Appendix A
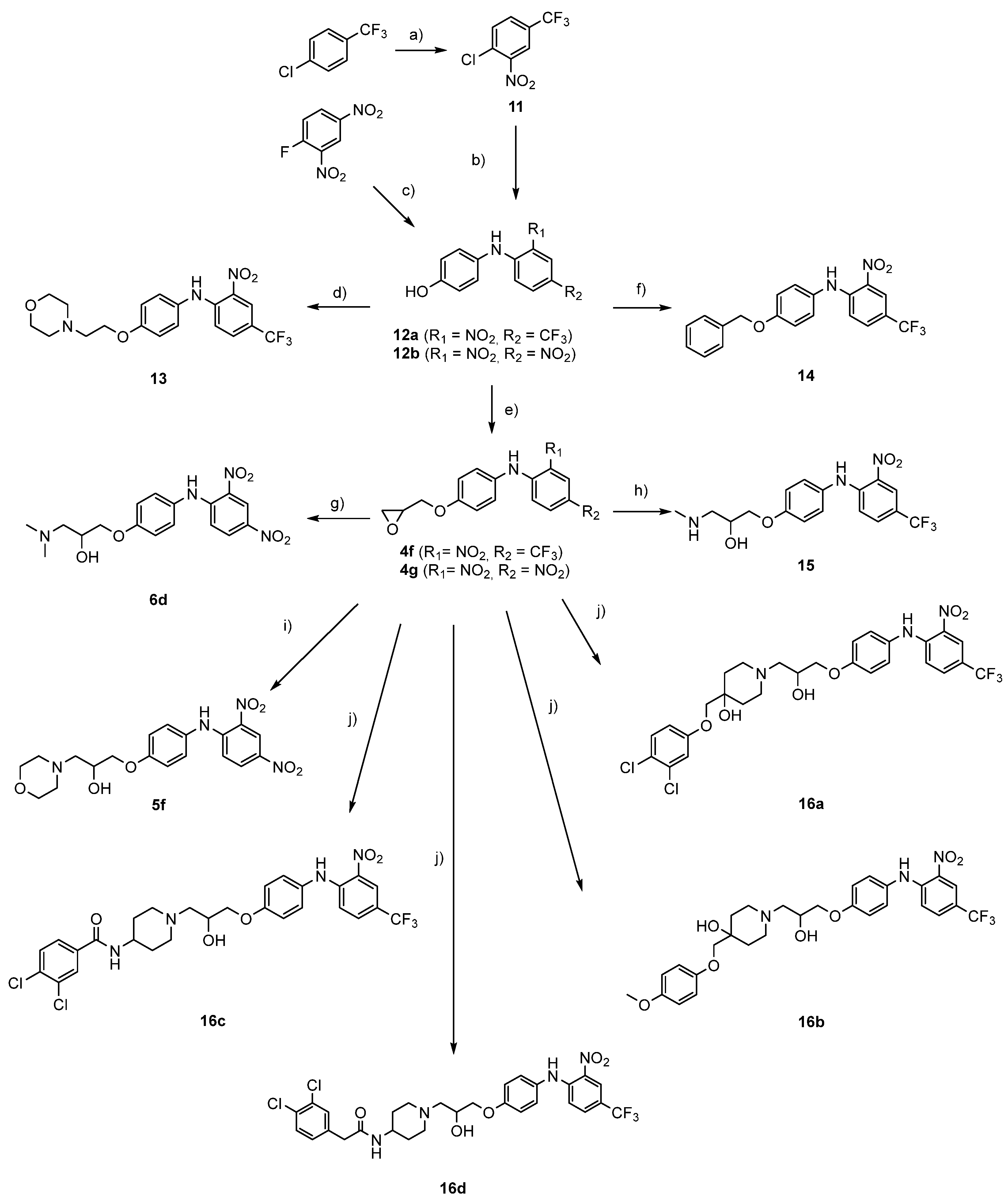
General procedure for synthesis of phenoxylphenol intermediates 3a-b (3a is given as example)
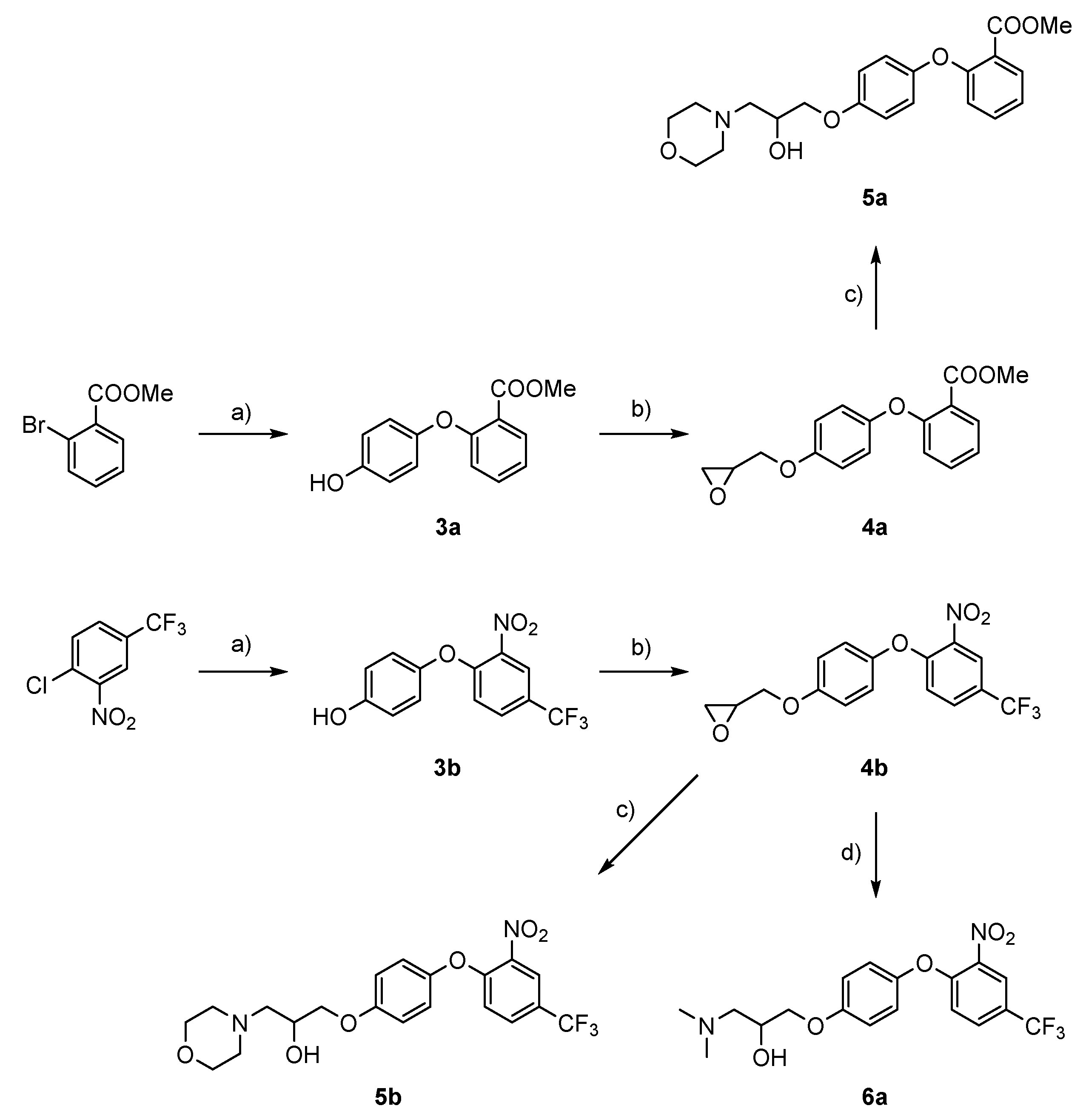
Synthesis of methyl 2-(4-hydroxyphenoxy)benzoate (3a): To a stirring solution of methyl 2-bromobenzoate (2.14 g, 9.95 mmol) in pyridine (50 mL), hydroquinone (1.10 g, 9.95 mmol), K2CO3 (2.75 g, 19.90 mmol), and CuO (80 mg, 1.00 mmol) were added. The batch was stirred at 110 °C for 72 h and then cooled to room temperature and filtered through Celite. The solvent was removed under reduced pressure and the crude product was purified by flash column chromatography using ethyl acetate/hexane = 1:1 as a mobile phase.
Methyl 2-(4-hydroxyphenoxy)benzoate (3a): Yield: 10%; white solid (250 mg); 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J1 = 7.8 Hz, J2 = 1.8 Hz, 1 H), 7.44–7.37 (m, 1 H), 7.11 (td, J1 = 7.7 Hz, J2 = 1.0 Hz, 1 H), 6.91–6.85 (m, 3 H), 6.83–6.78 (m, 2 H), 4.90 (brs, 1 H), and 3.86 (s, 3 H).
4-(2-Nitro-4-(trifluoromethyl)phenoxy)phenol (3b): Synthesised from 1-chloro-2-nitro-4-(trifluoromethyl)benzene (4.00 g, 17.73 mmol), hydroquinone (1.95 g, 17.73 mmol), K2CO3 (7.35 g, 53.20 mmol), and CuO (141 mg, 1.77 mmol) in pyridine (50 mL). Purified by flash column chromatography using ethyl acetate/hexane = 1:1 as mobile phase. Yield: 67%; yellow solid (3.565 g); 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 2.0 Hz, 1 H), 7.73–7.62 (m, 1 H), 7.03–6.96 (m, 3 H), 6.95–6.87 (m, 2 H), and 6.22 (brs, 1 H).
General procedure for binding of epichlorohydrin to phenol intermediates to obtain 4a–4g (4a is given as example)
To the argon-flushed solution of 3a (250 mg, 1.02 mmol) in MeCN (40 mL) on ice bath K2CO3 (423 mg, 3.06 mmol) and (±)-epichlorohydrin (0.94 g, 10.20 mmol, 0.80 mL) were added. Reaction mixture was stirred at 80 °C for 16 h. K2CO3 was filtered off and the solvent was removed under reduced pressure. Crude product was purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent to obtain 4a (428 mg).
Methyl 2-(4-(oxiran-2-ylmethoxy)phenoxy)benzoate (4a): Yield: 91%; white solid (280 mg); 1H NMR (400 MHz, CDCl3) δ 7.88 (dd, J1 = 7.8 Hz, J2 = 1.8 Hz, 1 H), 7.41 (ddd, J1 = 8.3 Hz, J2 = 7.4 Hz, J3 = 1.8 Hz, 1 H), 7.12 (td, J1 = 7.7 Hz, J2 = 1.1 Hz, 1 H), 6.98–6.85 (m, 5 H), 4.21 (dd, J1 = 11.0 Hz, J2 = 3.1 Hz, 1 H), 3.93 (dd, J1 = 11.0 Hz, J2 = 5.7 Hz, 1 H), 3.85 (s, 3 H), 3.35 (ddt, J1 = 5.7 Hz, J2 = 4.1 Hz, J3 = 3.0 Hz, 1 H), 2.91 (dd, J1 = 4.9 Hz, J2 = 4.2 Hz, 1 H), and 2.76 (dd, J1 = 4.9 Hz, J2 = 2.7 Hz, 1 H).
2-((4-(2-Nitro-4-(trifluoromethyl)phenoxy)phenoxy)methyl)oxirane (4b): Synthesised from 4-(2-nitro-4-(trifluoromethyl)phenoxy)phenol (3b) (3.55 g, 11.87 mmol, 1.0 equiv) with 10 equivalents of (±)-epichlorohydrin and 3 equivalents of K2CO3. Purified with flash column chromatography using ethyl acetate/hexane = 1:2 as mobile phase. Yield: 61.1%; yellow oil (1.45 g); 1H NMR (400 MHz, DMSO) δ 8.44 (d, J = 2.0 Hz, 1 H), 8.02–7.92 (m, 1 H), 7.24–7.15 (m, 2 H), 7.13–7.03 (m, 3 H), 4.36 (dd, J1 = 11.4 Hz, J2 = 2.6 Hz, 1 H), 3.86 (dd, J1 = 11.4 Hz, J2 = 6.6 Hz, 1 H), 3.37–3.33 (m, 1 H), 2.86 (dd, J1 = 5.0 Hz, J2 = 4.3 Hz, 1 H), and 2.72 (dd, J1 = 5.1 Hz, J2 = 2.7 Hz, 1 H).
2-Nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)benzamide (4c): Synthesised from N-(4-hydroxyphenyl)-2-nitro-4-(trifluoromethyl)benzamide (7) (1.60 g, 4.90 mmol) and 10 equivalents of (±)-epichlorohydrin and purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and with dry sample loading. Yield: 33%; red crystals (618 mg); 1H NMR (400 MHz, DMSO) δ 10.66 (s, 1 H), 8.50 (s, 1 H), 8.28 (dd, J1 = 8.0 Hz, J2 = 1.1 Hz, 1 H), 8.03 (d, J = 7.9 Hz, 1 H), 7.65–7.51 (m, 2 H), 7.04–6.92 (m, 2 H), 4.32 (dd, J1 = 11.4 Hz, J2 = 2.7 Hz, 1 H), 3.82 (dd, J1 = 11.4 Hz, J2 = 6.5 Hz, 1 H), 3.36–3.33 (m, 1 H), 2.87–2.81 (m, 1 H), and 2.71 (dd, J1 = 5.1 Hz, J2 = 2.7 Hz, 1 H).
Methyl 2-((4-(oxiran-2-ylmethoxy)phenyl)amino)benzoate (
4d): resynthesized according to described procedures [
26].
Methyl 2-((4-(oxiran-2-ylmethoxy)phenyl)amino)-5-(trifluoromethyl)benzoate (4e): Synthesised from methyl 2-((4-hydroxyphenyl)amino)-5-(trifluoromethyl)benzoate (9b) (330 mg, 1.06 mmol, 1.0 equiv.) with 10 equivalents of (±)-epichlorohydrin and purified with flash column chromatography using ethyl acetate/hexane = 1:4 as eluent. Yield: 72%; white solid (281 mg); 1H NMR (400 MHz, DMSO) δ 9.49 (s, 1 H), 8.10 (d, J = 1.7 Hz, 1 H), 7.63 (dd, J1 = 9.0 Hz, J2 = 2.3 Hz, 1 H), 7.29–7.19 (m, 2 H), 7.08–7.00 (m, 2 H), 6.97 (d, J = 9.0 Hz, 1 H), 4.35 (dd, J1 = 11.4 Hz, J2 = 2.6 Hz, 1 H), 3.89 (s, 3 H), 3.88–3.80 (m, 1 H), 3.40–3.33 (m, 1 H), 2.86 (dd, J1 = 5.0 Hz, J2 = 4.3 Hz, 1 H), and 2.72 (dd, J1 = 5.1 Hz, J2 = 2.7 Hz, 1 H).
2-Nitro-
N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (
4f): resynthesized according to described procedures [
26].
2,4-Dinitro-N-(4-(oxiran-2-ylmethoxy)phenyl)aniline (4g): Synthesised from 4-((2,4-dinitrophenyl)amino)phenol (12b) (1.93 g, 7.0 mmol, 1.0 equiv.), 10 equivalents of (±)-epichlorohydrin and K2CO3 (2.91 g, 21.1 mmol, 3.0 equiv.). Purified with flash column chromatography ethyl acetate/hexane = 1:1 as eluent. Yield: 78%; red solid (1.8 g); 1H NMR (400 MHz, DMSO) δ 10.10 (s, 1 H), 8.89 (d, J = 2.7 Hz, 1 H), 8.20 (dd, J1 = 9.6 Hz, J2 = 2.8 Hz, 1 H), 7.38–7.23 (m, 2 H), 7.16–7.04 (m, 2 H), 6.96 (d, J = 9.6 Hz, 1 H), 4.39 (dd, J1 = 11.4 Hz, J2 = 2.6 Hz, 1 H), 3.88 (dd, J1 = 11.4 Hz, J2 = 6.6 Hz, 1 H), 3.43–3.32 (m, 1 H), 2.87 (dd, J1 = 5.0 Hz, J2 = 4.3 Hz, 1 H), and 2.73 (dd, J1 = 5.1, J2 = 2.7 Hz, 1 H).
General procedure for opening of the epoxide with morpholine to obtain 5a-5f (5a is given as example)
To a stirring solution of methyl 2-(4-(oxiran-2-ylmethoxy)phenoxy)benzoate (4a) (100 mg, 0.333 mmol) in acetonitrile (20 mL), morpholine (87 mg, 0.999 mmol, 0.086 mL, 3.0 equiv.) was added and stirred for 72 h at room temperature. After 72 h, solvent was removed under reduced pressure and product was purified with flash column chromatography using dichloromethane/methanol/ammonium = 20:1:0.1.
Methyl 2-(4-(2-hydroxy-3-morpholinopropoxy)phenoxy)benzoate (5a): Yield: 40%; white solid (50 mg); 1H NMR (400 MHz, MeOD) δ 7.82 (dd, J1 = 7.8 Hz, J2 = 1.7 Hz, 1 H), 7.45 (ddd, J1 = 8.4 Hz, J2 = 7.4 Hz, J3 = 1.8 Hz, 1 H), 7.13 (td, J1 = 7.7 Hz, J2 = 1.0 Hz, 1 H), 6.99–6.88 (m, 4 H), 6.85 (dd, J1 = 8.3 Hz, J2 = 0.8 Hz, 1 H), 4.87 (brs, 1 H), 4.12 (ddd, J1 = 9.2 Hz, J2 = 7.4 Hz, J3 = 5.3 Hz, 1 H), 3.99 (dd, J1 = 9.7 Hz, J2 = 4.1 Hz, 1 H), 3.91 (dd, J1 = 9.8 Hz, J2 = 5.9 Hz, 1 H), 3.80 (s, 3 H), 3.74–3.60 (m, 4 H), 2.62–2.47 (m, 6 H); 13C NMR (101 MHz, MeOD) δ 168.01, 158.65, 156.71, 151.89, 134.71, 132.55, 123.76, 123.47, 121.21, 120.07, 116.78, 72.42, 68.11, 67.78, 62.57, 55.35, and 52.62; HRMS (ESI+) for C21H26NO6 ([M+H]+) calculated 388.1755 found 388.1745; HPLC retention time: 2.793 min (96.90% at 254 nm).
1-Morpholino-3-(4-(2-nitro-4-(trifluoromethyl)phenoxy)phenoxy)propan-2-ol (5b): Synthesised from 2-((4-(2-nitro-4-(trifluoromethyl)phenoxy)phenoxy)methyl)oxirane (4b) (500 mg, 1.41 mmol) in acetonitrile (30 mL) and three equivalents of morpholine (368 mg, 4.22 mmol, 0.364 mL). Purified with flash column chromatography using dichloromethane/methanol: 20/1 as eluent. Yield: 21%; yellow oil (130 mg); 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 1.9 Hz, 1 H), 7.71–7.64 (m, 1 H), 7.09–7.03 (m, 2 H), 6.99 (dt, J1 = 5.7 Hz, J2 = 3.3 Hz, 3 H), 4.18 (dq, J1 = 9.2 Hz, J2 = 4.7 Hz, 1 H), 4.05–3.95 (m, 2 H), 3.85–3.70 (m, 5 H), 2.81–2.71 (m, 2 H), 2.70–2.60 (m, 2 H), 2.60–2.52 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 156.71, 154.78, 147.72, 139.90, 130.86 (q, J = 3.4 Hz), 124.52 (q, J = 34.5 Hz), 123.64 (q, J = 3.9 Hz), 123.01 (q, J = 271.0 Hz), 121.80, 118.60, 116.27, 70.77, 66.86, 65.34, 61.18, and 53.87. HRMS (ESI+) for C20H22F3N2O6 ([M+H]+) calculated 443.1425 found 443.1415; HPLC retention time: 3.600 min (98.51% at 254 nm).
N-(4-(2-Hydroxy-3-morpholinopropoxy)phenyl)-2-nitro-4-(trifluoromethyl)benzamide (5c): Synthesised from 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)benzamide (4c) (300 mg, 0.78 mmol, 1.0 equiv.) and three equivalents of morpholine in dichloromethane (40 mL). Reaction was stopped after 72 h. Mobile phase for flash column chromatography was dichloromethane/methanol: 20/1. Yield: 41%; yellow solid (150 mg); 1H NMR (400 MHz, DMSO) δ 10.63 (s, 1 H), 8.50 (d, J = 1.0 Hz, 1 H), 8.28 (dd, J1 = 8.0 Hz, J2 = 1.1 Hz, 1 H), 8.02 (d, J = 7.9 Hz, 1 H), 7.59–7.49 (m, 2 H), 7.00–6.91 (m, 2 H), 4.87 (d, J = 4.5 Hz, 1 H), 4.00–3.92 (m, 2 H), 3.86 (dd, J1 = 11.0 Hz, J2 = 7.3 Hz, 1 H), 3.57 (t, J = 4.6 Hz, 4 H), 2.48–2.30 (m, 6 H); 13C NMR (101 MHz, MeOD) δ 165.61, 157.71, 147.99, 137.53, 133.66 (q, J = 34.2 Hz), 132.56, 131.82 (q, J = 3.4 Hz), 131.62, 124.20 (q, J = 271.0 Hz), 123.34, 122.88 (q, J = 3.8 Hz), 115.91, 72.19, 68.17, 67.84, 62.60, and 55.40. HRMS (ESI+) for C21H23F3N3O6 ([M+H]+) calculated 470.1534 found 470.1523; HPLC retention time: 2.930 min (99.76% at 254 nm).
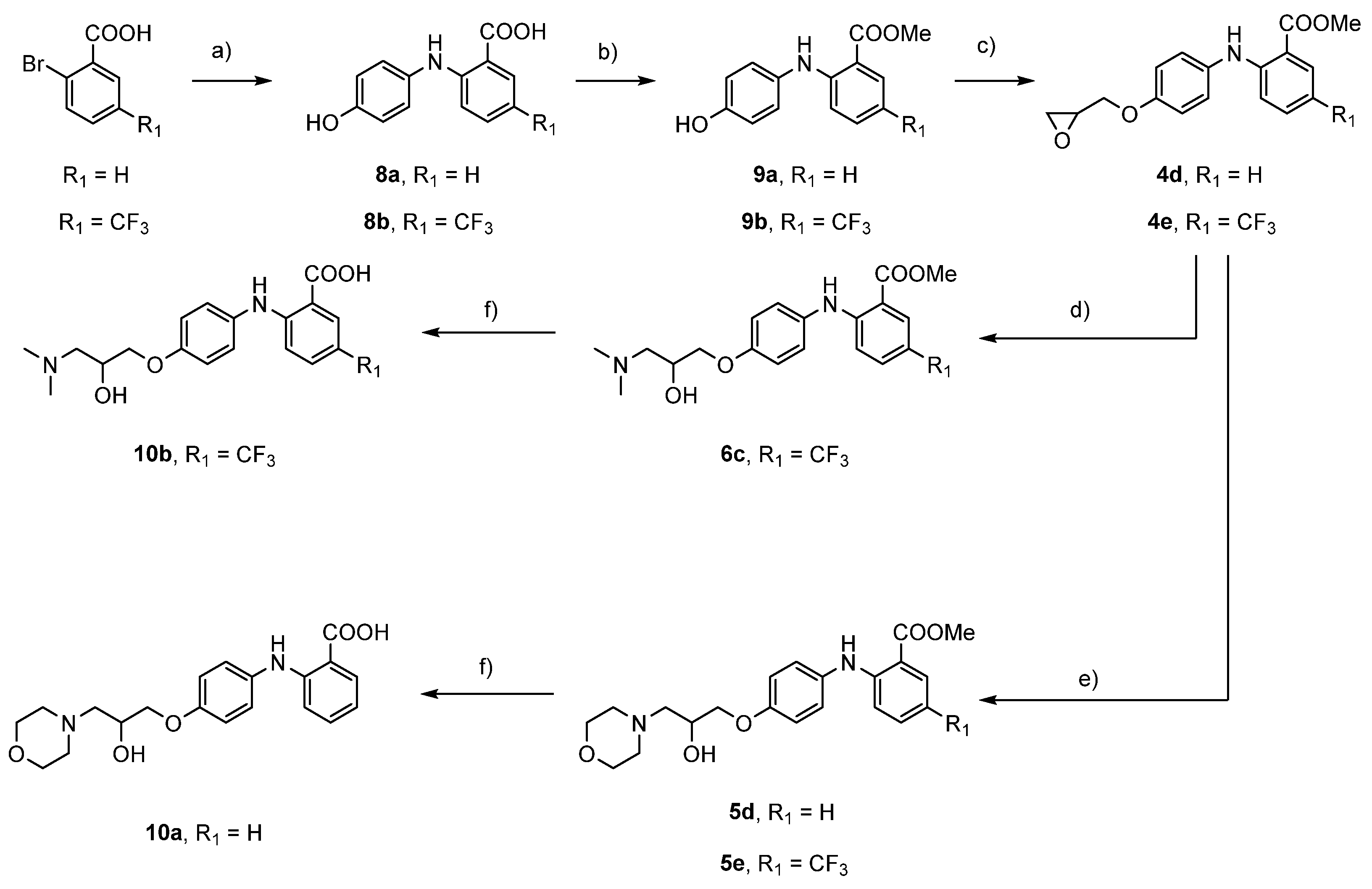
Methyl 2-((4-(2-hydroxy-3-morpholinopropoxy)phenyl)amino)benzoate (5d): Synthesised from methyl 2-((4-(oxiran-2-ylmethoxy)phenyl)amino)benzoate (4d) (590 mg, 1.97 mmol, 1.0 equiv.) and three equivalents of morpholine (515 mg, 5.91 mmol, 0.512 mL) for 72 h at room temperature. Purified with flash column chromatography using ethyl acetate/hexane = 2:1 and one more column with dichloromethane/methanol/ammonium = 20:1:0.1 as eluent and dry sample loading. Yield: 32%; yellow oil (240 mg); 1H NMR (400 MHz, DMSO) δ 9.15 (s, 1 H), 7.86 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1 H), 7.34 (ddd, J1 = 8.6 Hz, J2 = 7.2 Hz, J3 = 1.6 Hz, 1 H), 7.21–7.13 (m, 2 H), 7.00–6.95 (m, 2 H), 6.93 (dd, J1 = 8.6 Hz, J2 = 0.7 Hz, 1 H), 6.71 (ddd, J1 = 8.1 Hz, J2 = 7.1 Hz, J3 = 1.1 Hz, 1 H), 4.87 (d, J = 4.7 Hz, 1 H), 4.01–3.92 (m, 2 H), 3.87 (dd, J1 = 10.0 Hz, J2 = 6.4 Hz, 1 H), 3.85 (s, 3 H), 3.56 (t, J = 4.6 Hz, 4 H), 2.48–2.31 (m, 6 H). 13C NMR (101 MHz, MeOD) δ 170.24, 157.48, 150.64, 135.28, 134.88, 132.56, 126.63, 117.38, 116.53, 114.31, 112.04, 72.22, 68.14, 67.81, 62.61, 55.38, and 52.16; HRMS (ESI+) for C21H27N2O5 ([M+H]+) calculated 387.1915 found 387.1905; HPLC retention time: 3.940 min (98.44% at 254 nm).
Methyl 2-((4-(2-hydroxy-3-morpholinopropoxy)phenyl)amino)benzoate (5e): Synthesised from methyl 2-((4-(oxiran-2-ylmethoxy)phenyl)amino)-5-(trifluoromethyl)benzoate (4e) (590 mg, 1.97 mmol) with three equivalents of morpholine (100 mg, 1.14 mmol, 0.099 mL) at room temperature for 72 h. Purified with flash column chromatography using ethyl acetate/hexane = 1:4. Yield: 11%; yellow oil (100 mg); 1H NMR (400 MHz, CDCl3) δ 9.57 (s, 1 H), 8.21 (d, J = 1.4 Hz, 1 H), 7.43 (dd, J1 = 9.0 Hz, J2 = 2.2 Hz, 1 H), 7.21–7.12 (m, 2 H), 7.02–6.89 (m, 3 H), 4.13 (tt, J1 = 9.5 Hz, J2 = 4.9 Hz, 1 H), 4.05–3.97 (m, 2 H), 3.93 (s, 3 H), 3.81–3.66 (m, 4 H), 2.76–2.64 (m, 2 H), 2.64–2.53 (m, 2 H), 2.54–2.43 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 168.38, 156.75, 151.92, 132.51, 130.71 (q, J = 3.3 Hz), 129.46 (q, J = 3.9 Hz), 124.50 (q, J = 270.6 Hz), 126.74, 117.98 (q, J = 33.2 Hz), 115.69, 113.42, 109.99, 70.64, 67.15, 65.52, 61.14, 53.90, and 52.16; HRMS (ESI+) for C22H26F3N2O5 ([M+H]+) calculated 455.1788 found 455.1777; HPLC retention time: 3.953 min (96.49% at 254 nm).
1-(4-((2,4-Dinitrophenyl)amino)phenoxy)-3-morpholinopropan-2-ol (5f): Synthesised from 2,4-dinitro-N-(4-(oxiran-2-ylmethoxy)phenyl)aniline (4g) (442 mg, 1.34 mmol, 1.0 equiv.) with morpholine (465 mg, 5.34 mmol, 0.470 mL, 4.0 equiv.) in acetonitrile (50 mL) at room temperature for 72 h. Purified with flash column chromatography using dichloromethane/methanol/NH3 = 20:1:0.01 as eluent and Biotage Isolera One System reversed-phase chromatography (Biotage SNAP Cartridge KP-C18-HS 12 g column), MF: gradient 0,1% trifluoroacetic acid in H2O/acetonitrile). Yield: 18%; red amorph solid (100 mg); 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1 H), 9.17 (d, J = 2.6 Hz, 1 H), 8.21–8.07 (m, 1 H), 7.25–7.19 (m, 2 H), 7.09–6.94 (m, 3 H), 4.16 (td, J1 = 9.4 Hz, J2 = 4.5 Hz, 1 H), 4.09–3.98 (m, 2 H), 3.85–3.68 (m, 4 H), 2.79–2.44 (m, 6 H); 13C NMR (101 MHz, CDCl3) δ 158.35, 148.03, 137.24, 130.88, 130.01, 129.64, 127.59, 124.25, 116.19, 116.09, 70.69, 67.10, 65.39, 61.03, and 53.86; HRMS (ESI+) for C19H23N4O7 ([M+H]+) calculated 419.1561 found 419.1550; HPLC retention time: 3.430 min (99.08% at 254 nm).
General procedure for opening of the epoxide with dimethylamine to obtain 6a–6d (6a is given as example)
To a stirring solution of methyl 2-(4-(oxiran-2-ylmethoxy)phenoxy)benzoate (4b) (500 mg, 1.4 mmol, 1.0 equiv) in acetonitrile (30 mL), Me2NH × HCl (460 mg, 5.63 mmol, 4.0 equiv) and K2CO3 (580 mg, 4.2 mmol, 3.0 equiv) were added and the batch was stirred at 70 °C for 24 h. The solvent was removed under reduced pressure and the crude product was purified with Biotage Isolera One System reversed-phase chromatography (Biotage SNAP Cartridge KP-C18-HS 12 g column), MF: gradient 0,1% trifluoroacetic acid in H2O/acetonitrile). Fractions containing 1-(dimethylamino)-3-(4-(2-nitro-4-(trifluoromethyl)phenoxy)phenoxy)propan-2-ol (6a) were combined, acetonitrile was removed under reduced pressure and pH of remaining water phase was set to 8, and product was extracted into ethyl acetate (2 × 25 mL). Organic phase was washed with brine (2 × 25 mL) dried over Na2SO4 filtered and the solvent removed under reduced pressure to obtain 6a (100 mg).
1-(Dimethylamino)-3-(4-(2-nitro-4-(trifluoromethyl)phenoxy)phenoxy)propan-2-ol (6a): Yield: 18%; yellow oil (100 mg); 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 1.8 Hz, 1 H), 7.68 (dd, J1 = 8.9 Hz, J2 = 2.0 Hz, 1 H), 7.09–6.93 (m, 5 H), 4.36 (brs, 1 H), 4.22–4.13 (m, 1 H), 4.01 (d, J = 4.9 Hz, 2 H), 2.77–2.67 (m, 1 H), 2.55 (dd, J1 = 12.4 Hz, J2 = 2.7 Hz, 1 H), 2.45 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 156.43, 154.67, 147.80, 139.85, 130.95 (q, J = 3.7 Hz), 124.55 (q, J = 34.5 Hz), 123.60 (q, J = 3.8 Hz), 123.02 (q, J = 271.0 Hz), 121.75, 118.73, 116.30, 70.54, 65.41, 61.62, and 45.07; HRMS (ESI+) for C18H20F3N2O5 ([M+H]+) calculated 401.1319 found 401.1310; HPLC retention time: 3.593 min (95.01% at 254 nm).
N-(4-(3-(Dimethylamino)-2-hydroxypropoxy)phenyl)-2-nitro-4-(trifluoromethyl)benzamide (6b): Synthesised from 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)benzamide (4c) (300 mg, 0.78 mmol) and six equivalents of Me2NH × HCl in acetonitrile (40 mL). Purified with Biotage Isolera One System reversed-phase chromatography (Biotage SNAP Cartridge KP-C18-HS 12 g column), MF: gradient 0,1% trifluoroacetic acid in H2O/acetonitrile). Yield: 27%; yellow solid (90 mg); 1H NMR (400 MHz, DMSO) δ 10.77 (s, 1 H), 9.82 (s, 1 H), 8.50 (d, J = 0.9 Hz, 1 H), 8.28 (dd, J1 = 8.0 Hz, J2 = 1.1 Hz, 1 H), 8.03 (d, J = 7.9 Hz, 1 H), 7.64–7.53 (m, 2 H), 7.04–6.91 (m, 2 H), 6.00 (d, J = 3.9 Hz, 1 H), 4.29 (d, J = 2.5 Hz, 1 H), 4.02–3.88 (m, 2 H), 3.40–3.13 (m, 2 H), 2.84 (d, J = 11.5 Hz, 6 H); 13C NMR (101 MHz, MeOD) δ 165.70, 157.18, 148.00, 137.49, 133.73 (q, J = 34.2 Hz), 133.05, 131.86 (q, J = 3.5 Hz), 130.79, 123.76 (q, J = 271.0 Hz), 123.40, 122.90 (q, J = 3.3 Hz), 115.94, 71.30, 65.37, and 61.01. HRMS (ESI+) for C19H21F3N3O5 ([M+H]+) calculated 428.1428 found 428.1420; HPLC retention time: 2.893 min (97.37% at 254 nm).
Methyl 2-((4-(3-(dimethylamino)-2-hydroxypropoxy)phenyl)amino)-5-(trifluoromethyl)benzoate (6c): Synthesised from methyl 2-((4-(oxiran-2-ylmethoxy)phenyl)amino)-5-(trifluoromethyl)benzoate (4e) (140 mg, 0.381 mmol), K2CO3 (157 mg, 1.14 mmol, 3.0 equiv.) and Me2NH × HCl (186 mg, 2.28 mmol, 6.0 equiv) in acetonitrile (35 mL) at 70 °C overnight. Yield: 64%; yellow oil (100 mg); 1H NMR (400 MHz, CDCl3) δ 9.56 (s, 1 H), 8.21 (d, J = 1.4 Hz, 1 H), 7.42 (dd, J1 = 9.0 Hz, J2 = 2.3 Hz, 1 H), 7.21–7.10 (m, 2 H), 7.02–6.90 (m, 3 H), 4.12–4.04 (m, 1 H), 4.03–3.96 (m, 2 H), 3.93 (s, 3 H), 2.63–2.50 (m, 1 H), 2.40 (dd, J1 = 12.2 Hz, J2 = 3.8 Hz, 1 H), 2.34 (s, 6 H). 13C NMR (101 MHz, CDCl3) δ 168.32, 156.81, 151.90, 132.36, 130.66 (q, J = 3.2 Hz), 129.40 (q, J = 3.9 Hz), 126.68, 124.48 (q, J = 270.6 Hz), 117.87 (q, J = 33.2 Hz), 115.66, 113.40, 109.92, 70.84, 66.31, 61.92, 52.11, and 45.67; HRMS (ESI+) C20H24F3N2O4 ([M+H]+) calculated 413.1683 found 413.1672; HPLC retention time: 3.950 min (98.29% at 254 nm).
1-(Dimethylamino)-3-(4-((2,4-dinitrophenyl)amino)phenoxy)propan-2-ol (6d): Synthesised from 2,4-dinitro-N-(4-(oxiran-2-ylmethoxy)phenyl)aniline (4g) (170 mg, 0.706 mmol, 1.0 equiv.), Me2NH × HCl (344 mg, 4.220 mmol, 6.0 equiv.) and K2CO3 (291 mg, 2.110 mmol, 3.0 equiv.) in acetonitrile (30 mL) at 70 °C for 24 h. Purified with flash column chromatography using dichloromethane/methanol/NH3 = 20:1:0.01 as eluent and Biotage Isolera One System reversed-phase chromatography (Biotage SNAP Cartridge KP-C18-HS 12 g column), MF: gradient 0,1% trifluoroacetic acid in H2O/acetonitrile). Yield: 30%; red solid (80 mg); 1H NMR (400 MHz, DMSO) δ 10.09 (s, 1 H), 8.89 (d, J = 2.7 Hz, 1 H), 8.20 (dd, J1 = 9.6 Hz, J2 = 2.8 Hz, 1 H), 7.33–7.22 (m, 2 H), 7.12–7.04 (m, 2 H), 6.97 (d, J = 9.6 Hz, 1 H), 4.88 (d, J = 3.8 Hz, 1 H), 4.02 (dd, J1 = 9.0 Hz, J2 = 2.9 Hz, 1 H), 3.97–3.86 (m, 2 H), 2.46–2.27 (m, 2 H), and 2.21 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 158.39, 148.03, 137.16, 130.80, 129.97, 129.51, 127.53, 124.19, 116.16, 116.11, 70.86, 66.15, 61.78, and 45.65. HRMS (ESI+) for C17H21N4O6 ([M+H]+) calculated 377.1456 found 377.1447; HPLC retention time: 3.460 min (99.44% at 254 nm).
Synthesis of amide intermediate 7
To a solution of 2-nitro-4-(trifluoromethyl)benzoic acid (3.00 g, 12.76 mmol, 1.0 equiv.) in dichloromethane (100 mL), two drops of DMF and oxalyl chloride (4.86 g, 38.28 mmol, 3.28 mL, 3.0 equiv.) were added while ice cooling and the batch was stirred at 40 °C for 24 h. Solvent was removed under reduced pressure and crude product was resolved in the acetonitrile (20 mL) on the ice bath. To the ice cooled solution, 4-aminophenol (1.67 g, 15.31 mmol, 1.2 equiv.) and triethylamine (3.87 g, 51.04 mmol, 5.33 mL, 3.0 equiv.) were added dropwise and then reaction mixture was heated at reflux overnight. The solvent was removed under reduced pressure. Product was further purified using flash column chromatography with ethyl acetate/hexane = 1:2 as mobile phase and with dry sample loading.
N-(4-Hydroxyphenyl)-2-nitro-4-(trifluoromethyl)benzamide (7): Yield: 38%; red solid (1.6 g); 1H NMR (400 MHz, DMSO) δ 10.52 (s, 1 H), 9.34 (s, 1 H), 8.49 (d, J = 0.8 Hz, 1 H), 8.26 (dd, J1 = 8.0 Hz, J2 = 1.1 Hz, 1 H), 8.00 (d, J = 7.9 Hz, 1 H), 7.49–7.36 (m, 2 H), and 6.83–6.68 (m, 2 H).
General procedure for synthesis of benzoic acid derivatives 8a–b (8a is given as example)
To an argon-flushed solution of 2-bromobenzoic acid (4.00 g, 19.90 mmol, 1.0 equiv.) in 2-ethoxyethanol (20 mL), CuO (63 mg, 0.80 mmol, 0.04 equiv.), Cu (114 mg, 1.79 mmol, 0.09 equiv.), K2CO3 (5.50 g, 39.80 mmol), and 4-aminophenol (2.28 g, 20.89 mmol, 1.05 equiv.) were added and the batch was stirred at 130 °C for 16 h. Solution was cooled to room temperature and then water (100 mL) was added. Mixture was filtered through Celite and then pH was set to 5 to precipitate black solid that was filtered off. Black precipitate was dissolved in the 5% solution of Na2CO3 (250 mL) and solution was filtered through Celite. pH in solution was then set to 5 to precipitate black solid that was filtered off and dried overnight.
2-((4-Hydroxyphenyl)amino)benzoic acid (8a): Yield: 50%; black solid (2.28 g); 1H NMR (400 MHz, DMSO) δ 12.90 (s, 1 H), 9.42 (brs, 1 H), 9.34 (brs, 1 H), 7.84 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1 H), 7.30 (ddd, J1 = 8.7 Hz, J2 = 7.1 Hz, J3 = 1.7 Hz, 1 H), 7.08–7.02 (m, 2 H), 6.86 (dd, J1 = 8.5 Hz, J2 = 0.7 Hz, 1 H), 6.83–6.77 (m, 2 H), and 6.69–6.62 (m, 1 H).
2-((4-Hydroxyphenyl)amino)-5-(trifluoromethyl)benzoic acid (8b): Synthesised from 2-bromo-5-(trifluoromethyl)benzoic acid (3.00 g, 11.15 mmol, 1.0 equiv.), 4-aminophenol (1.46 g, 13.38 mmol, 1.2 equiv.), K2CO3 (4.62 g, 33.46 mmol, 3.0 equiv.), CuO (35 mg, 0.45 mmol, 0.04 equiv.), and Cu (64 mg, 1.00 mmol, 0.09 equiv.) in 2-ethoxyethanol (20 mL). Yield: 60%; black solid (2 g); 1H NMR (400 MHz, DMSO) δ 9.69 (s, 1 H), 9.54 (brs, 1 H), 8.08 (d, J = 1.7 Hz, 1 H), 7.58 (dd, J1 = 9.0 Hz, J2 = 2.2 Hz, 1 H), 7.14–7.06 (m, 2 H), 6.91 (d, J = 8.9 Hz, 1 H), and 6.86–6.79 (m, 2 H).
General procedure for synthesis of methyl ester derivatives 9a–b (9a is given as an example)
2-((4-Hydroxyphenyl)amino)benzoic acid (8a) (2.28 g, 9.96 mmol, 1.0 equiv.) was dissolved in anhydrous methanol (100 mL) and then thionyl chloride (11.85 g, 99.59 mmol, 7.22 mL, 10.0 equiv) was slowly added while ice cooling. The batch was stirred overnight at 70 °C. The solvent was removed under reduced pressure, crude product was purified with flash column chromatography using ethyl acetate/hexane = 1:4 as mobile phase and with dry sample loading.
Methyl 2-((4-hydroxyphenyl)amino)benzoate (9a): Yield: 55%; white solid (1.33 g); 1H NMR (400 MHz, DMSO) δ 9.38 (s, 1 H), 9.08 (s, 1 H), 7.85 (dd, J1 = 8.0 Hz, J2 = 1.6 Hz, 1 H), 7.32 (ddd, J1 = 8.6 Hz, J2 = 7.2 Hz, J3 = 1.6 Hz, 1 H), 7.09–7.02 (m, 2 H), 6.86 (dd, J1 = 8.6 Hz, J2 = 0.8 Hz, 1 H), 6.83–6.76 (m, 2 H), 6.67 (ddd, J = 8.1 Hz, J2 = 7.1 Hz, J3 = 1.1 Hz, 1 H), and 3.84 (s, 3 H).
Methyl 2-((4-hydroxyphenyl)amino)-5-(trifluoromethyl)benzoate (9b): Synthesised from 2-((4-hydroxyphenyl)amino)-5-(trifluoromethyl)benzoic acid (7b) (2.00 g, 6.73 mmol, 1.0 equiv.) with thionyl chloride (8.00 g, 67.29 mmol, 4.88 mL, 10.0 equiv.) in anhydrous methanol (75 mL). Purified with flash column chromatography using ethyl acetate/hexane = 1:4 as mobile phase. Yield: 16%; white solid (330 mg); 1H NMR (400 MHz, DMSO) δ 9.54 (s, 1 H), 9.41 (s, 1 H), 8.09 (d, J = 1.7 Hz, 1 H), 7.61 (dd, J1 = 9.1 Hz, J2 = 2.2 Hz, 1 H), 7.10 (d, J = 8.7 Hz, 2 H), 6.91 (d, J = 9.0 Hz, 1 H), 6.86–6.73 (m, 2 H), and 3.88 (s, 3 H).
General procedure for hydrolysis of methyl ester to obtain 10a–b (10a is given as an example)
Methyl 2-((4-(2-hydroxy-3-morpholinopropoxy)phenyl)amino)benzoate (5d) (123 mg, 0.318 mmol, 1.0 equiv.) was dissolved in ethanol (20 mL) and then solution of NaOH (19 mg, 0.477 mmol, 1.5 equiv.) in 1 mL of water was added and stirred at room temperature for 72 h. Solvent was removed under reduced pressure to obtain product 10a.
2-((4-(2-Hydroxy-3-morpholinopropoxy)phenyl)amino)benzoic acid (10a): Yield: 100%; white solid (118 mg); 1H NMR (400 MHz, DMSO) δ 11.50 (s, 1 H), 7.90 (dd, J1 = 7.7 Hz, J2 = 1.6 Hz, 1 H), 7.11–7.02 (m, 3 H), 6.98 (d, J = 7.6 Hz, 1 H), 6.88 (d, J = 8.9 Hz, 2 H), 6.62–6.49 (m, 1 H), 4.95 (brs, 1 H), 4.01–3.89 (m, 2 H), 3.88–3.75 (m, 1 H), 3.56 (t, J = 4.6 Hz, 4 H), 2.48–2.28 (m, 6 H); 13C NMR (101 MHz, DMSO) δ 171.88, 153.58, 146.66, 135.84, 132.10, 129.92, 122.22, 121.71, 115.62, 115.33, 111.85, 71.34, 66.45, 66.29, 61.60, and 54.09; HRMS (ESI+) for C20H25N2O5 ([M+H]+) calculated 373.1758 found 373.1751; HPLC retention time: 3.033 min (99.37% at 254 nm).
2-((4-(3-(Dimethylamino)-2-hydroxypropoxy)phenyl)amino)-5-(trifluoromethyl)benzoic acid (10b): Synthesised from methyl 2-((4-(3-(dimethylamino)-2-hydroxypropoxy)phenyl)amino)-5-(trifluoromethyl)benzoate (6c) (28 mg, 0.07 mmol, 1.0 equiv.) with solution of NaOH (4.2 mg, 0.11 mmol, 1.5 equiv.) in 1 mL of water and in ethanol (5 mL) at room temperature for 72 h. Yield: 100%; white solid (28 mg); 1H NMR (400 MHz, DMSO) δ 12.10 (s, 1 H), 8.16 (d, J = 2.2 Hz, 1 H), 7.33 (dd, J1 = 8.7 Hz, J2 = 2.3 Hz, 1 H), 7.10 (d, J = 8.8 Hz, 2 H), 6.98 (d, J = 8.6 Hz, 1 H), 6.93 (d, J = 8.9 Hz, 2 H), 4.87 (brs, 1 H), 4.00–3.77 (m, 3 H), 2.44–2.24 (m, 2 H), 2.18 (s, 6 H). 13C NMR (101 MHz, MeOD) δ 174.97, 157.11, 151.89, 135.41, 130.67 (q, J = 3.7 Hz), 128.77 (q, J = 3.5 Hz), 126.55 (q, J = 269.4 Hz), 125.95, 120.53, 118.11 (q, J = 32.4 Hz), 116.51, 112.99, 72.34, 68.80, 63.31, and 46.21; HRMS (ESI+) for C19H22F3N2O4 ([M+H]+) calculated 399.1526, found 399.1519; HPLC retention time: 3.830 min (95.01% at 254 nm).
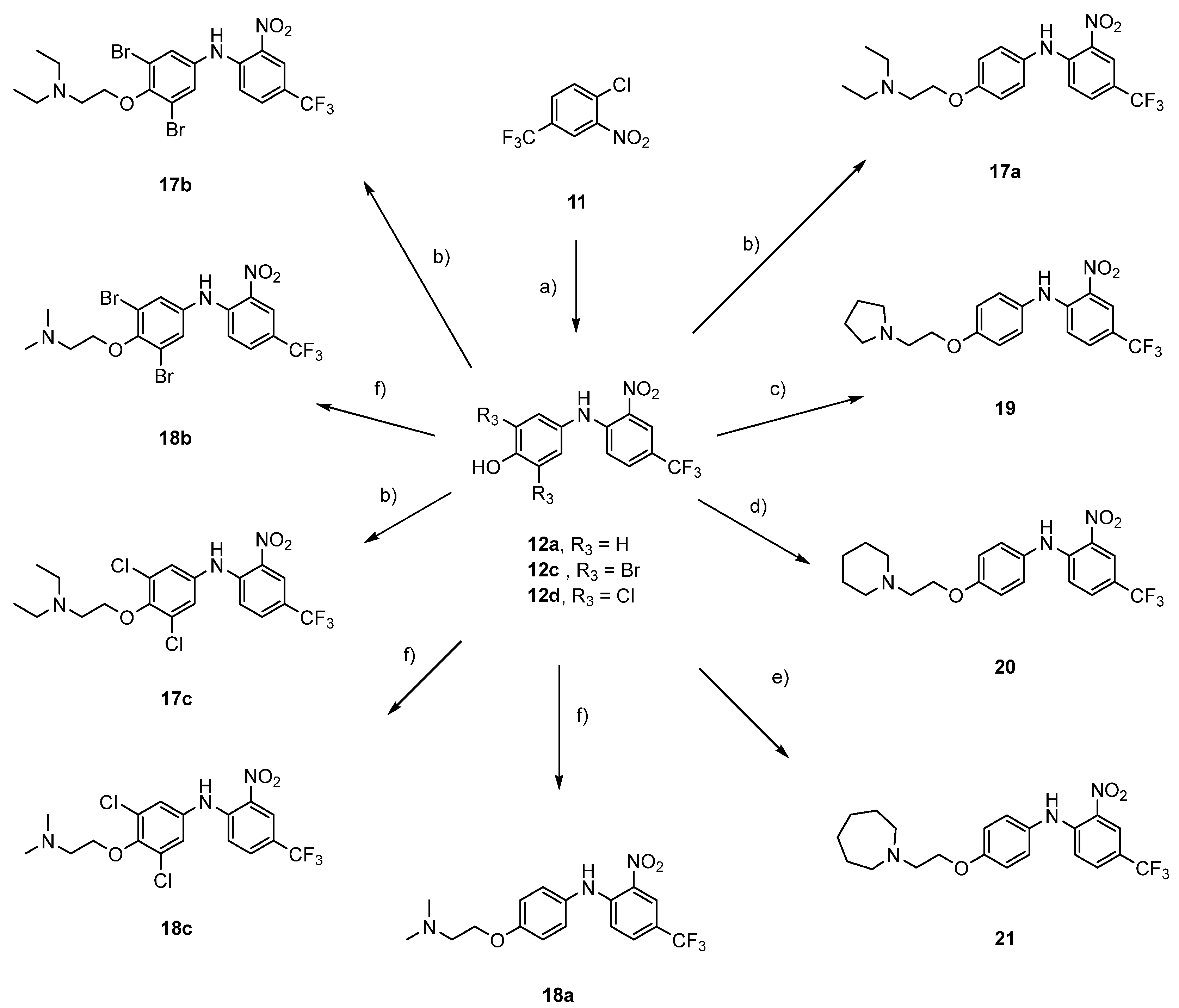
1-Chloro-2-nitro-4-(trifluoromethyl)benzene (
11): resynthesized according to described procedure [
26].
General procedure for synthesis of phenol intermediates 12a–d (12b is given as an example)
To a stirred solution of 1-fluoro-2,4-dinitrobenzene (3.00 g, 14.80 mmol, 1.0 equiv.) in acetonitrile (150 mL), NaHCO3 (3.73 g, 44.40 mmol, 3.0 equiv.) and 4-aminophenol (1.94 g, 17.77 mmol, 1.2 equiv.) were added and the mixture was stirred at 90 °C for 24 h. After reaction mixture was cooled to room temperature and NaHCO3 was filtered off, the solvent was removed under reduced pressure. The crude product was purified with flash column chromatography using dichloromethane/methanol = 20:1) as eluent and dry sampling loading to obtain 12b.
4-((2-Nitro-4-(trifluoromethyl)phenyl)amino)phenol (
12a): resynthesized according to described procedure [
26].
4-((2,4-Dinitrophenyl)amino)phenol (12b): Yield: 47%; red solid (1.93 g); 1H NMR (400 MHz, DMSO) δ 10.04 (s, 1 H), 9.73 (s, 1 H), 8.86 (d, J = 2.7 Hz, 1 H), 8.17 (dd, J1 = 9.6 Hz, J2 = 2.8 Hz, 1 H), 7.21–7.08 (m, 2 H), 6.94 (d, J = 9.6 Hz, 1 H), and 6.91–6.79 (m, 2 H).
2,6-Dibromo-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12c)
Synthesised from 1-chloro-2-nitro-4-(trifluoromethyl)benzene (11) (2.5 g, 11.1 mmol, 1.0 equiv), 4-amino-2,6-dibromophenol (3.55 g, 13.3 mmol, 1.2 equiv), and NaHCO3 (2 g, 22.2 mmol, 2.0 equiv) in acetonitrile (125 mL) at 90 °C. Purified with flash column chromatography using ethyl acetate/hexane = 1:3 as eluent. Yield: 31%; orange solid (1.59 g); 1H NMR (400 MHz, CDCl3) δ 9.51 (s, 1 H), 8.56–8.45 (m, 1 H), 7.59 (dd, J1 = 9.0, J2 = 2.1 Hz, 1 H), 7.49–7.41 (m, 2 H), 7.08 (d, J = 9.0 Hz, 1 H), and 5.98 (s, 1 H).
2,6-Dichloro-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12d)
Synthesised from 1-chloro-2-nitro-4-(trifluoromethyl)benzene (11) (2.5 g, 11.1 mmol, 1.0 equiv), 4-amino-2,6-dichlorophenol (2.37 g, 13.3 mmol, 1.2 equiv), and NaHCO3 (2 g, 22.2 mmol, 2.0 equiv) in acetonitrile (125 mL) at 90 °C. Purified with flash column chromatography using ethyl acetate/hexane = 1:3 as eluent. Yield: 25%; orange solid (1.00 g); 1H NMR (400 MHz, CDCl3) δ 9.50 (s, 1 H), 8.61–8.35 (m, 1 H), 7.59 (dd, J1 = 9.0 Hz, J2 = 2.1 Hz, 1 H), 7.26 (s, 2 H), 7.09 (d, J = 9.0 Hz, 1 H), and 5.93 (s, 1 H).
N-(4-(2-Morpholinoethoxy)phenyl)-2-nitro-4-(trifluoromethyl)aniline (13). To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.01 mmol, 1.0 equiv.) in ethanol (30 mL), 4-(2-chloroethyl)morpholine hydrochloride (223 mg, 1.21 mmol, 1.2 equiv.) and KOH (414 mg, 3.03 mmol, 3.0 equiv.) were added to reaction mixture while stirring. The batch was stirred at 100 °C overnight. The next day, solvent was removed under reduced pressure and crude product was purified with flash column chromatography using dichloromethane/methanol/NH3 = 20:1:0.01 as eluent. Yield: 29%; red solid (120 mg); Rf (dichloromethane/methanol = 20:1) = 0.55; 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1 H), 8.50 (d, J = 1.1 Hz, 1 H), 7.49 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.22–7.15 (m, 2 H), 7.05 (d, J = 9.1 Hz, 1 H), 7.02–6.96 (m, 2 H), 4.15 (t, J = 5.7 Hz, 2 H), 3.84–3.68 (m, 4 H), 2.84 (t, J = 5.7 Hz, 2 H), 2.66–2.53 (m, 4 H); 13C NMR (101 MHz, CDCl3) δ 157.90, 146.45, 131.84 (q, J = 3.0 Hz), 131.47, 130.32, 127.56, 124.88 (q, J = 4.5 Hz), 123.60 (q, J = 271.2 Hz), 118.96 (q, J = 34.4 Hz), 116.62, 116.03, 67.08, 66.34, 57.73, and 54.27; HRMS (ESI+) for C19H21F3N3O4 ([M+H]+) calculated 412.1479 found 412.1473; HPLC retention time: 4.247 min (99.01% at 254 nm).
N-(4-(Benzyloxy)phenyl)-2-nitro-4-(trifluoromethyl)aniline (14): To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (200 mg, 0.8 mmol, 1.0 equiv.) in toluene (50 mL), NaHCO3 (134 mg, 1.6 mmol, 2.0 equiv) and 4-benzyloxy aniline were added (150 mg, 1.6 mmol, 2.0 equiv.). The batch was stirred at 100 °C for 24 h. Solvent was removed under reduced pressure and crude product was purified with Biotage Isolera One System reversed-phase chromatography (Biotage SNAP Cartridge KP-C18-HS 12 g column), MF: gradient water in H2O/acetonitrile. Yield: 31%; red solid (95 mg); 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1 H), 8.50 (s, 1 H), 7.53–7.32 (m, 6 H), 7.19 (d, J = 8.8 Hz, 2 H), 7.06 (d, J = 8.9 Hz, 3 H), 5.11 (s, 2 H); 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ 157.92, 146.43, 136.65, 131.83 (q, J = 3.0 Hz), 131.47, 130.38, 128.84, 128.34, 127.63, 127.56, 124.87 (q, J = 4.3 Hz), 123.65 (q, J = 268.0 Hz), 118.95 (q, J = 34.4 Hz), 116.65, 116.33, and 70.50; HRMS (ESI-) for C20H14F3N2O3 ([M]-) calculated 387.0962 found 387.0964; HPLC retention time: 7.033 min (99.77% at 254 nm).
1-(Methylamino)-3-(4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenoxy)propan-2-ol (15): To a stirring solution of 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (4f) (500 mg, 1.41 mmol, 1.0 equiv.) in acetonitrile (50 mL), MeNH2 (263 mg, 8.47 mmol, 6.0 equiv.) and K2CO3 (581 mg. 4.23 mmol, 3.0 equiv.) were added. The batch was stirred at 70 °C for 24 h. Solvent was removed under reduced pressure and crude product was purified with flash column chromatography using dichloromethane/methanol/NH3 = 20:1:0. as mobile phase. Yield: 22%; red solid (120 mg); 1H NMR (400 MHz, DMSO) δ 9.73 (s, 1 H), 8.35 (d, J = 1.3 Hz, 1 H), 7.72 (dd, J1 = 9.2 Hz, J2 = 2.2 Hz, 1 H), 7.35–7.20 (m, 2 H), 7.10–6.95 (m, 3 H), 3.99 (q, J = 7.0 Hz, 1 H), 3.91 (t, J = 5.9 Hz, 2 H), 2.69–2.51 (m, 2 H), 2.31 (s, 3 H). 13C NMR (101 MHz, CDCl3) δ 157.84, 146.41, 131.85 (q, J = 2.9 Hz), 131.46, 130.46, 127.56, 124.86 (q, J = 4.9 Hz), 123.54 (q, J = 270.0 Hz), 118.95 (q, J = 34.4 Hz), 116.61, 116.01, 73.35, 70.99, 68.10, 63.07, 53.98, 47.99, 36.48, and 31.09; HRMS (ESI+) for C17H19F3N3O4 ([M+H]+) calculated 386.1322 found 386.1318; HPLC retention time: 4.063 min (95.47% at 254 nm).
General procedure for opening of the epoxide with various aromatic amines to obtain 16a–d (16a is given as example)
To a stirring solution of 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (4f) (100 mg, 0.28 mmol, 1.0 equiv.) in acetonitrile (20 mL), 4-((3,4-dichlorophenoxy)methyl)piperidin-4-ol (78 mg, 0.28 mmol, 1.0 equiv) was added. The batch was stirred for 24 h at 70 °C. The next day, solvent was removed under reduced pressure and crude product was purified with flash column chromatography using dichloromethane/methanol = 100:1.
4-((3,4-Dichlorophenoxy)methyl)-1-(2-hydroxy-3-(4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenoxy)propyl)piperidin-4-ol (16a): Yield: 17%; red solid (30 mg); 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1 H), 8.49 (d, J = 1.0 Hz, 1 H), 7.49 (dd, J1 = 9.1 Hz, J2 = 2.0 Hz, 1 H), 7.34 (d, J = 8.9 Hz, 1 H), 7.19 (t, J = 5.9 Hz, 2 H), 7.11–6.95 (m, 4 H), 6.79 (dd, J1 = 8.9 Hz, J2 = 2.9 Hz, 1 H), 4.16 (td, J1 = 9.3 Hz, J2 = 4.8 Hz, 1 H), 4.09–3.97 (m, 2 H), 3.81 (s, 2 H), 2.87 (d, J = 11.3 Hz, 1 H), 2.83–2.57 (m, 4 H), 2.51 (td, J1 = 11.3 Hz, J2 = 3.0 Hz, 1 H), 1.92–1.66 (m, 4 H); 13C NMR (101 MHz, CDCl3) δ 157.90, 157.71, 146.39, 133.09, 131.82 (q, J = 3.0 Hz), 131.42, 130.90, 130.41, 127.51, 124.82 (q, J = 4.2 Hz), 124.69, 123.58 (q, J = 271.0 Hz), 118.90 (q, J = 34.4 Hz), 116.62, 116.60, 116.00, 114.67, 76.24, 70.79, 68.80, 65.64, 60.50, 50.41, 47.90, 34.21, and 34.00; HRMS (ESI+) for C28H29Cl2F3N3O6 ([M+H]+) calculated 630.1380 found 630.1365; HPLC retention time: 5.577 min (98.11% at 254 nm).
1-(2-Hydroxy-3-(4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenoxy)propyl)-4-((4-methoxyphenoxy)methyl)piperidin-4-ol (16b): Synthesised from 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (4f) (100 mg, 0.282 mmol, 1.0 equiv.) and 4-((4-methoxyphenoxy)methyl)piperidin-4-ol (67 mg, 0.282 mmol, 1.0 equiv) in acetonitrile (20 mL). Purified with flash column chromatography using dichloromethane/methanol = 100:1. Yield: 14%; red solid (23 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.49 (d, J = 1.1 Hz, 1 H), 7.49 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.18 (t, J = 6.0 Hz, 2 H), 7.08–6.96 (m, 3 H), 6.92–6.77 (m, 4 H), 4.23–4.12 (m, 1 H), 4.10–3.97 (m, 2 H), 3.78 (s, 2 H), 3.77 (s, 3 H), 2.91–2.75 (m, 2 H), 2.74–2.57 (m, 3 H), 2.52 (td, J1 = 11.3 Hz, J2 = 2.9 Hz, 1 H), 1.88–1.67 (m, 4 H); 13C NMR (101 MHz, CDCl3) δ 157.92, 154.30, 152.87, 146.40, 131.80 (q, J = 2.9 Hz), 131.41, 130.37, 127.50, 124.81 (q, J = 4.2 Hz), 123.58 (q, J = 270.9 Hz), 118.87 (q, J = 34.3 Hz), 116.60, 116.00, 115.68, 114.81, 76.43, 70.82, 68.85, 65.60, 60.52, 55.83, 50.55, 48.06, 34.29, and 34.08; HRMS (ESI+) for C29H33F3N3O7 ([M+H]+) calculated 592.2265 found 592.2251; HPLC retention time: 5.073 min (97.04% at 254 nm).
3,4-Dichloro-N-(1-(2-hydroxy-3-(4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenoxy)propyl)piperidin-4-yl)benzamide (16c): Synthesised from 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (4f) (100 mg, 0.282 mmol, 1.0 equiv.) and 3,4-dichloro-N-(piperidin-4-yl)benzamide (77 mg, 0.282 mmol, 1.0 equiv) in acetonitrile (20 mL). Purified with flash column chromatography using dichloromethane/methanol = 100:1. Yield: 17%; red solid (30 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.50 (d, J = 1.1 Hz, 1 H), 7.85 (d, J = 2.0 Hz, 1 H), 7.59 (dd, J1 = 8.3 Hz, J2 = 2.0 Hz, 1 H), 7.54–7.46 (m, 2 H), 7.24–7.16 (m, 2 H), 7.08–6.96 (m, 3 H), 6.07 (d, J = 7.7 Hz, 1 H), 4.13 (td, J1 = 9.3 Hz, J2 = 4.4 Hz, 1 H), 4.09–3.96 (m, 3 H), 3.07 (d, J = 11.6 Hz, 1 H), 2.90 (d, J = 11.7 Hz, 1 H), 2.70–2.48 (m, 3 H), 2.29–2.19 (m, 1 H), 2.09 (d, J = 11.2 Hz, 2 H), 1.72–1.50 (m, 2 H); 13C NMR (101 MHz, CDCl3) δ 164.84, 157.85, 146.40, 136.06, 134.53, 133.21, 131.85 (q, J = 2.9 Hz), 131.47, 130.77, 130.47, 129.25, 127.54, 126.21, 124.87 (q, J = 4.6 Hz), 123.59 (q, J = 271.1 Hz), 118.97 (q, J = 34.4 Hz), 116.60, 115.99, 70.67, 65.74, 60.36, 54.15, 51.34, 47.40, 32.63, and 32.33; HRMS (ESI+) for C28H28Cl2F3N4O5 ([M+H]+) calculated 627.1383 found 627.1369; HPLC retention time: 5.313 min (99.57% at 254 nm).
2-(3,4-Dichlorophenyl)-N-(1-(2-hydroxy-3-(4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenoxy)propyl)piperidin-4-yl)acetamide (16d): Synthesised from 2-nitro-N-(4-(oxiran-2-ylmethoxy)phenyl)-4-(trifluoromethyl)aniline (4f) (100 mg, 0.282 mmol, 1.0 equiv.) and 2-(3,4-dichlorophenyl)-N-(piperidin-4-yl)acetamide (81 mg, 0.282 mmol, 1.0 equiv) in acetonitrile (20 mL). Purified with flash column chromatography using dichloromethane/methanol = 100:1. Yield: 22%; red solid (40 mg); 1H NMR (400 MHz, DMSO) δ 9.74 (s, 1 H), 8.35 (d, J = 1.3 Hz, 1 H), 8.04 (d, J = 7.6 Hz, 1 H), 7.72 (dd, J1 = 9.2 Hz, J2 = 2.2 Hz, 1 H), 7.55 (d, J = 8.2 Hz, 1 H), 7.50 (d, J = 2.0 Hz, 1 H), 7.31–7.25 (m, 2 H), 7.22 (dd, J1 = 8.3 Hz, J2 = 2.0 Hz, 1 H), 7.13–6.98 (m, 3 H), 4.87 (d, J = 4.2 Hz, 1 H), 4.01 (dd, J = 9.2, 3.1 Hz, 1 H), 3.98–3.85 (m, 1 H), 3.56–3.44 (m, 1 H), 3.41 (s, 1 H), 2.91–2.77 (m, 1 H), 2.40 (ddd, J = 33.4, 12.7, 5.9 Hz, 1 H), 2.08 (dd, J = 19.9, 9.4 Hz, 1 H), 1.76–1.63 (m, 1 H), 1.39 (dd, J = 21.8, 10.8 Hz, 1 H); 13C NMR (101 MHz, CDCl3) δ 169.14, 157.87, 146.40, 135.13, 132.98, 131.84 (q, J = 2.9 Hz), 131.68, 131.45, 131.31, 130.91, 130.44, 128.74, 127.54, 124.86 (q, J = 4.7 Hz), 123.55 (q, J = 277.7 Hz), 118.96 (q, J = 34.5 Hz), 116.60, 116.00, 70.69, 65.69, 60.32, 54.02, 51.25, 46.90, 42.85, 32.50, and 32.21; HRMS (ESI+) for C29H29Cl2F3N4O5 ([M+H]+) calculated 641.1540 found 641.1524; HPLC retention time: 5.280 min (97.83% at 254 nm).
General procedure for synthesis of (diethylamino)ethoxy compounds 17a–c (17a is given as example)
To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.00 mmol, 1.0 equiv.) in ethanol (30 mL), 2-chloro-N,N-diethylethan-1-amine hydrochloride (206 mg, 1.2 mmol, 1.2 equiv) and KOH (414 mg, 7.38 mmol, 7.3 equiv) were added. Batch was stirred overnight at 100 °C. KOH was filtered off and solvent was removed under reduced pressure. Crude product was purified with flash column chromatography using dichloromethane/methanol/NH3 = 10:1:0.01 and dry sample loading.
N-(4-(2-(Diethylamino)ethoxy)phenyl)-2-nitro-4-(trifluoromethyl)aniline (17a): Yield: 38%; red solid (150 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.49 (d, J =, 1.3 Hz, 1 H), 7.48 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.20–7.14 (m, 2 H), 7.04 (d, J = 9.1 Hz, 1 H), 7.01–6.95 (m, 2 H), 4.07 (t, J = 6.3 Hz, 2 H), 2.90 (t, J = 6.3 Hz, 2 H), 2.66 (q, J = 7.1 Hz, 4 H), 1.09 (t, J = 7.1 Hz, 6 H); 13C NMR (101 MHz, CDCl3) δ 158.11, 146.52, 131.82 (q, J = 3.0 Hz), 131.41, 130.06, 127.54, 124.86 (q, J = 4.3 Hz), 123.62 (q, J = 270.8 Hz), 118.87 (q, J = 34.4 Hz), 116.65, 115.96, 67.20, 51.87, 48.05, and 12.01; HRMS (ESI+) for C19H22F3N3O3 ([M+H]+) calculated 398.1686 found 398.1682; HPLC retention time: 4.323 min (98.90% at 254 nm).
3,5-Dibromo-4-(2-(diethylamino)ethoxy)-N-(2-nitro-4-(trifluoromethyl)phenyl)aniline (17b): Synthesised from 2,6-dibromo-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12c) (300 mg, 0.66 mmol, 1.0 equiv), 2-chloro-N,N-diethylethan-1-amine hydrochloride (227 mg, 1.32 mmol, 2.0 equiv), and K2CO3 (273 mg, 2.0 mmol, 3.0 equiv) at 90 °C in acetonitrile (40 mL) overnight. Purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 15%; orange solid (55 mg); 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1 H), 8.49 (d, J = 1.4 Hz, 1 H), 7.63 (dd, J1 = 9.0 Hz, J2 = 2.0 Hz, 1 H), 7.48 (s, 2 H), 7.21 (d, J = 9.0 Hz, 1 H), 4.37 (t, J = 5.0 Hz, 2 H), 3.54 (t, J = 9.2 Hz, 2 H), 3.29 (q, J = 6.8 Hz, 4 H), 1.36 (t, J = 7.2 Hz, 6 H); 13C NMR (101 MHz, CDCl3) δ 151.00, 144.11, 136.23, 132.75, 132.30 (q, J = 3.1 Hz), 129.08, 124.88 (q, J = 4.1 Hz), 123.25 (q, J = 271.8 Hz), 120.85 (q, J = 34.6 Hz), 119.20, 116.58, 68.46, 50.73, 47.52, and 9.53; HRMS (ESI+) for C19H20Br2F3N3O3 ([M+H]+) calculated 553.9896 found 553.9894; HPLC retention time: 4.717 min (95.89% at 254 nm).
3,5-Dichloro-4-(2-(diethylamino)ethoxy)-N-(2-nitro-4-(trifluoromethyl)phenyl)aniline (17c): Synthesised from 2,6-dichloro-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12d) (240 mg, 0.65 mmol, 1.0 equiv), 2-chloro-N,N-diethylethan-1-amine hydrochloride (190 mg,1.3 mmol, 2.0 equiv), and K2CO3 (271 mg, 2.0 mmol, 3.0 equiv.) at 90 °C in acetonitrile (40 mL) overnight. Purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 15%; orange solid (40 mg); 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1 H), 8.51 (d, J = 1.3 Hz, 1 H), 7.62 (dd, J1 = 9.0 Hz, J2 = 2.1 Hz, 1 H), 7.26 (s, 2 H), 7.21 (d, J = 9.0 Hz, 1 H), 4.14 (t, J = 6.5 Hz, 2 H), 3.00 (t, J = 6.5 Hz, 2 H), 2.69 (q, J = 7.1 Hz, 4 H), 1.09 (t, J = 7.1 Hz, 6 H); 13C NMR (101 MHz, CDCl3) δ 150.59, 144.51, 134.30, 132.56, 132.27 (q, J = 3.0 Hz), 130.74, 125.63, 124.95 (q, J = 4.1 Hz), 123.35 (q, J = 271.5 Hz), 120.58 (q, J = 34.6 Hz), 116.63, 72.00, 52.37, 47.64, and 11.87; HRMS (ESI+) for C19H20Cl2F3N3O3 ([M+H]+) calculated 466.0903 found 466.0907; HPLC retention time: 4.603 min (95.04% at 254 nm).
General procedure for synthesis of (dimethylamino)ethoxy compounds 18a–c (18a is given as an example)
To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.01 mmol, 1.0 equiv.) in ethanol (30 mL), 2-chloro-N,N-dimethylethan-1-amine hydrochloride (174 mg, 1.21 mmol) and KOH (414 mg, 7.38 mmol, 7.3 equiv.) were added. Batch was stirred overnight at 100 °C. Solvent was removed under reduced pressure and product was purified with flash column chromatography using dichloromethane/methanol/NH3 = 10:1:0.01 and dry sample loading.
N-(4-(2-(Dimethylamino)ethoxy)phenyl)-2-nitro-4-(trifluoromethyl)aniline (18a): Yield: 27%; red solid (100 mg); Rf (dichloromethane/methanol = 20:1) = 0.05; 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1 H), 8.50 (d, J = 1.1 Hz, 1 H), 7.49 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.22–7.14 (m, 2 H), 7.08–6.96 (m, 3 H), 4.11 (t, J = 5.7 Hz, 2 H), 2.78 (t, J = 5.7 Hz, 2 H), 2.38 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 158.01, 146.48, 131.83 (q, J = 3.1 Hz), 131.42, 130.19, 127.52, 124.85 (q, J = 4.3 Hz), 123.61 (q, J = 271.0 Hz), 118.89 (q, J = 34.4 Hz), 116.65, 116.00, 66.46, 58.37, and 46.05; HRMS (ESI+) for C17H19F3N3O3 ([M+H]+) calculated 370.1373 found 370.1368; HPLC retention time: 4.200 min (97.85% at 254 nm).
3,5-Dibromo-4-(2-(dimethylamino)ethoxy)-N-(2-nitro-4-(trifluoromethyl)phenyl)aniline (18b): Synthesised from 2,6-dibromo-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12c) (300 mg, 0.66 mmol, 1.0 equiv), 2-chloro-N,N-dimethylethan-1-amine hydrochloride (112 mg, 0.8 mmol, 1.2 equiv) and K2CO3 (273 mg, 2.0 mmol, 3.0 equiv) in acetonitrile (40 mL) at 90 °C overnight. Purified with flash column chromatography using flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 17%; orange solid (60 mg); 1H NMR (400 MHz, CDCl3) δ 9.54 (s, 1 H), 8.51 (d, J = 1.3 Hz, 1 H), 7.62 (dd, J1 = 9.0 Hz, J2 = 2.0 Hz, 1 H), 7.47 (s, 2 H), 7.20 (d, J = 9.0 Hz, 1 H), 4.14 (t, J = 5.9 Hz, 2 H), 2.85 (t, J = 5.9 Hz, 2 H), 2.39 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 152.41, 144.49, 135.27, 132.54, 132.27 (q, J = 3.1 Hz), 129.30, 124.93 (q, J = 4.1 Hz), 123.34 (d, J = 271.2 Hz), 120.59 (q, J = 34.6 Hz), 119.31, 116.61, 71.35, 58.87, and 46.00; HRMS (ESI+) for C17H16Br2F3N3O3 ([M+H]+) calculated 525.9583 found 525.9580; HPLC retention time: 4.417 min (99.88% at 254 nm).
3,5-Dichloro-4-(2-(dimethylamino)ethoxy)-N-(2-nitro-4-(trifluoromethyl)phenyl)aniline (18c): Synthesized from 2,6-dichloro-4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12d) (300 mg, 0.82 mmol, 1.0 equiv), 2-chloro-N,N-dimethylethan-1-amine hydrochloride (235 mg, 1.6 mmol, 2.0 equiv), and K2CO3 (338 mg, 2.45 mmol, 3.0 equiv) in acetonitrile (40 mL) at 90 °C overnight. Purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 16%; orange solid (57 mg); 1H NMR (400 MHz, CDCl3) δ 9.53 (s, 1 H), 8.51 (d, J = 1.4 Hz, 1 H), 7.62 (dd, J1 = 9.0 Hz, J2 = 2.1 Hz, 1 H), 7.26 (s, 2 H), 7.21 (d, J = 9.0 Hz, 1 H), 4.15 (t, J = 5.8 Hz, 2 H), 2.82 (t, J = 5.8 Hz, 2 H), 2.38 (s, 6 H); 13C NMR (101 MHz, CDCl3) δ 150.46, 144.45, 134.38, 132.55, 132.24 (q, J = 3.1 Hz), 130.76, 125.57, 124.92 (q, J = 4.2 Hz), 123.33 (q, J = 271.3 Hz), 120.56 (q, J = 34.6 Hz), 116.63, 71.51, 58.89, and 45.93; HRMS (ESI+) for C17H16Cl2F3N3O3 ([M+H]+) calculated 438.0594 found 438.0591; HPLC retention time: 4.307 min (98.68% at 254 nm).
2-Nitro-N-(4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)-4-(trifluoromethyl)aniline (19): To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.00 mmol, 1.0 equiv.) in acetonitrile (40 mL), 1-(2-chloroethyl)pyrrolidine × HCl (204 mg, 1.2 mmol, 1.2 equiv) and K2CO3 (414 mg, 3.0 mmol, 3.0 equiv) were added. Batch was stirred overnight at 90 °C. K2CO3 was filtered off and solvent was removed under reduced pressure. Crude product was purified with flash column chromatography using dichloromethane/methanol = 40:1 and dry sample loading. Yield: 30%; red solid (120 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.49 (d, J = 1.6 Hz, 1 H), 7.48 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.21–7.13 (m, 2 H), 7.04 (d, J = 9.1 Hz, 1 H), 7.02–6.93 (m, 2 H), 4.15 (t, J = 5.9 Hz, 2 H), 2.95 (t, J = 5.9 Hz, 2 H), 2.67 (t, J = 6.6 Hz, 4 H), 1.84 (h, J = 3.2 Hz, 4 H); 13C NMR (101 MHz, CDCl3) δ 158.00, 146.47, 131.81 (q, J = 3.1 Hz), 131.40, 130.14, 127.52, 124.93 (d, J = 271.6 Hz), 124.83 (q, J = 4.3 Hz), 118.86 (q, J = 34.4 Hz), 116.64, 116.00, 67.52, 55.14, 54.90, and 23.63; HRMS (ESI+) for C19H20F3N3O3 ([M+H]+) calculated 396.15295 found 396.15260; HPLC retention time: 4.190 min (98.91% at 254 nm).
2-Nitro-N-(4-(2-(piperidin-1-yl)ethoxy)phenyl)-4-(trifluoromethyl)aniline (20): To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.00 mmol, 1.0 equiv.) in acetonitrile (40 mL), 1-(2-chloroethyl)piperidine × HCl (370 mg, 2.0 mmol, 2.0 equiv) and K2CO3 (414 mg, 3.0 mmol, 3.0 equiv) were added. Batch was stirred overnight at 90 °C. K2CO3 was filtered off and solvent was removed under reduced pressure. Crude product was purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 27%; red solid (109 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.49 (d, J = 1.3 Hz, 1 H), 7.48 (dd, J1 = 9.1 Hz, J2 =2.2 Hz, 1 H), 7.22–7.13 (m, 2 H), 7.04 (d, J = 9.1 Hz, 1 H), 7.01–6.94 (m, 2 H), 4.13 (t, J = 6.0 Hz, 2 H), 2.79 (t, J = 6.0 Hz, 2 H), 2.52 (s, 4 H), 1.62 (p, J = 5.6 Hz, 4 H), 1.46 (q, J = 5.9 Hz, 2 H); 13C NMR (101 MHz, CDCl3) δ 158.04, 146.49, 131.81 (q, J = 3.1 Hz), 131.41, 130.11, 127.52, 124.85 (q, J = 4.3 Hz), 123.61 (q, J = 271.0 Hz), 118.88 (q, J = 34.4 Hz), 116.65, 116.02, 66.53, 58.03, 55.26, 26.09, and 24.32; HRMS (ESI+) for C20H22F3N3O3 ([M+H]+) calculated 410.1686 found 410.1682; HPLC retention time: 4.283 min (99.95% at 254 nm).
N-(4-(2-(Azepan-1-yl)ethoxy)phenyl)-2-nitro-4-(trifluoromethyl)aniline (21): To a stirring solution of 4-((2-nitro-4-(trifluoromethyl)phenyl)amino)phenol (12a) (300 mg, 1.00 mmol, 1.0 equiv.) in acetonitrile (40 mL), 1-(2-chloroethyl)azepane hydrochloride × HCl (400 mg, 2.0 mmol, 2.0 equiv) and K2CO3 (414 mg, 3.0 mmol, 3.0 equiv) were added. Batch was stirred overnight at 90 °C. K2CO3 was filtered off and solvent was removed under reduced pressure. Crude product was purified with flash column chromatography using ethyl acetate/hexane = 1:1 as eluent and dry sample loading. Yield: 26%; red solid (112 mg); 1H NMR (400 MHz, CDCl3) δ 9.60 (s, 1 H), 8.49 (d, J = 1.4 Hz, 1 H), 7.48 (dd, J1 = 9.1 Hz, J2 = 2.1 Hz, 1 H), 7.21–7.15 (m, 2 H), 7.04 (d, J = 9.1 Hz, 1 H), 7.01–6.96 (m, 2 H), 4.09 (t, J = 6.2 Hz, 2 H), 2.98 (t, J = 6.2 Hz, 2 H), 2.84–2.74 (m, 4 H), 1.75–1.57 (m, 8 H); 13C NMR (101 MHz, CDCl3) δ 158.14, 146.51, 131.81 (q, J = 3.0 Hz), 131.41, 130.05, 127.52, 124.85 (q, J = 4.3 Hz), 123.61 (q, J = 271.0 Hz), 118.87 (q, J = 34.4 Hz), 116.65, 116.02, 67.09, 56.41, 56.05, 28.10, and 27.21; HRMS (ESI+) for C21H24F3N3O3 ([M+H]+) calculated 424.1843 found 424.1837; HPLC retention time: 4.450 min (99.58% at 254 nm).



 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





















