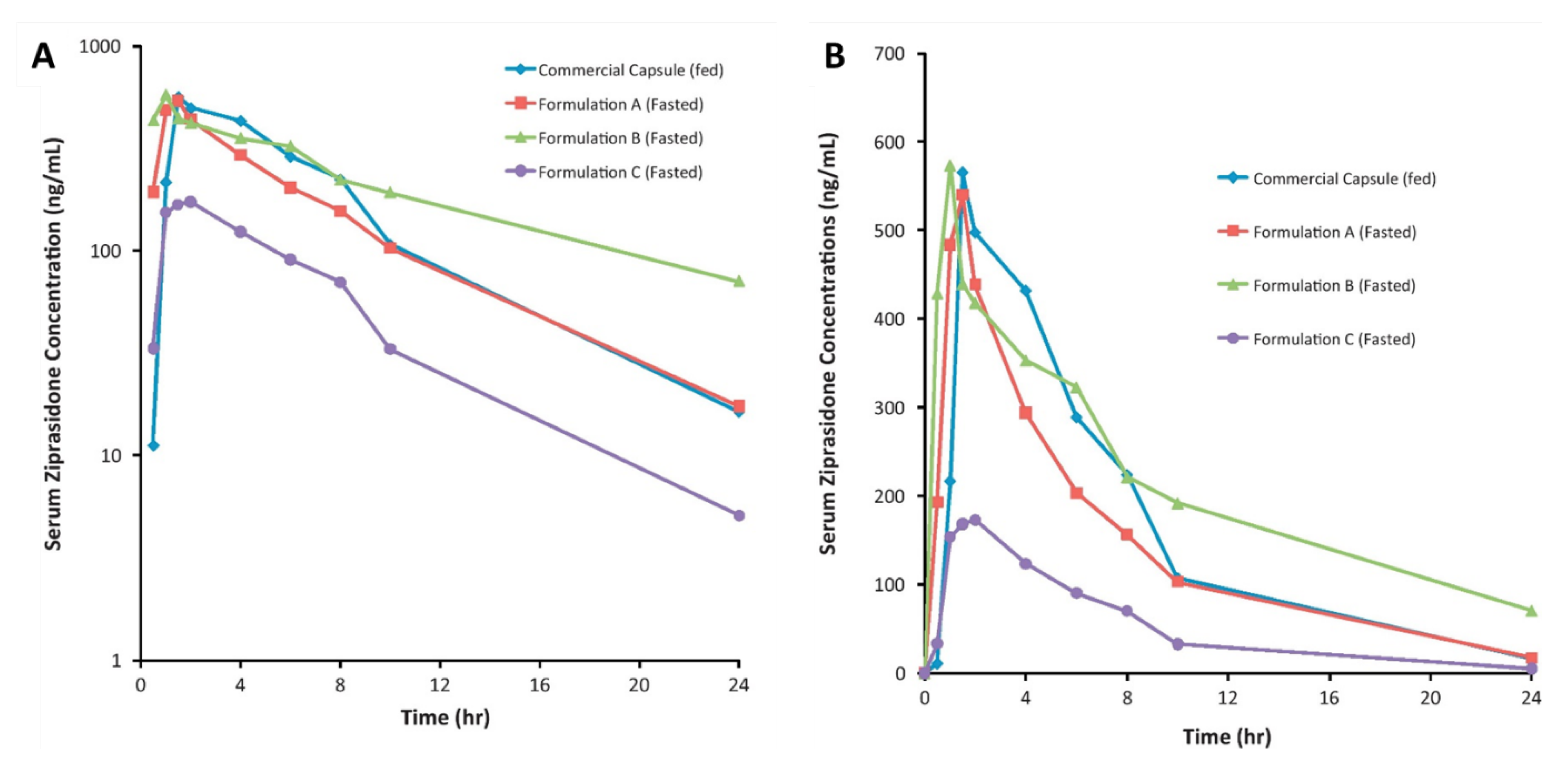
- Yanfei, M.; Guoguang, C.; Lili, R.; Pingkai, O. Controlled release of ziprasidone solid dispersion systems from osmotic pump tablets with enhanced bioavailability in the fasted state. Drug Dev. Ind. Pharm. 2015, 41, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Patel, N.; Lin, S. Solubility and dissolution enhancement strategies: Current understanding and recent trends. Drug Dev. Ind. Pharm. 2015, 41, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Van Speybroeck, M.; Mellaerts, R.; Mols, R.; Thi, T.D.; Martens, J.A.; Van Humbeeck, J.; Annaert, P.; Van den Mooter, G.; Augustijns, P. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur. J. Pharm. Sci. 2010, 41, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.S.; Kumar, S.; Zhou, Q. Pharmaceutical amorphous solid dispersion: A review of manufacturing strategies. Acta Pharm. Sin. B 2021, 11, 2505–2536. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643. [Google Scholar] [CrossRef]
- Klein, C.E.; Chiu, Y.L.; Awni, W.; Zhu, T.; Heuser, R.S.; Doan, T.; Breitenbach, J.; Morris, J.B.; Brun, S.C.; Hanna, G.J. The tablet formulation of lopinavir/ritonavir provides similar bioavailability to the soft-gelatin capsule formulation with less pharmacokinetic variability and diminished food effect. J. Acquir. Immune Defic. Syndr. 2007, 44, 401–410. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Manufacturing Routes to Recently Approved PARP Inhibitors: Olaparib, Rucaparib, and Niraparib. Org. Process Res. Dev. 2017, 21, 1227–1244. [Google Scholar] [CrossRef]
- Banerjee, S.; Shankar, K.R.; Prasad, Y.R. Formulation development and systematic optimization of stabilized ziprasidone hydrochloride capsules devoid of any food effect. Pharm. Dev. Technol. 2016, 21, 775–786. [Google Scholar] [CrossRef]
- Othman, A.A.; Cheskin, H.; Locke, C.; Nothaft, W.; Dutta, S. A Phase 1 Study to Evaluate the Bioavailability and Food Effect of 2 Solid-Dispersion Formulations of the TRPV1 Antagonist ABT-102, Relative to the Oral Solution Formulation, in Healthy Human Volunteers. Clin. Pharmacol. Drug Dev. 2012, 1, 24–31. [Google Scholar] [CrossRef]
- Junnuthula, V.; Boroujeni, A.S.; Cao, S.; Tavakoli, S.; Ridolfo, R.; Toropainen, E.; Ruponen, M.; van Hest, J.C.M.; Urtti, A. Intravitreal polymeric nanocarriers with long ocular retention and targeted delivery to the retina and optic nerve head region. Pharmaceutics 2021, 13, 445. [Google Scholar] [CrossRef] [PubMed]
- Devassy, G.; Ramachandran, R.; Jeena, K.; Junnuthula, V.R.; Gopinatha, V.K.; Manju, C.; Manohar, M.; Nair, S.V.; Raghavan, S.C.; Koyakutty, M. Simultaneous release of two drugs from polymer nano-implant inhibits recurrence in glioblastoma spheroids. Precis. Nanomed. 2019, 2, 218–229. [Google Scholar] [CrossRef]
- Sarkar, A.; Junnuthula, V.; Dyawanapelly, S. Ocular therapeutics and molecular delivery strategies for neovascular age-related macular degeneration (Namd). Int. J. Mol. Sci. 2021, 22, 10594. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.S.; Pawar, P.S.; Sarkar, A.; Junnuthula, V.; Dyawanapelly, S. Bionanofactories for green synthesis of silver nanoparticles: Toward antimicrobial applications. Int. J. Mol. Sci. 2021, 22, 11993. [Google Scholar] [CrossRef]
- Sarkar, A.; Sodha, S.J.; Junnuthula, V.; Kolimi, P.; Dyawanapelly, S. Novel and investigational therapies for wet and dry age-related macular degeneration. Drug Discov. Today 2022, 27, 2322–2332. [Google Scholar] [CrossRef]
- Ridolfo, R.; Tavakoli, S.; Junnuthula, V.; Williams, D.S.; Urtti, A.; Van Hest, J.C.M. Exploring the Impact of Morphology on the Properties of Biodegradable Nanoparticles and Their Diffusion in Complex Biological Medium. Biomacromolecules 2021, 22, 126–133. [Google Scholar] [CrossRef]
- Ramachandran, R.; Junnuthula, V.R.; Gowd, G.S.; Ashokan, A.; Thomas, J.; Peethambaran, R.; Thomas, A.; Unni, A.K.K.; Panikar, D.; Nair, S.V.; et al. Theranostic 3-Dimensional nano brain-implant for prolonged and localized treatment of recurrent glioma. Sci. Rep. 2017, 7, srep43271. [Google Scholar] [CrossRef]
- Dyawanapelly, S.; Junnuthula, V.; Singh, A. The Holy Grail of Polymer Therapeutics for Cancer Therapy: An Overview on the Pharmacokinetics and Bio Distribution. Curr. Drug Metab. 2015, 16, 522–537. [Google Scholar] [CrossRef]
- Pailla, S.; Sampathi, S.; Junnuthula, V.; Maddukuri, S.; Dodoala, S.; Dyawanapelly, S. Brain-Targeted Intranasal Delivery of Zotepine Microemulsion: Pharmacokinetics and Pharmacodynamics. Pharmaceutics 2022, 14, 978. [Google Scholar] [CrossRef]
- Gera, S.; Sampathi, S.; Maddukuri, S.; Dodoala, S.; Junnuthula, V.; Dyawanapelly, S. Therapeutic Potential of Naringenin Nanosuspension: In Vitro and In Vivo Anti-Osteoporotic Studies. Pharmaceutics 2022, 14, 1449. [Google Scholar] [CrossRef]
- Khatik, R.; Dwivedi, P.; Junnuthula, V.R.; Sharma, K.; Chuttani, K.; Mishra, A.K.; Dwivedi, A.K. Potential in vitro and in vivo colon specific anticancer activity in a HCT-116 xenograft nude mice model: Targeted delivery using enteric coated folate modified nanoparticles. RSC Adv. 2015, 5, 16507–16520. [Google Scholar] [CrossRef]
- Al-Kassas, R.; Bansal, M.; Shaw, J. Nanosizing techniques for improving bioavailability of drugs. J. Control. Release 2017, 260, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; Müller, R.H.; Möschwitzer, J.P. Bottom-up approaches for preparing drug nanocrystals: Formulations and factors affecting particle size. Int. J. Pharm. 2013, 453, 126–141. [Google Scholar] [CrossRef]
- Rangaraj, N.; Pailla, S.R.; Chowta, P.; Sampathi, S. Fabrication of Ibrutinib Nanosuspension by Quality by Design Approach: Intended for Enhanced Oral Bioavailability and Diminished Fast Fed Variability. AAPS PharmSciTech 2019, 20, 326. [Google Scholar] [CrossRef] [PubMed]
- Sauron, R.; Wilkins, M.; Jessent, V.; Dubois, A.; Maillot, C.; Weil, A. Absence of a food effect with a 145 mg nanoparticle fenofibrate tablet formulation. Int. J. Clin. Pharmacol. Ther. 2006, 44, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Guivarc’h, P.H.; Vachon, M.G.; Fordyce, D. A new fenofibrate formulation: Results of six single-dose, clinical studies of bioavailability under fed and fasting conditions. Clin. Ther. 2004, 26, 1456–1469. [Google Scholar] [CrossRef]
- Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U.; et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: A Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004, 285, 135–146. [Google Scholar] [CrossRef]
- Deschamps, B.; Musaji, N.; Gillespie, J.A. Food effect on the bioavailability of two distinct formulations of megestrol acetate oral suspension. Int. J. Nanomed. 2009, 4, 185–192. [Google Scholar] [CrossRef]
- AboulFotouh, K.; Allam, A.A.; El-Badry, M.; El-Sayed, A.M. Role of self-emulsifying drug delivery systems in optimizing the oral delivery of hydrophilic macromolecules and reducing interindividual variability. Colloids Surf. B Biointerfaces 2018, 167, 82–92. [Google Scholar] [CrossRef]
- Cao, M.; Xue, X.; Pei, X.; Qian, Y.; Liu, L.; Ren, L.; Chen, G. Formulation optimization and pharmacokinetics evaluation of oral self-microemulsifying drug delivery system for poorly water soluble drug cinacalcet and no food effect. Drug Dev. Ind. Pharm. 2018, 44, 969–981. [Google Scholar] [CrossRef]
- Xue, X.; Cao, M.; Ren, L.; Qian, Y.; Chen, G. Preparation and Optimization of Rivaroxaban by Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Enhanced Oral Bioavailability and No Food Effect. AAPS PharmSciTech 2018, 19, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.P.; Holm, R.; O’Driscoll, C.M.; Griffin, B.T. Food for thought: Formulating away the food effect—A PEARRL review. J. Pharm. Pharmacol. 2019, 71, 510–535. [Google Scholar] [CrossRef] [PubMed]
- Michaelsen, M.H.; Wasan, K.M.; Sivak, O.; Müllertz, A.; Rades, T. The Effect of Digestion and Drug Load on Halofantrine Absorption from Self-nanoemulsifying Drug Delivery System (SNEDDS). AAPS J. 2016, 18, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Imada, C.; Takahashi, T.; Kuramoto, M.; Masuda, K.; Ogawara, K.I.; Sato, A.; Wataya, Y.; Kim, H.S.; Higaki, K. Improvement of oral bioavailability of n-251, a novel antimalarial drug, by increasing lymphatic transport with long-chain fatty acid-based self-nanoemulsifying drug delivery system. Pharm. Res. 2015, 32, 2595–2608. [Google Scholar] [CrossRef]
- Tong, Y.; Zhang, Q.; Shi, W.; Wang, J. Mechanisms of oral absorption improvement for insoluble drugs by the combination of phospholipid complex and SNEDDS. Drug Deliv. 2019, 26, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discov. 2007, 6, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Cherniakov, I.; Domb, A.J.; Hoffman, A. Self-nano-emulsifying drug delivery systems: An update of the biopharmaceutical aspects. Expert Opin. Drug Deliv. 2015, 12, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
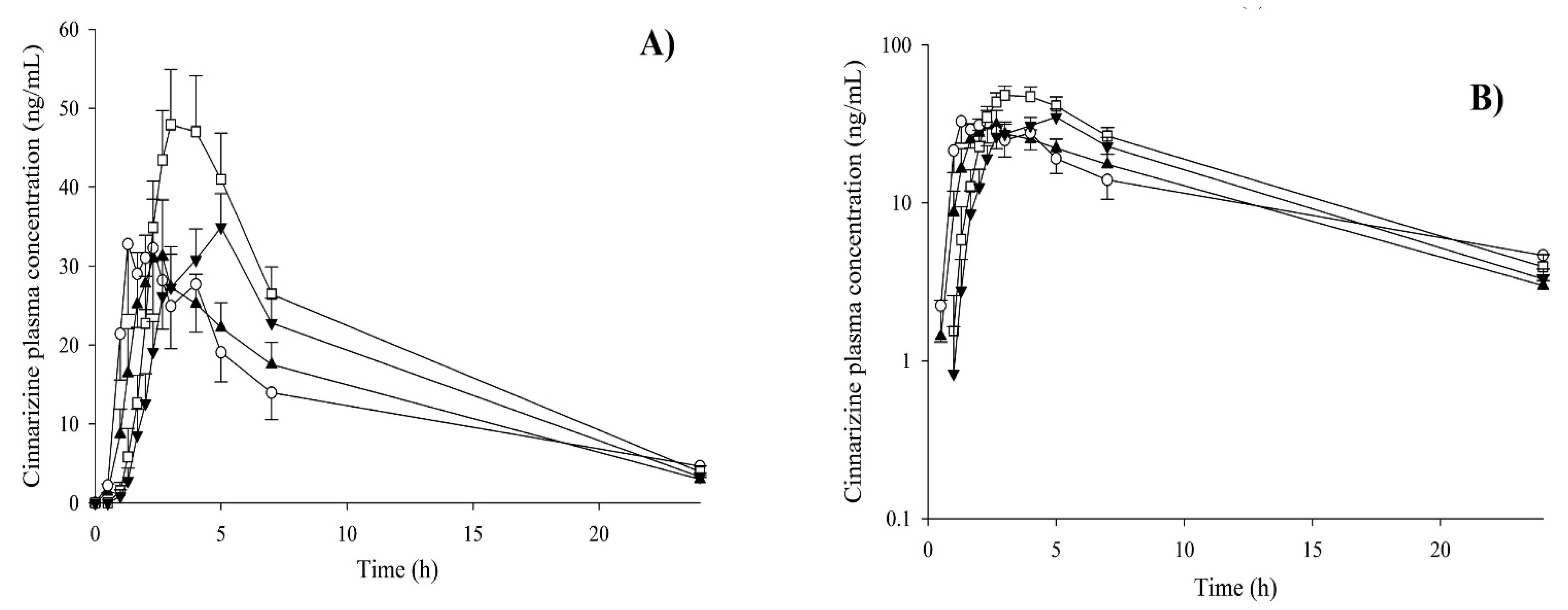
- Christiansen, M.L.; Holm, R.; Abrahamsson, B.; Jacobsen, J.; Kristensen, J.; Andersen, J.R.; Müllertz, A. Effect of food intake and co-administration of placebo self-nanoemulsifying drug delivery systems on the absorption of cinnarizine in healthy human volunteers. Eur. J. Pharm. Sci. 2016, 84, 77–82. [Google Scholar] [CrossRef]
- Miao, Y.; Sun, J.; Chen, G.; Lili, R.; Ouyang, P. Enhanced oral bioavailability of lurasidone by self-nanoemulsifying drug delivery system in fasted state. Drug Dev. Ind. Pharm. 2015, 42, 1234–1240. [Google Scholar] [CrossRef]
- Miao, Y.; Chen, G.; Ren, L.; Pingkai, O. Characterization and evaluation of self-nanoemulsifying sustained-release pellet formulation of ziprasidone with enhanced bioavailability and no food effect. Drug Deliv. 2016, 23, 2163–2172. [Google Scholar] [CrossRef]
- Hong, J.Y.; Kim, J.K.; Song, Y.K.; Park, J.S.; Kim, C.K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control. Release 2006, 110, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Dening, T.J.; Rao, S.; Thomas, N.; Prestidge, C.A. Silica encapsulated lipid-based drug delivery systems for reducing the fed/fasted variations of ziprasidone in vitro. Eur. J. Pharm. Biopharm. 2016, 101, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.M. The universality of the solubility parameter. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 2–11. [Google Scholar] [CrossRef]
- Bushra, R.; Aslam, N.; Khan, A.Y. Food-drug interactions. Oman Med. J. 2011, 26, 77–83. [Google Scholar] [CrossRef] [PubMed]
- ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media; ACR: Reston, VA, USA, 2013; Volume 105, p. 128. [Google Scholar]
- FDA. Food-Effect Bioavailability and Fed Bioequivalence Studies. J. Korean Pharm. Sci. 2004, 34, 223–228. [Google Scholar] [CrossRef]
- Chen, S.Q. Controlled Release Compositions with Reduced Food Effect. CN103037849A, 10 April 2013. [Google Scholar]
- NOAA. United States: United States. European Journal of Political Research Political Data Yearbook; NOAA: Washington, DC, USA, 2020; pp. 2–4.
- Zheng, A.P. Puerarin Nanocrystalline Medical Composition and Preparation Method Thereof. CN103211759B, 8 July 2015. [Google Scholar]
- Genovéva, F. Nanostructured Sildenafil Base, Its Pharmaceutically Acceptable Salts and Co-Crystals, Compositions of Them, Process for the Preparation Thereof and Pharmaceutical Compositions Containing Them. US9504652B2, 18 June 2010. [Google Scholar]
- Malhotra, G. Pharmaceutical Composition Comprising Abiraterone. WO2015114314A1, 6 August 2015. [Google Scholar]
- Givalucci, P. Fibrate-Statin Combination with Reduced Fed-Fasting Effect. JP2004523552A, 5 August 2014. [Google Scholar]
- Rudy, S.B. Nanoparticulate Fibrate Formulations. KR101300654B1, 28 August 2013. [Google Scholar]
- Filipcsei, G.; Ötvös, Z. Nanostructured Aprepitant Compositions, Process for the Preparation Thereof and Pharmaceutical Compositions Containing Them. U.S. Patent 9,504,652, 29 November 2018. [Google Scholar]
- Guivarch, P.H. Micropartículas de Fibrato Estabilizadas. ES2372746T3, 26 January 2012. [Google Scholar]
- Woelfel, K. Cannabinoid Compositions and Methods of Preparation Thereof. US20190015383A1, 17 January 2019. [Google Scholar]
- Grahek, R. Nanosuspension of Abiraterone Acetate. WO2014009436A1, 16 January 2014. [Google Scholar]
- Kallem, V.R. Reduced Dose Pharmaceutical Compositions of Fenofibrate. CH707330A2, 13 June 2014. [Google Scholar]
- Neville, D. Nanoparticulate Cinacalcet Compositions. US9012511B2, 21 April 2015. [Google Scholar]
- Nilsson, G. Controlled Food Effect Composition. US20080044486A1, 21 February 2008. [Google Scholar]
- Filipcsei, G. Nanoparticulate Telmisartan Compositions and Process for the Preparation Thereof. US20120135053A1, 31 May 2012. [Google Scholar]
- Rizzarelli, E. Carnosine Derivatives, A Process for the Preparation Thereof and Pharmaceutical Compositions Containing Them. EP1176154A1, 30 January 2002. [Google Scholar]
- Livesage, G. Nanoparticulate Corticosteroid and Antihistamine Formulations. CN101180038A, 14 May 2008. [Google Scholar]
- Douglas, H. Nanoparticulate Megestrol Formulations. KR20080024213A, 17 March 2008. [Google Scholar]
- Genoveva, P. Nanoparticulate Olmesartan Medoxomil Composition, Method for Its Preparation, and Pharmaceutical Composition Containing Them. JP2012530126A, 29 November 2012. [Google Scholar]
- Bosch, W.H. Compositions of Nanoparticles of Inhibitors of Protein Kinase ACTIVATED by mitogen (Map). ES2302925T3, 1 August 2008. [Google Scholar]
- Shaw, K. Nanoparticulate Formulations and Methods for the Making and Use Therof. US20090004262A1, 1 January 2009. [Google Scholar]
- Awadesh, M.K. Oral dosage forms comprising fenofibrate. JP2005535582A, 12 August 2010. [Google Scholar]
- Jenkins, S. Nanoparticulate Tacrolimus Formulations. CN101132768A, 27 February 2008. [Google Scholar]
- Dhingra, O.; Bernstein, J.S. Emulsion Formulations. US20130303495A1, 14 November 2013. [Google Scholar]
- Dong, X. Situ Self-Assembling pro-Nanoparticle Compositions and Methods of Preparation and Use Thereof. US20170112775A1, 27 April 2017. [Google Scholar]
- Hustvedt, S.O. A Composition Comprising A Lipid Compound, a TRIGLYCERIDE, and a Surfactant, and Methods of Using the Same. WO2014132134A1, 4 September 2014. [Google Scholar]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. NO. | Gastrointestinal Tract | Length (m) | Surface Area (m2) | Residence Time |
|---|---|---|---|---|
| 1. | Esophagus | 0.3 | 0.02 | 30 s |
| 2. | Stomach | 0.2 | 0.2 | 1–5 h |
| 3. | Duodenum | 0.3 | 0.02 | 5 min |
| 4. | Jejunum | 3 | 100 | 1–2 h |
| 5. | Ileum | 4 | 100 | 2–3 h |
| 6. | Colon | 1.5 | 3 | 15–48 |
| S. NO. | Branded Name | Drug | Formulation | Manufacturer |
|---|---|---|---|---|
| 1. | Prograf® | Tacrolimus | Amorphous solid dispersion | Astellas Pharma US, Inc., Northbrook, IL, USA |
| 2. | Kaletra® | Ritonavir/lopinavir | Amorphous solid dispersion | AbbVie Inc., North Chicago, IL, USA |
| 3. | Zortress®/Certican® | Everolimus | Amorphous solid dispersion | Novartis Pharmaceuticals Corporation East Hanover, NJ, USA |
| 4. | Zelboraf® | Vemurafenib | Amorphous solid dispersion | Genentech, Inc., South San Francisco, CA, USA |
| 5. | Ceftin® | Cefuroxime axetil | Amorphous form of drug | GlaxoSmithKline Inc., Collegeville, PA, USA |
| 6. | Accupril® | Quinapril HCl | Amorphous form of drug | Pfizer Inc., New York, NY, USA |
| 7. | Crestor® | Rosuvastatin Calcium | Amorphous form of drug | AstraZeneca Pharmaceuticals LP, Wilmington, DE, USA |
| 8. | Zepatier® | Elbasvir/Grazoprevir | Amorphous form of drug | Merck & Co., Inc., Rahway, NJ, USA |
| 9. | Agenerase® | Amprenavir | Lipid based formulation | GlaxoSmithKline Inc., Collegeville, PA, USA |
| 10. | Avodart® | Dutasteride | Lipid based formulation | GlaxoSmithKline Inc., Collegeville, PA, USA |
| 11. | Procardia® | Nifedipine | Lipid based formulation | Pfizer Inc., New York, NY, USA |
| 12. | Rapamune® | Sirolimus | Lipid based formulation | Pfizer Inc., New York, NY, USA |
| 13. | Amitiza® | Lubiprostone | Lipid based formulation | Sucampo Pharma Americas LLC, Bedminster, NJ, USA and Takeda Pharmaceuticals U.S.A., Inc., Lexington, MA, USA |
| 14. | Hycamtin® | Topotecan HCl | Lipid based formulation | Novartis Pharmaceuticals Corporation East Hanover, NJ, USA |
| 15. | Akynzeo® | Netupitant | Lipid based formulation | Helsinn Therapeutics (U.S.), Inc. Iselin, NJ, USA |
| 16. | Prometrium® | Progesterone | Lipid based formulation | Virtus Pharmaceuticals, LLC, Langhorne, PA, USA |
| 17. | Absorica® | Isotretinoin | Lipid based formulation | Sun Pharmaceutical Industries, Inc., Princeton, NJ, USA |
| 18. | Zemplar® | Paricalcitol | Lipid based formulation | AbbVie Inc. North Chicago, IL, USA |
| 19. | Vyndaqel® | Tafamidismeglumine | Lipid based formulation | Pfizer Inc., New York, NY, USA |
| 20. | Xtandi® | Enzalutamide | Lipid based formulation | Astellas Pharma US, Inc. Northbrook, IL, USA |
| 21. | Lipantil Supra® | Fenofibrate | Nanocrystal | AbbVie Inc. North Chicago, IL, USA |
| 22. | Emend® | Aprepitant | Nanocrystal | Merck & Co., Inc., Rahway, NJ, USA |
| 23. | Triglide® | Fenofibrate | Nanocrystal | Skye Pharma Inc., San Diego, CA, USA |
| 24. | Rapamune® | Sirolimus | Nanocrystal | Pfizer Inc., New York, NY, USA |
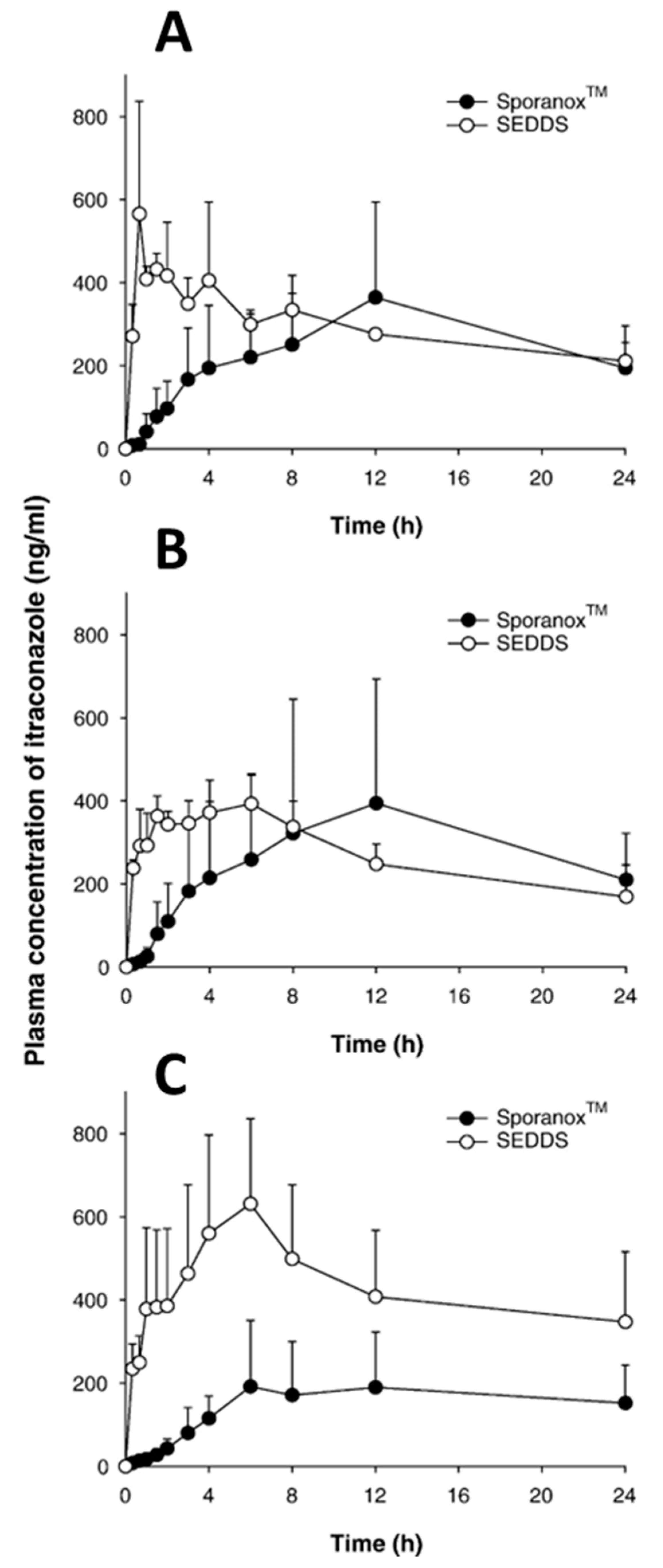
| 25. | Sporanox® | Itraconazole | Cyclodextrin | Janssen Pharmaceuticals, Inc. Titusville, NJ, USA |
| 26. | Lynparza® (capsule) | Olaparib | Crystalline solid dispersion | AstraZeneca Pharmaceuticals LP, Wilmington, DE, USA and Merck & Co., Inc., Rahway, NJ, USA |
| 27. | Lynparza® (tablet) | Olaparib | Hot-melt extrusion followed by compression of crystalline solid dispersion | AstraZeneca Pharmaceuticals LP, Wilmington, DE, USA and Merck & Co., Inc., Rahway, NJ, USA |
| Formulation Approaches | Drug | Fasted State | Fed State | Refs | ||
|---|---|---|---|---|---|---|
| AUC0–∞ (ng.h/mL) | Cmax (ng/mL) | AUC0–∞ (ng.h/mL) | Cmax (ng/mL) | |||
| Prodrug approach | Enalapril | 1209 ± 203 | 154 ± 39 | 1173 ±212 | 147 ± 36 | [96] |
| Cyclodextrin complexation | Amiodarone HCl | 1788 ± 121 | 3.024 ± 0.6631 | 1911 ± 141 | 3.314 ± 0.6139 | [104] |
| Osmotic delivery system | Methylphenidate HCl | 1857 ± 224 | 112.6 ± 15.6 | 1872 ± 242 | 124.9 ± 17.9 | [107] |
| Solid dispersion | Ziprasidone HCl | 874.265 ± 3.908 | 122.116 ± 2.081 | 988.67 ±4.234 | 123.457 ± 1.987 | [116] |
| Nanocrystal technology | Lurasidone HCl | 4718.81 ± 638.37 | 353.72 ± 21.83 | 4796.30 ± 562.44 | 360.70 ± 20.71 | [10] |
| SNEDDS | Cinnarizine | 1386 ± 474 | 372 ± 101 | 1961 ± 324 | 389 ± 57.0 | [145] |
| S. NO. | Marketed Drugs with High Fast-Fed Variability | pH-Dependent Solubility | pKa | Partition Coefficient | Molecular Weight | BCS Class | AUCfed/AUCfast | Cmaxfed/Cmaxfast |
|---|---|---|---|---|---|---|---|---|
| 1. | Tacrolimus | Acidic | 9.96 | 3.19 | 804.08 | II | 0.63 | 0.23 |
| 2. | Ritonavir | Acidic | 13.68 | 3.9 | 720.946 | II | 0.79 | 0.78 |
| 3. | Everolimus | Acidic | 9.96 | 7.4 | 958.224 | III | 0.84 | 0.40 |
| 4. | Vemurafenib | No | 7.1 | 4.62 | 489.92 | IV | 4.6 | 2.5 |
| 5. | Cefuroxime axetil | Acidic | 10.92 | 0.89 | 510.475 | II | 1.41 | 1.43 |
| 6. | Quinapril HCl | Acidic | 5.2 | 1.96 | 438.516 | II | 0.75 | - |
| 7. | Rosuvastatin Calcium | Basic | 4.6 | 1.92 | 1001.14 | II | 1 | 0.8 |
| 8. | Elbasvir/Grazoprevir | Basic | 3.77 | 3.34 | 882.05 | II | 1.5 | 2.8 |
| 9. | Amprenavir | Acidic | 13.61 | 2.2 | 505.628 | II | 0.79 | 0.64 |
| 10. | Dutasteride | Acidic | 12.56 | 6.8 | 528.53 | II | - | 0.85 |
| 11. | Nifedipine | No | 3.93 | 2.5 | 346.335 | II | 1 | 0.74 |
| 12. | Sirolimus | Acidic | 9.96 | 4.85 | 914.172 | II | 1.35 | - |
| 13. | Lubiprostone | No | 4.3 | 2.76 | 390.462 | II | 1 | 0.45 |
| 14. | Topotecan HCl | Acidic | 10.50 | −0.88 | 457.9 | IV | 1 | 1 |
| 15. | Netupitant | Acidic | 9 | 7.26 | 578.59 | II | 1.1 | 1.2 |
| 16. | Progesterone | Acidic | 18.92 | 3.87 | 314.46 | II | 1.99 | 5.19 |
| 17. | Isotretinoin | Basic | 5 | 6.3 | 300.44 | II | 1.5 | 1.26 |
| 18. | Paricalcitol | No | 14.81 | 4.5 | 416.36 | III | 1 | 1 |
| 19. | Tafamidismeglumine | Basic | 3.6 | 4.21 | 503.33 | IV | 1 | 1 |
| 20. | Fenofibrate | Basic | 3.1 | 5.24 | 360.831 | II | 1.58 | - |
| 21. | Aprepitant | Acidic | 9.7 | 4.8 | 534.427 | IV | 1.4 | - |
| 22. | Itraconazole | Acidic | 3.7 | 5.56 | 705.64 | II | 0.76 | 0.42 |
| 23. | Olaparib | No | 12.07 | 1.49 | 435.08 | IV | 1.2 | 1 |
| S. NO. | Patent | Title | Formulation Approach Used | Refs |
|---|---|---|---|---|
| 1. | US20110311594A1 | Controlled release compositions with reduced food effect. | Bilayered controlled release. | [154] |
| 2. | US20140212491A1 | Combination formulation of two antiviral compounds. | Solid dispersion. | [155] |
| 3. | CN103211759B | Puerarin nanocrystalline medical composition and preparation method thereof. | Nanocrystal. | [156] |
| 4. | CN102497857A | Nanostructured sildenafil base, its pharmaceutically acceptable salts and cocrystals, compositions of them, process for the preparation thereof, and pharmaceutical compositions containing them. | Cocrystals. | [157] |
| 5. | WO2015145157A1 | Pharmaceutical composition comprising pazopanib. | Nanoparticles. | [158] |
| 6. | JP2004523552A | Reduced food intake, fibrates with a fasting effect, the combination of statins. | Microparticles. | [159] |
| 7. | KR101300654B1 | Nanoparticulate fibrate formulations. | Nanoparticles. | [160] |
| 8. | US9504652B2 | Nanostructured aprepitant compositions, process for the preparation thereof, and pharmaceutical compositions containing them. | Polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol graft copolymer nanoparticles. | [161] |
| 9. | ES2372746T3 | Stabilized microparticles fibrate. | Microparticles stabilized by surface active phospholipids. | [162] |
| 10. | US20090028935A1 | Carvedilol forms, compositions, and methods of preparation thereof. | Amorphous carvedilol phosphate salt and a complexing agent and controlled release of amorphous form. | [163] |
| 11. | WO2014009436A1 | Nanosuspension of abiraterone acetate. | Nanosuspension of abiraterone acetate. | [164] |
| 12. | CH707330A2 | Pharmaceutical compositions with reduced dose of fenofibrate. | A mixture of fenofibrate nanoparticles and micronized fenofibrate. | [165] |
| 13. | US9012511B2 | Nanoparticulate cinacalcet compositions. | Cinacacalcet nanoparticles. | [166] |
| 14. | US20080044486A1 | Controlled food effect composition. | Membrane lipids for controlled release. | [167] |
| 15. | WO2015145145A1 | Pharmaceutical composition comprising lapatinib. | Nanoparticles. | [158] |
| 16. | US20120135053A1 | Nanoparticulate telmisartan compositions and process for the preparation thereof. | Nanostructured Telmisartan. | [168] |
| 17. | US20130210794A1 | Nanostructured ezetimibe compositions, process for the preparation thereof, and pharmaceutical compositions containing them. | Nanostructured ezetimibe. | [169] |
| 18. | CN101180038A | Nanoparticulate corticosteroid and antihistamine formulations. | Antihistamine corticosteroid nanoparticles. | [170] |
| 19. | KR20080024213A | Nanoparticulate megestrol formulations. | Megesterol acetate nanoparticles. | [171] |
| 20. | JP2012530126A | Nanoparticulate Olmesartan medoxomil composition, method for its preparation, and pharmaceutical composition containing them. | Nano cocrystals. | [172] |
| 21. | ES2302925T3 | Nanoparticle compositions, kinase inhibitors, mitogen activated protein (MAP). | Nanoparticles. | [173] |
| 22. | US20090004262A1 | Nanoparticulate formulations and methods for the making and use thereof. | Cyclodextrin inclusion complex | [174] |
| 23. | JP2005535582A | Coated tablets. | Phospholipid applied to the surface of the fenofibrate microparticles. | [175] |
| 24. | CN101132768A | Nanoparticulate tacrolimus formulations. | Nanoparticles. | [176] |
| 25. | US20130303495A1 | Emulsion formulations. | SNEDDS, SMEDDS, and SEDDS | [177] |
| 26. | US20170112775A1 | Situ self-assembling pro-nanoparticle compositions and methods of preparation and use thereof. | Self-assembling pronanoparticles. | [178] |
| 27. | WO2014132134A1 | A composition comprising a lipid compound, a triglyceride, and a surfactant, and methods of using the same. | SNEDDS, SMEDDS, and SEDDS | [179] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangaraj, N.; Sampathi, S.; Junnuthula, V.; Kolimi, P.; Mandati, P.; Narala, S.; Nyavanandi, D.; Dyawanapelly, S. Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects. Pharmaceutics 2022, 14, 1807. https://doi.org/10.3390/pharmaceutics14091807
Rangaraj N, Sampathi S, Junnuthula V, Kolimi P, Mandati P, Narala S, Nyavanandi D, Dyawanapelly S. Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects. Pharmaceutics. 2022; 14(9):1807. https://doi.org/10.3390/pharmaceutics14091807
Chicago/Turabian StyleRangaraj, Nagarjun, Sunitha Sampathi, Vijayabhaskarreddy Junnuthula, Praveen Kolimi, Preethi Mandati, Sagar Narala, Dinesh Nyavanandi, and Sathish Dyawanapelly. 2022. "Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects" Pharmaceutics 14, no. 9: 1807. https://doi.org/10.3390/pharmaceutics14091807
APA StyleRangaraj, N., Sampathi, S., Junnuthula, V., Kolimi, P., Mandati, P., Narala, S., Nyavanandi, D., & Dyawanapelly, S. (2022). Fast-Fed Variability: Insights into Drug Delivery, Molecular Manifestations, and Regulatory Aspects. Pharmaceutics, 14(9), 1807. https://doi.org/10.3390/pharmaceutics14091807