

Optimizing Antiviral Dosing for HSV and CMV Treatment in Immunocompromised Patients
 , , , ,
, , , ,
Abstract
:1. Introduction


2. Acyclovir
2.1. Background
2.2. Mechanism of Action
2.3. Activity against HSV and CMV
2.4. Pharmacokinetics
2.5. Pharmacodynamics
2.6. Side Effects

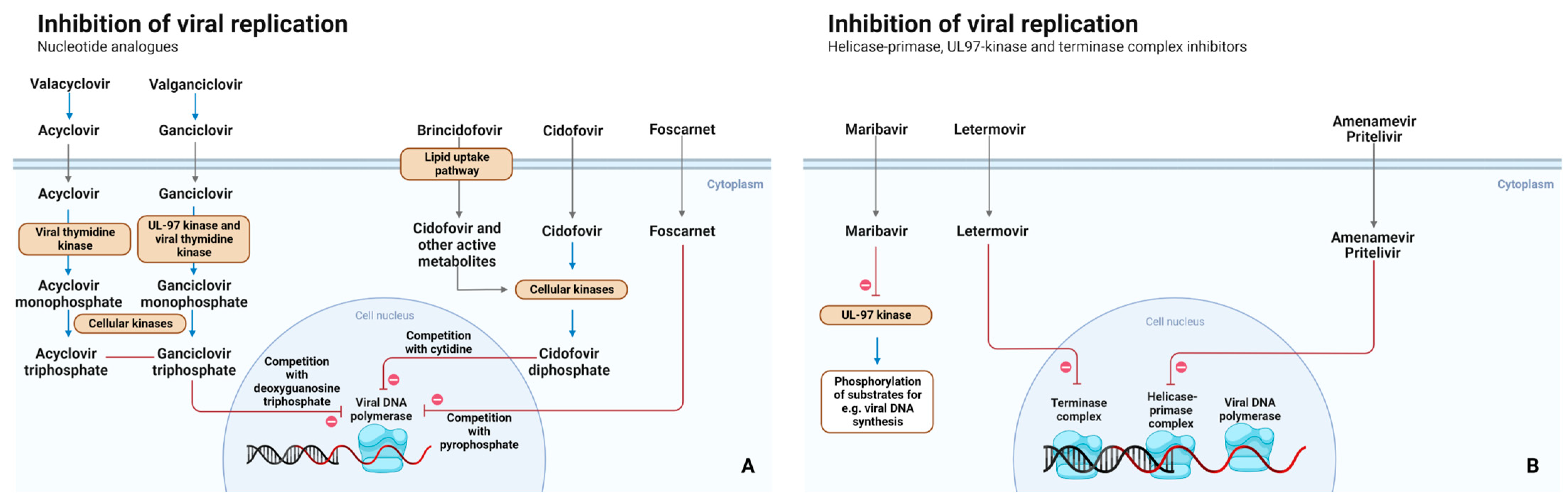{kind=link}
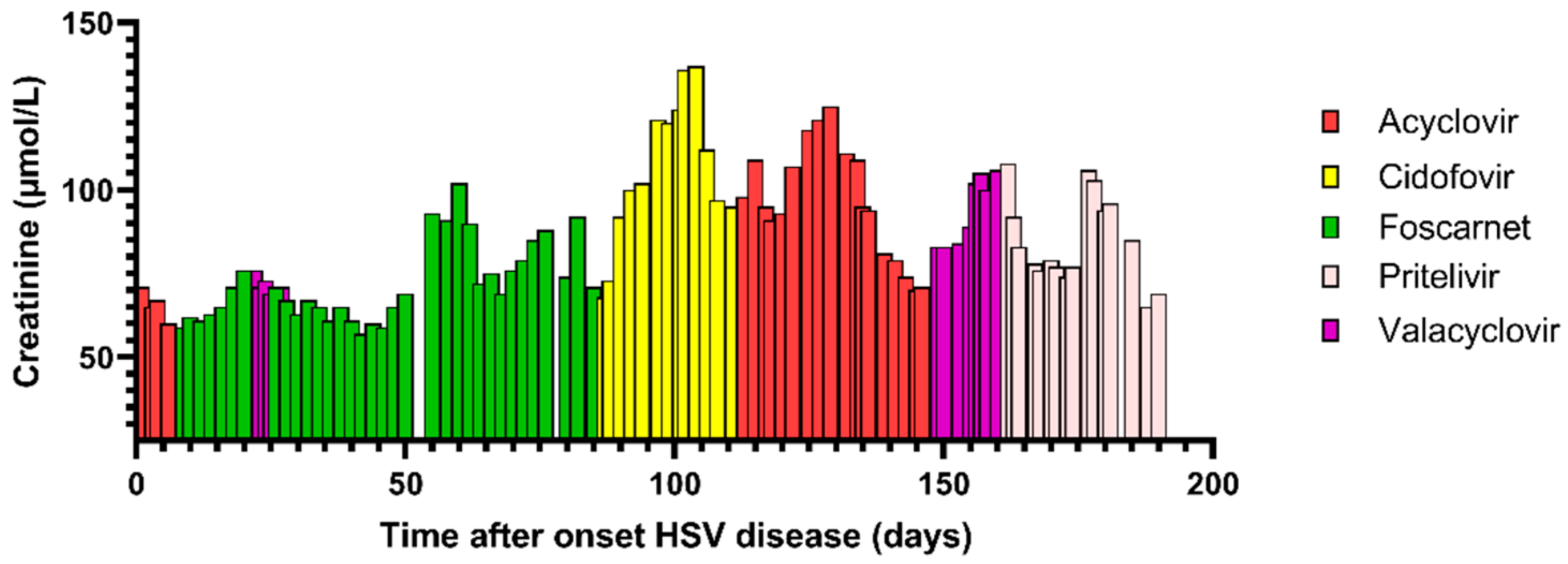{kind=link}
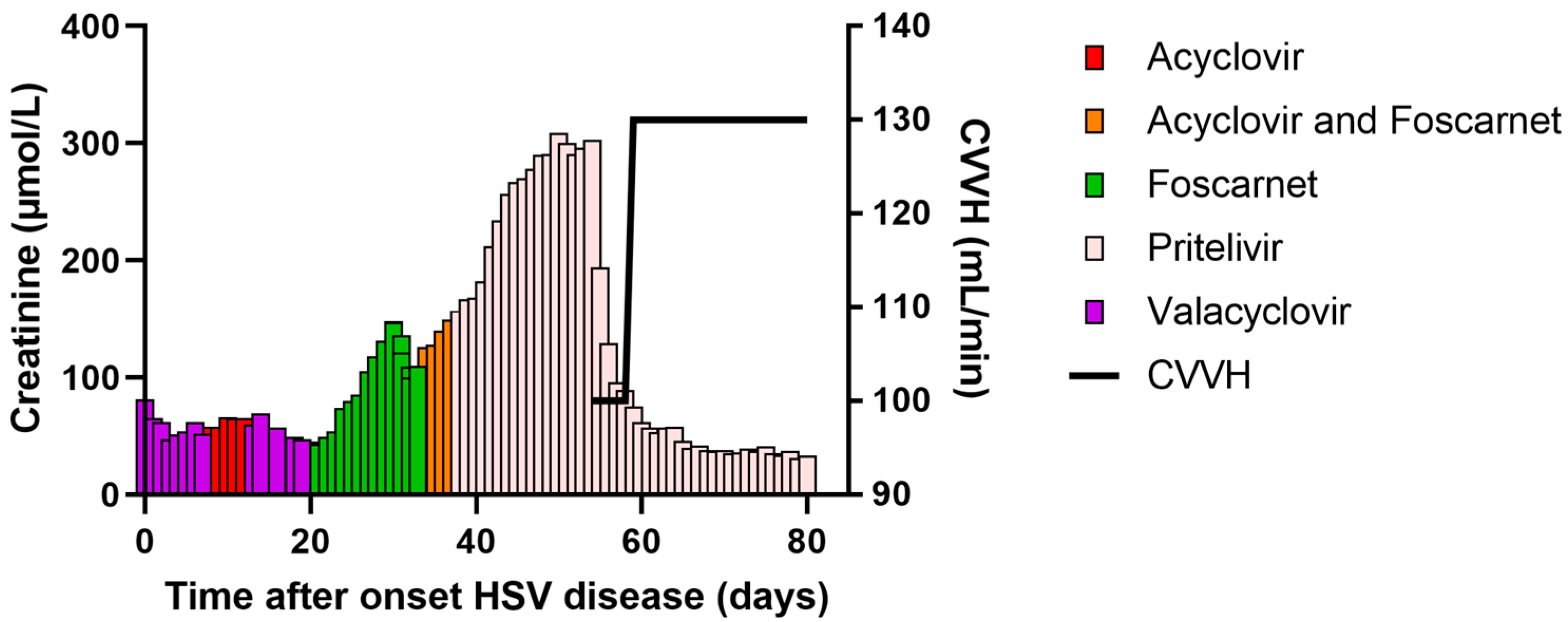{kind=link}
| Drug | EC50 HSV (μM) [18] | EC50 CMV (μM) [18] | Mean Observed Total Css (μM) | Mean Predicted Unbound Css (μM) | Toxic Total Concentration (μM) | Proposed Total Plasma Target Range (mg/L) |
|---|---|---|---|---|---|---|
| Acyclovir | 0.1–20 | n.a. $ | 8.9 | 6.8–9.3 | 20.0 | Ctrough 1.0–5.0 |
| Ganciclovir | 0.2–2.5 | 0.1–37 | 7.8 | 7.8 | 15.6–47 | Ctrough 2.0–4.0 |
| Foscarnet | 50–250 | 27–300 | 150 | 135 | Unknown | Ctrough > 20.0 |
| Cidofovir | 0.6–9 | 0.6–9.5 | 41 # [26] | 41 # [26] | Unknown | Cmax > 11.5 |
| Letermovir | n.a.$ | 7–61 * 10−4 [27] | 4.5–38 # [27] | 4.45–38 * 10−2 # | 38 # | Cmax 2.5–22.0 |
| Pritelivir | 2.6–2.9 * 10−2 [28] | n.a. $ | 1.66 [29] | 0.04 [29] | Unknown | Ctrough 0.83–5.0 |
| Brincidovovir | 0.9–2.9 * 10−2 | 0.1–3 * 10−2 | 0.85 # [30] | 8.5 * 10−3 # | Unknown | Cmax > 0.48 |
| Maribavir | Inactive | 0.08–0.32 | 44 | 0.66 | Unknown | Cmax > 15 |
3. Ganciclovir
3.1. Background
3.2. Mechanism of Action
3.3. Activity against HSV and CMV
3.4. Pharmacokinetics
3.5. Pharmacodynamics
3.6. Side Effects
4. Foscarnet
4.1. Background
4.2. Mechanism of Action
4.3. Activity against HSV and CMV
4.4. Pharmacokinetics
4.5. Pharmacodynamics
4.6. Side Effects
5. Cidofovir
5.1. Background
5.2. Mechanism of Action
5.3. Activity against HSV and CMV
5.4. Pharmacokinetics
5.5. Pharmacodynamics
5.6. Side Effects
6. Letermovir
6.1. Background
6.2. Mechanism of Action
6.3. Activity against HSV and CMV
6.4. Pharmacokinetics
6.5. Pharmacodynamics
6.6. Side Effects
7. Pritelivir and Amenamevir
7.1. Background
7.2. Mechanism of Action
7.3. Activity against HSV and CMV
7.4. Pharmacokinetics
7.5. Pharmacodynamics
7.6. Side Effects
8. Maribavir
8.1. Background
8.2. Mechanism of Action
8.3. Activity against CMV
8.4. Pharmacokinetics
8.5. Pharmacodynamics
8.6. Side Effects
9. Brincidovovir
9.1. Background
9.2. Pharmacokinetics
9.3. Pharmacodynamics
10. Optimized HSV and CMV Treatment Guidelines
11. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Herpes Simplex Virus. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 27 December 2022).
- Centers for Disease Control and Prevention. About Cytomegalovirus (CMV). Available online: https://www.cdc.gov/cmv/overview.html (accessed on 27 December 2022).
- Lin, R.; Liu, Q. Diagnosis and treatment of viral diseases in recipients of allogeneic hematopoietic stem cell transplantation. J. Hematol. Oncol. 2013, 6, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styczynski, J. Management of Herpesvirus Infections in Hematopoietic Cell Transplant Recipients. Transplantology 2021, 2, 8–21. [Google Scholar] [CrossRef]
- Johnston, C.; Wald, A. Epidemiology, Clinical Manifestations, and Diagnosis of Herpes Simplex Virus Type 1 Infection; Post, T.W., Ed.; UpToDate Inc.: Waltham, MA, USA, 2019; Available online: http://www.uptodate.com (accessed on 1 November 2022).
- Ljungman, P.; Brand, R.; Einsele, H.; Frassoni, F.; Niederwieser, D.; Cordonnier, C. Donor CMV serologic status and outcome of CMV-seropositive recipients after unrelated donor stem cell transplantation: An EBMT megafile analysis. Blood 2003, 102, 4255–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasfeld, L.; Chou, S. Antiviral drug resistance: Mechanisms and clinical implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnann, J.W., Jr.; Barton, N.H.; Whitley, R.J. Acyclovir: Mechanism of action, pharmacokinetics, safety and clinical applications. Pharmacotherapy 1983, 3, 275–283. [Google Scholar] [CrossRef] [PubMed]
- MacDougall, C.; Guglielmo, B.J. Pharmacokinetics of valaciclovir. J. Antimicrob. Chemother. 2004, 53, 899–901. [Google Scholar] [CrossRef] [Green Version]
- Kausar, S.; Said Khan, F.; Ishaq Mujeeb Ur Rehman, M.; Akram, M.; Riaz, M.; Rasool, G.; Hamid Khan, A.; Saleem, I.; Shamim, S.; Malik, A. A review: Mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211002621. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Faulds, D.; Goa, K.L. Aciclovir. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1994, 47, 153–205. [Google Scholar] [CrossRef]
- Andrei, G.; Snoeck, R. Herpes simplex virus drug-resistance: New mutations and insights. Curr. Opin. Infect. Dis. 2013, 26, 551–560. [Google Scholar] [CrossRef]
- Danve-Szatanek, C.; Aymard, M.; Thouvenot, D.; Morfin, F.; Agius, G.; Bertin, I.; Billaudel, S.; Chanzy, B.; Coste-Burel, M.; Finkielsztejn, L.; et al. Surveillance network for herpes simplex virus resistance to antiviral drugs: 3-year follow-up. J. Clin. Microbiol. 2004, 42, 242–249. [Google Scholar] [CrossRef]
- Plotkin, S.A.; Starr, S.E.; Bryan, C.K. In vitro and in vivo responses of cytomegalovirus to acyclovir. Am. J. Med. 1982, 73, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Talarico, C.L.; Burnette, T.C.; Miller, W.H.; Smith, S.L.; Davis, M.G.; Stanat, S.C.; Ng, T.I.; He, Z.; Coen, D.M.; Roizman, B.; et al. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein. Antimicrob. Agents Chemother. 1999, 43, 1941–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owers, D.S.; Webster, A.C.; Strippoli, G.F.; Kable, K.; Hodson, E.M. Pre-emptive treatment for cytomegalovirus viraemia to prevent cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2013, 2013, Cd005133. [Google Scholar] [CrossRef] [PubMed]
- Helldén, A.; Odar-Cederlöf, I.; Diener, P.; Barkholt, L.; Medin, C.; Svensson, J.O.; Säwe, J.; Ståhle, L. High serum concentrations of the acyclovir main metabolite 9-carboxymethoxymethylguanine in renal failure patients with acyclovir-related neuropsychiatric side effects: An observational study. Nephrol. Dial. Transplant. 2003, 18, 1135–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemaly, R.F.; Hill, J.A.; Voigt, S.; Peggs, K.S. In vitro comparison of currently available and investigational antiviral agents against pathogenic human double-stranded DNA viruses: A systematic literature review. Antivir. Res. 2019, 163, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, M.H.; Alffenaar, J.C.; Bassetti, M.; Bracht, H.; Dimopoulos, G.; Marriott, D.; Neely, M.N.; Paiva, J.A.; Pea, F.; Sjovall, F.; et al. Antimicrobial therapeutic drug monitoring in critically ill adult patients: A Position Paper. Intensive Care Med. 2020, 46, 1127–1153. [Google Scholar] [CrossRef]
- Helldén, A.; Lycke, J.; Vander, T.; Svensson, J.O.; Odar-Cederlöf, I.; Ståhle, L. The aciclovir metabolite CMMG is detectable in the CSF of subjects with neuropsychiatric symptoms during aciclovir and valaciclovir treatment. J. Antimicrob. Chemother. 2006, 57, 945–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perazella, M.A. Crystal-induced acute renal failure. Am. J. Med. 1999, 106, 459–465. [Google Scholar] [CrossRef]
- Fleischer, R.; Johnson, M. Acyclovir nephrotoxicity: A case report highlighting the importance of prevention, detection, and treatment of acyclovir-induced nephropathy. Case Rep. Med. 2010, 2010, 602783. [Google Scholar] [CrossRef] [Green Version]
- Bean, B.; Aeppli, D. Adverse effects of high-dose intravenous acyclovir in ambulatory patients with acute herpes zoster. J. Infect. Dis. 1985, 151, 362–365. [Google Scholar] [CrossRef]
- Haefeli, W.E.; Schoenenberger, R.A.; Weiss, P.; Ritz, R.F. Acyclovir-induced neurotoxicity: Concentration-side effect relationship in acyclovir overdose. Am. J. Med. 1993, 94, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Aoki, F.Y. 45-Antivirals against Herpes Viruses. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2015; pp. 546–562.e547. [Google Scholar]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical pharmacokinetics of cidofovir in human immunodeficiency virus-infected patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohme, B.V.; Merck, S. Prevymis (Letermovir) SmPC. Available online: https://www.ema.europa.eu/en/documents/product-information/prevymis-epar-product-information_en.pdf (accessed on 1 November 2022).
- Field, H.J.; Mickleburgh, I. The helicase-primase complex as a target for effective herpesvirus antivirals. Adv. Exp. Med. Biol. 2013, 767, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Sukla, S.; Goldner, T.; Field, H.J.; Kropeit, D.; Paulsen, D.; Welbers, A.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Birkmann, A. Pharmacokinetics-pharmacodynamics of the helicase-primase inhibitor pritelivir following treatment of wild-type or pritelivir-resistant virus infection in a murine herpes simplex virus 1 infection model. Antimicrob. Agents Chemother. 2014, 58, 3843–3852. [Google Scholar] [CrossRef] [Green Version]
- Tembexa (Brincidofovir) SmPC. PRESCRIBING INFORMATION of TEMBEXA. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214460s000,214461s000lbl.pdf (accessed on 15 October 2022).
- Selby, P.R.; Shakib, S.; Peake, S.L.; Warner, M.S.; Yeung, D.; Hahn, U.; Roberts, J.A. A Systematic Review of the Clinical Pharmacokinetics, Pharmacodynamics and Toxicodynamics of Ganciclovir/Valganciclovir in Allogeneic Haematopoietic Stem Cell Transplant Patients. Clin. Pharmacokinet. 2021, 60, 727–739. [Google Scholar] [CrossRef]
- Franck, B.; Autmizguine, J.; Marquet, P.; Ovetchkine, P.; Woillard, J.B. Pharmacokinetics, Pharmacodynamics, and Therapeutic Drug Monitoring of Valganciclovir and Ganciclovir in Transplantation. Clin. Pharmacol. Ther. 2022, 112, 233–276. [Google Scholar] [CrossRef]
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplantation 2018, 102, 900–931. [Google Scholar] [CrossRef] [Green Version]
- Ljungman, P.; de la Camara, R.; Robin, C.; Crocchiolo, R.; Einsele, H.; Hill, J.A.; Hubacek, P.; Navarro, D.; Cordonnier, C.; Ward, K.N. Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European Conference on Infections in Leukaemia (ECIL 7). Lancet Infect. Dis. 2019, 19, e260–e272. [Google Scholar] [CrossRef]
- Crumpacker, C.S. Ganciclovir. N. Engl. J. Med. 1996, 335, 721–729. [Google Scholar] [CrossRef]
- Sullivan, V.; Talarico, C.L.; Stanat, S.C.; Davis, M.; Coen, D.M.; Biron, K.K. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 358, 162–164. [Google Scholar] [CrossRef]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Märtson, A.G.; Edwina, A.E.; Kim, H.Y.; Knoester, M.; Touw, D.J.; Sturkenboom, M.G.G.; Alffenaar, J.C. Therapeutic Drug Monitoring of Ganciclovir: Where Are We? Ther. Drug Monit. 2022, 44, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Cymevene (Ganciclovir) SmPC. Available online: https://www.ema.europa.eu/en/documents/referral/cymevene-article-30-referral-annex-iii_en.pdf (accessed on 16 October 2022).
- Biron, K.K. Antiviral drugs for cytomegalovirus diseases. Antivir. Res. 2006, 71, 154–163. [Google Scholar] [CrossRef]
- Paintsil, E.; Cheng, Y.C. Antiviral Agents. Encycl. Microbiol. 2019, 176–225. [Google Scholar] [CrossRef]
- Reusser, P.; Einsele, H.; Lee, J.; Volin, L.; Rovira, M.; Engelhard, D.; Finke, J.; Cordonnier, C.; Link, H.; Ljungman, P. Randomized multicenter trial of foscarnet versus ganciclovir for preemptive therapy of cytomegalovirus infection after allogeneic stem cell transplantation. Blood 2002, 99, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhou, T.; Huang, M.; Gu, G.; Xia, Q. Prevention of cytomegalovirus infection after solid organ transplantation: A Bayesian network analysis. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Boivin, G. Human cytomegalovirus resistance to antiviral drugs. Antimicrob. Agents Chemother. 2005, 49, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.E.; Knudsen, J.L.; Lease, E.D.; Jerome, K.R.; Rakita, R.M.; Boeckh, M.; Limaye, A.P. Risk Factors and Outcomes of Ganciclovir-Resistant Cytomegalovirus Infection in Solid Organ Transplant Recipients. Clin. Infect. Dis. 2017, 65, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Ganciclovir (Systemic): Drug Information. Available online: https://www.uptodate.com/contents/ganciclovir-systemic-drug-information?search=ganciclovir&source=panel_search_result&selectedTitle=1~101&usage_type=panel&showDrugLabel=true&display_rank=1 (accessed on 8 November 2022).
- Ritchie, B.M.; Barreto, J.N.; Barreto, E.F.; Crow, S.A.; Dierkhising, R.A.; Jannetto, P.J.; Tosh, P.K.; Razonable, R.R. Relationship of Ganciclovir Therapeutic Drug Monitoring with Clinical Efficacy and Patient Safety. Antimicrob. Agents Chemother. 2019, 63, e01855-18. [Google Scholar] [CrossRef] [Green Version]
- Bedino, G.; Esposito, P.; Bosio, F.; Corradetti, V.; Valsania, T.; Rocca, C.; Pattonieri, E.F.; Gregorini, M.; Rampino, T.; Dal Canton, A. The role of therapeutic drug monitoring in the treatment of cytomegalovirus disease in kidney transplantation. Int. Urol. Nephrol. 2013, 45, 1809–1813. [Google Scholar] [CrossRef]
- McGavin, J.K.; Goa, K.L. Ganciclovir. Drugs 2001, 61, 1153–1183. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.N.; Schulman, G. Nephrotoxicity of antiviral therapies. Curr. Opin. Nephrol. Hypertens. 1996, 5, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Naesens, M.; Kuypers, D.R.; Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol. 2009, 4, 481–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrisp, P.; Clissold, S.P. Foscarnet. A review of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with cytomegalovirus retinitis. Drugs 1991, 41, 104–129. [Google Scholar] [CrossRef]
- Noormohamed, F.H.; Youle, M.S.; Higgs, C.J.; Martin-Munley, S.; Gazzard, B.G.; Lant, A.F. Pharmacokinetics and absolute bioavailability of oral foscarnet in human immunodeficiency virus-seropositive patients. Antimicrob. Agents Chemother. 1998, 42, 293–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon-Céron, D.; Fillet, A.M.; Aboulker, J.P.; Gérard, L.; Houhou, N.; Carrière, I.; Ostinelli, J.; Vildé, J.L.; Brun-Vézinet, F.; Leport, C. Effect of a 14-day course of foscarnet on cytomegalovirus (CMV) blood markers in a randomized study of human immunodeficiency virus-infected patients with persistent CMV viremia. Agence National de Recherche du SIDA 023 Study Group. Clin. Infect. Dis. 1999, 28, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, 3s–7s. [Google Scholar] [CrossRef]
- Ogata, M.; Takano, K.; Moriuchi, Y.; Kondo, T.; Ueki, T.; Nakano, N.; Mori, T.; Uoshima, N.; Nagafuji, K.; Yamasaki, S.; et al. Effects of Prophylactic Foscarnet on Human Herpesvirus-6 Reactivation and Encephalitis in Cord Blood Transplant Recipients: A Prospective Multicenter Trial with an Historical Control Group. Biol. Blood Marrow Transplant. 2018, 24, 1264–1273. [Google Scholar] [CrossRef] [Green Version]
- Ippoliti, C.; Morgan, A.; Warkentin, D.; van Besien, K.; Mehra, R.; Khouri, I.; Giralt, S.; Gajewski, J.; Champlin, R.; Andersson, B.; et al. Foscarnet for prevention of cytomegalovirus infection in allogeneic marrow transplant recipients unable to receive ganciclovir. Bone Marrow Transplant. 1997, 20, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Foscavir (Foscarnet) SmPC. Available online: https://www.geneesmiddeleninformatiebank.nl/smpc/h13057_smpc.pdf (accessed on 1 November 2022).
- Delanaye, P.; Björk, J.; Courbebaisse, M.; Couzi, L.; Ebert, N.; Eriksen, B.O.; Dalton, R.N.; Dubourg, L.; Gaillard, F.; Garrouste, C.; et al. Performance of creatinine-based equations to estimate glomerular filtration rate with a methodology adapted to the context of drug dosage adjustment. Br. J. Clin. Pharmacol. 2022, 88, 2118–2127. [Google Scholar] [CrossRef]
- Donker, E.M.; Bet, P.; Nurmohamed, A.; Serné, E.; Burchell, G.L.; Friedman, A.N.; Bouquegneau, A.; Lemoine, S.; Ebert, N.; Cirillo, M.; et al. Estimation of glomerular filtration rate for drug dosing in patients with very high or low body mass index. Clin. Transl. Sci. 2022, 15, 2206–2217. [Google Scholar] [CrossRef] [PubMed]
- Castelli, F.; Tomasoni, L.; Zeroli, C.; Suter, F.; Trespi, G.; Carosi, G.; Hedman, A. Comparison of pharmacokinetics and dynamics of two dosage regimens of foscarnet in AIDS patients with Cytomegalovirus retinitis. Eur. J. Clin. Pharmacol. 1997, 52, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L.; Aweeka, F.; Gambertoglio, J.; Jacobson, M.; Polis, M.; Lane, C.H.; Eaton, C.; Martin-Munley, S. Relationship between foscarnet exposure, baseline cytomegalovirus (CMV) blood culture and the time to progression of CMV retinitis in HIV-positive patients. AIDS 1996, 10, 1113–1119. [Google Scholar] [PubMed]
- Bregante, S.; Bertilson, S.; Tedone, E.; Van Lint, M.T.; Trespi, G.; Mordini, N.; Berisso, G.; Gualandi, F.; Lamparelli, T.; Figari, O.; et al. Foscarnet prophylaxis of cytomegalovirus infections in patients undergoing allogeneic bone marrow transplantation (BMT): A dose-finding study. Bone Marrow Transplant. 2000, 26, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Moretti, S.; Zikos, P.; Van Lint, M.T.; Tedone, E.; Occhini, D.; Gualandi, F.; Lamparelli, T.; Mordini, N.; Berisso, G.; Bregante, S.; et al. Forscarnet vs ganciclovir for cytomegalovirus (CMV) antigenemia after allogeneic hemopoietic stem cell transplantation (HSCT): A randomised study. Bone Marrow Transplant. 1998, 22, 175–180. [Google Scholar] [CrossRef]
- Ishiyama, K.; Katagiri, T.; Ohata, K.; Hosokawa, K.; Kondo, Y.; Yamazaki, H.; Takami, A.; Nakao, S. Safety of pre-engraftment prophylactic foscarnet administration after allogeneic stem cell transplantation. Transpl. Infect. Dis. 2012, 14, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Lalezari, J.P.; Holland, G.N.; Kramer, F.; McKinley, G.F.; Kemper, C.A.; Ives, D.V.; Nelson, R.; Hardy, W.D.; Kuppermann, B.D.; Northfelt, D.W.; et al. Randomized, controlled study of the safety and efficacy of intravenous cidofovir for the treatment of relapsing cytomegalovirus retinitis in patients with AIDS. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 339–344. [Google Scholar] [CrossRef]
- Lalezari, J.P.; Stagg, R.J.; Kuppermann, B.D.; Holland, G.N.; Kramer, F.; Ives, D.V.; Youle, M.; Robinson, M.R.; Drew, W.L.; Jaffe, H.S. Intravenous cidofovir for peripheral cytomegalovirus retinitis in patients with AIDS. A randomized, controlled trial. Ann. Intern. Med. 1997, 126, 257–263. [Google Scholar] [CrossRef]
- De Clercq, E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 2003, 16, 569–596. [Google Scholar] [CrossRef] [Green Version]
- Ljungman, P.; Deliliers, G.L.; Platzbecker, U.; Matthes-Martin, S.; Bacigalupo, A.; Einsele, H.; Ullmann, J.; Musso, M.; Trenschel, R.; Ribaud, P.; et al. Cidofovir for cytomegalovirus infection and disease in allogeneic stem cell transplant recipients. The Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Blood 2001, 97, 388–392. [Google Scholar] [CrossRef]
- Kopp, T.; Geusau, A.; Rieger, A.; Stingl, G. Successful treatment of an aciclovir-resistant herpes simplex type 2 infection with cidofovir in an AIDS patient. Br. J. Dermatol. 2002, 147, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.A.; Hitchcock, M.J.; Lee, W.A.; Tellier, P.; Cundy, K.C. Effect of oral probenecid coadministration on the chronic toxicity and pharmacokinetics of intravenous cidofovir in cynomolgus monkeys. Toxicol. Sci. 1998, 44, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Polis, M.A.; Spooner, K.M.; Baird, B.F.; Manischewitz, J.F.; Jaffe, H.S.; Fisher, P.E.; Falloon, J.; Davey, R.T., Jr.; Kovacs, J.A.; Walker, R.E.; et al. Anticytomegaloviral activity and safety of cidofovir in patients with human immunodeficiency virus infection and cytomegalovirus viruria. Antimicrob. Agents Chemother. 1995, 39, 882–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, P.; Dautheville-Guibal, S.; Ronco, P.M.; Rossert, J. Cidofovir-induced end-stage renal failure. Nephrol. Dial. Transplant. 2002, 17, 148–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, A.; Alonso, C.D.; Garcia-Vidal, C.; Cardozo, C.; Slavin, M.; Yong, M.K.; Ho, S.A.; Mehta Steinke, S.; Avery, R.K.; Koehler, P.; et al. Safety and efficacy of intravenously administered cidofovir in adult haematopoietic cell transplant recipients: A retrospective multicentre cohort study. J. Antimicrob. Chemother. 2021, 76, 3020–3028. [Google Scholar] [CrossRef]
- Majewska, A.; Mlynarczyk-Bonikowska, B. 40 Years after the Registration of Acyclovir: Do We Need New Anti-Herpetic Drugs? Int. J. Mol. Sci. 2022, 23, 3431. [Google Scholar] [CrossRef]
- Kim, E.S. Letermovir: First Global Approval. Drugs 2018, 78, 147–152. [Google Scholar] [CrossRef]
- Letermovir: Drug Information. Available online: https://www.uptodate.com/contents/letermovir-drug-information?search=letermovir&source=panel_search_result&selectedTitle=1~17&usage_type=panel&kp_tab=drug_general&display_rank=1 (accessed on 6 October 2022).
- Marschall, M.; Stamminger, T.; Urban, A.; Wildum, S.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob. Agents Chemother. 2012, 56, 1135–1137. [Google Scholar] [CrossRef] [Green Version]
- Trial on Efficacy and Safety of Pritelivir Tablets for Treatment of Acyclovir-Resistant Mucocutaneous HSV (Herpes Simplex Virus) Infections in Immunocompromised Subjects (PRIOH-1). Available online: https://clinicaltrials.gov/ct2/show/NCT03073967 (accessed on 2 November 2022).
- Crute, J.J.; Tsurumi, T.; Zhu, L.A.; Weller, S.K.; Olivo, P.D.; Challberg, M.D.; Mocarski, E.S.; Lehman, I.R. Herpes simplex virus 1 helicase-primase: A complex of three herpes-encoded gene products. Proc. Natl. Acad. Sci. USA 1989, 86, 2186–2189. [Google Scholar] [CrossRef] [Green Version]
- Chono, K.; Katsumata, K.; Kontani, T.; Shiraki, K.; Suzuki, H. Characterization of virus strains resistant to the herpes virus helicase-primase inhibitor ASP2151 (Amenamevir). Biochem. Pharmacol. 2012, 84, 459–467. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Zhu, D.; Zhao, X.; Chen, S.; et al. Alpha-Herpesvirus Thymidine Kinase Genes Mediate Viral Virulence and Are Potential Therapeutic Targets. Front. Microbiol. 2019, 10, 941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quenelle, D.C.; Birkmann, A.; Goldner, T.; Pfaff, T.; Zimmermann, H.; Bonsmann, S.; Collins, D.J.; Rice, T.L.; Prichard, M.N. Efficacy of pritelivir and acyclovir in the treatment of herpes simplex virus infections in a mouse model of herpes simplex encephalitis. Antivir. Res. 2018, 149, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, A.; Kropeit, D.; McCormick, D.; Zimmermann, H.; Ruebsamen-Schaeff, H. Safety and Human Pharmacokinetics of AIC316, a Potent Helicase-Primase Inhibitor of Herpes Simplex Virus (HSV). Antivir. Res. 2011, 90, A25. [Google Scholar] [CrossRef]
- Birkmann, A.; Bonsmann, S.; Kropeit, D.; Pfaff, T.; Rangaraju, M.; Sumner, M.; Timmler, B.; Zimmermann, H.; Buschmann, H.; Ruebsamen-Schaeff, H. Discovery, Chemistry, and Preclinical Development of Pritelivir, a Novel Treatment Option for Acyclovir-Resistant Herpes Simplex Virus Infections. J. Med. Chem. 2022, 65, 13614–13628. [Google Scholar] [CrossRef]
- Kusawake, T.; Keirns, J.J.; Kowalski, D.; den Adel, M.; Groenendaal-van de Meent, D.; Takada, A.; Ohtsu, Y.; Katashima, M. Pharmacokinetics and Safety of Amenamevir in Healthy Subjects: Analysis of Four Randomized Phase 1 Studies. Adv. Ther. 2017, 34, 2625–2637. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, K.; Yasumoto, S.; Toyama, N.; Fukuda, H. Amenamevir, a Helicase-Primase Inhibitor, for the Optimal Treatment of Herpes Zoster. Viruses 2021, 13, 1547. [Google Scholar] [CrossRef]
- Wald, A.; Corey, L.; Timmler, B.; Magaret, A.; Warren, T.; Tyring, S.; Johnston, C.; Kriesel, J.; Fife, K.; Galitz, L.; et al. Helicase–Primase Inhibitor Pritelivir for HSV-2 Infection. N. Engl. J. Med. 2014, 370, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Wald, A.; Timmler, B.; Magaret, A.; Warren, T.; Tyring, S.; Johnston, C.; Fife, K.; Selke, S.; Huang, M.L.; Stobernack, H.P.; et al. Effect of Pritelivir Compared with Valacyclovir on Genital HSV-2 Shedding in Patients With Frequent Recurrences: A Randomized Clinical Trial. JAMA 2016, 316, 2495–2503. [Google Scholar] [CrossRef] [Green Version]
- Cannon, L.; Tholouli, E.; Ward, C.; Farooq, H.; Kingston, M. Use of pritelivir in refractory aciclovir-resistant herpes simplex virus type 2. Int. J. STD AIDS 2021, 32, 978–980. [Google Scholar] [CrossRef]
- Katsumata, K.; Chono, K.; Suzuki, H. Antiviral efficacy of the helicase-primase inhibitor amenamevir in murine models of severe herpesvirus infection. Biochem. Pharmacol. 2018, 158, 201–206. [Google Scholar] [CrossRef]
- Kawashima, M.; Nemoto, O.; Honda, M.; Watanabe, D.; Nakayama, J.; Imafuku, S.; Kato, T.; Katsuramaki, T. Amenamevir, a novel helicase-primase inhibitor, for treatment of herpes zoster: A randomized, double-blind, valaciclovir-controlled phase 3 study. J. Dermatol. 2017, 44, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trofe, J.; Pote, L.; Wade, E.; Blumberg, E.; Bloom, R.D. Maribavir: A novel antiviral agent with activity against cytomegalovirus. Ann. Pharmacother. 2008, 42, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Peck, R.W.; Yin, Y.; Allanson, J.; Wiggs, R.; Wire, M.B. Phase I safety and pharmacokinetic trials of 1263W94, a novel oral anti-human cytomegalovirus agent, in healthy and human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 2003, 47, 1334–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.H.; Ilic, K.; Murphy, J.; Lasseter, K.; Martin, P. Effects of Maribavir on P-Glycoprotein and CYP2D6 in Healthy Volunteers. J. Clin. Pharmacol. 2020, 60, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Ljungman, P.; Papanicolaou, G.A.; Winston, D.J.; Chemaly, R.F.; Strasfeld, L.; Young, J.A.; Rodriguez, T.; Maertens, J.; Schmitt, M.; et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: A phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2011, 11, 284–292. [Google Scholar] [CrossRef]
- Papanicolaou, G.A.; Silveira, F.P.; Langston, A.A.; Pereira, M.R.; Avery, R.K.; Uknis, M.; Wijatyk, A.; Wu, J.; Boeckh, M.; Marty, F.M.; et al. Maribavir for Refractory or Resistant Cytomegalovirus Infections in Hematopoietic-cell or Solid-organ Transplant Recipients: A Randomized, Dose-ranging, Double-blind, Phase 2 Study. Clin. Infect. Dis. 2019, 68, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Chittick, G.; Morrison, M.; Brundage, T.; Nichols, W.G. Short-term clinical safety profile of brincidofovir: A favorable benefit-risk proposition in the treatment of smallpox. Antivir. Res. 2017, 143, 269–277. [Google Scholar] [CrossRef]
- Naderer, O.; Schuck, V.; Morrison, M.; Anderson, M.; Arumugham, T.; Dunn, J. 1421. IV Brincidofovir (BCV): Pharmacokinetics (PK) and Safety of Multiple Ascending Doses (MAD) in Healthy Subjects. Open Forum Infect. Dis. 2018, 5, S438–S439. [Google Scholar] [CrossRef] [Green Version]
- El-Haddad, D.; El Chaer, F.; Vanichanan, J.; Shah, D.P.; Ariza-Heredia, E.J.; Mulanovich, V.E.; Gulbis, A.M.; Shpall, E.J.; Chemaly, R.F. Brincidofovir (CMX-001) for refractory and resistant CMV and HSV infections in immunocompromised cancer patients: A single-center experience. Antivir. Res. 2016, 134, 58–62. [Google Scholar] [CrossRef]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef]
- Lee, Y.J.; Neofytos, D.; Kim, S.J.; Cheteyan, L.; Huang, Y.T.; Papadopoulos, E.B.; Jakubowski, A.A.; Papanicolaou, G.A. Efficacy of brincidofovir as prophylaxis against HSV and VZV in hematopoietic cell transplant recipients. Transpl. Infect. Dis. 2018, 20, e12977. [Google Scholar] [CrossRef] [PubMed]
- Adler, H.; Gould, S.; Hine, P.; Snell, L.B.; Wong, W.; Houlihan, C.F.; Osborne, J.C.; Rampling, T.; Beadsworth, M.B.; Duncan, C.J.; et al. Clinical features and management of human monkeypox: A retrospective observational study in the UK. Lancet Infect. Dis. 2022, 22, 1153–1162. [Google Scholar] [CrossRef]
- Wagner, J.N.; Leibetseder, A.; Troescher, A.; Panholzer, J.; von Oertzen, T.J. Efficacy and safety of intravenous immunoglobulins for the treatment of viral encephalitis: A systematic literature review. J. Neurol. 2022, 269, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Cernik, C.; Gallina, K.; Brodell, R.T. The treatment of herpes simplex infections: An evidence-based review. Arch. Intern. Med. 2008, 168, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Wingard, J. Prevention of Viral Infections in Hematopoietic Cell Transplant Recipients; Post, T.W., Ed.; UptoDate Inc.: Waltham, MA, USA, 2020; Available online: http://www.uptodate.com (accessed on 1 November 2022).
- Anton-Vazquez, V.; Mehra, V.; Mbisa, J.L.; Bradshaw, D.; Basu, T.N.; Daly, M.L.; Mufti, G.J.; Pagliuca, A.; Potter, V.; Zuckerman, M. Challenges of aciclovir-resistant HSV infection in allogeneic bone marrow transplant recipients. J. Clin. Virol. 2020, 128, 104421. [Google Scholar] [CrossRef] [PubMed]
- Houghtelin, A.; Bollard, C.M. Virus-Specific T Cells for the Immunocompromised Patient. Front. Immunol. 2017, 8, 1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, M.D.; Bollard, C.M. Virus-specific T-cell therapies for patients with primary immune deficiency. Blood 2020, 135, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Torii, Y.; Horiba, K.; Kawada, J.-i.; Haruta, K.; Yamaguchi, M.; Suzuki, T.; Uryu, H.; Kashiwa, N.; Goishi, K.; Ogi, T.; et al. Detection of antiviral drug resistance in patients with congenital cytomegalovirus infection using long-read sequencing: A retrospective observational study. BMC Infect. Dis. 2022, 22, 568. [Google Scholar] [CrossRef]
- Hardy, W.D. Foscarnet treatment of acyclovir-resistant herpes simplex virus infection in patients with acquired immunodeficiency syndrome: Preliminary results of a controlled, randomized, regimen-comparative trial. Am. J. Med. 1992, 92, 30S–35S. [Google Scholar] [CrossRef]
- Sauerbrei, A.; Bohn-Wippert, K. Phenotypic and Genotypic Testing of HSV-1 and HSV-2 Resistance to Antivirals. In Herpes Simplex Virus: Methods and Protocols; Diefenbach, R.J., Fraefel, C., Eds.; Springer: New York, NY, USA, 2020; pp. 241–261. [Google Scholar]
- Kim, S.-H. Interferon-γ Release Assay for Cytomegalovirus (IGRA-CMV) for Risk Stratification of Posttransplant CMV Infection: Is It Time to Apply IGRA-CMV in Routine Clinical Practice? Clin. Infect. Dis. 2020, 71, 2386–2388. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huntjens, D.W.; Dijkstra, J.A.; Verwiel, L.N.; Slijkhuis, M.; Elbers, P.; Welkers, M.R.A.; Veldkamp, A.I.; Kuijvenhoven, M.A.; de Leeuw, D.C.; Abdullah-Koolmees, H.; et al. Optimizing Antiviral Dosing for HSV and CMV Treatment in Immunocompromised Patients. Pharmaceutics 2023, 15, 163. https://doi.org/10.3390/pharmaceutics15010163
Huntjens DW, Dijkstra JA, Verwiel LN, Slijkhuis M, Elbers P, Welkers MRA, Veldkamp AI, Kuijvenhoven MA, de Leeuw DC, Abdullah-Koolmees H, et al. Optimizing Antiviral Dosing for HSV and CMV Treatment in Immunocompromised Patients. Pharmaceutics. 2023; 15(1):163. https://doi.org/10.3390/pharmaceutics15010163
Chicago/Turabian StyleHuntjens, Daan W., Jacob A. Dijkstra, Lisanne N. Verwiel, Mirjam Slijkhuis, Paul Elbers, Matthijs R. A. Welkers, Agnes I. Veldkamp, Marianne A. Kuijvenhoven, David C. de Leeuw, Heshu Abdullah-Koolmees, and et al. 2023. "Optimizing Antiviral Dosing for HSV and CMV Treatment in Immunocompromised Patients" Pharmaceutics 15, no. 1: 163. https://doi.org/10.3390/pharmaceutics15010163
APA StyleHuntjens, D. W., Dijkstra, J. A., Verwiel, L. N., Slijkhuis, M., Elbers, P., Welkers, M. R. A., Veldkamp, A. I., Kuijvenhoven, M. A., de Leeuw, D. C., Abdullah-Koolmees, H., Kuipers, M. T., & Bartelink, I. H. (2023). Optimizing Antiviral Dosing for HSV and CMV Treatment in Immunocompromised Patients. Pharmaceutics, 15(1), 163. https://doi.org/10.3390/pharmaceutics15010163