
The Effect of Necrosis Inhibitor on Dextran Sulfate Sodium Induced Chronic Colitis Model in Mice

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Ethics Statement
2.2. Dextran Sulfate Sodium Induced Chronic Colitis Model
2.3. Disease Activity Index (DAI)
2.4. Histological Evaluations
2.5. Evaluation of Pro-Inflammatory Cytokines Using Real-Time PCR
2.6. Western Blot Assay
2.7. Immunohistochemistry
2.8. Statistical Analysis
3. Results
3.1. NI Effect on the DAI
3.2. Colon Length and Histological Score
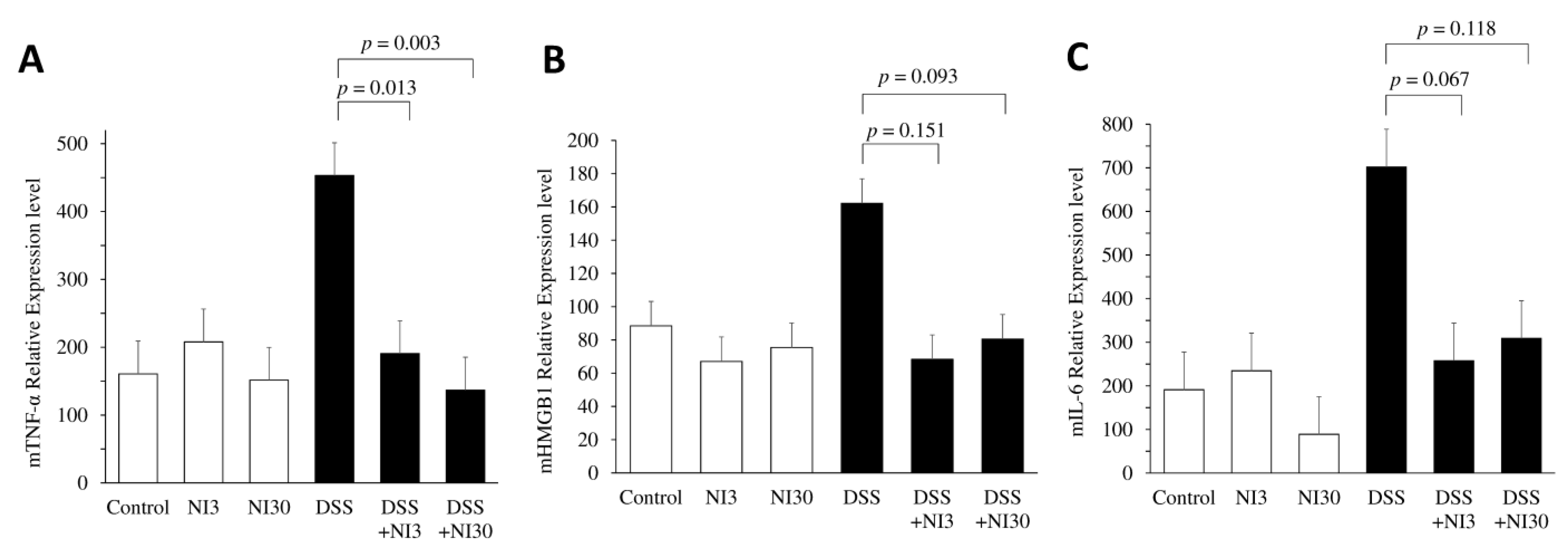
3.3. Pro-Inflammatory Cytokine Quantitative Analysis Using Real-Time PCR
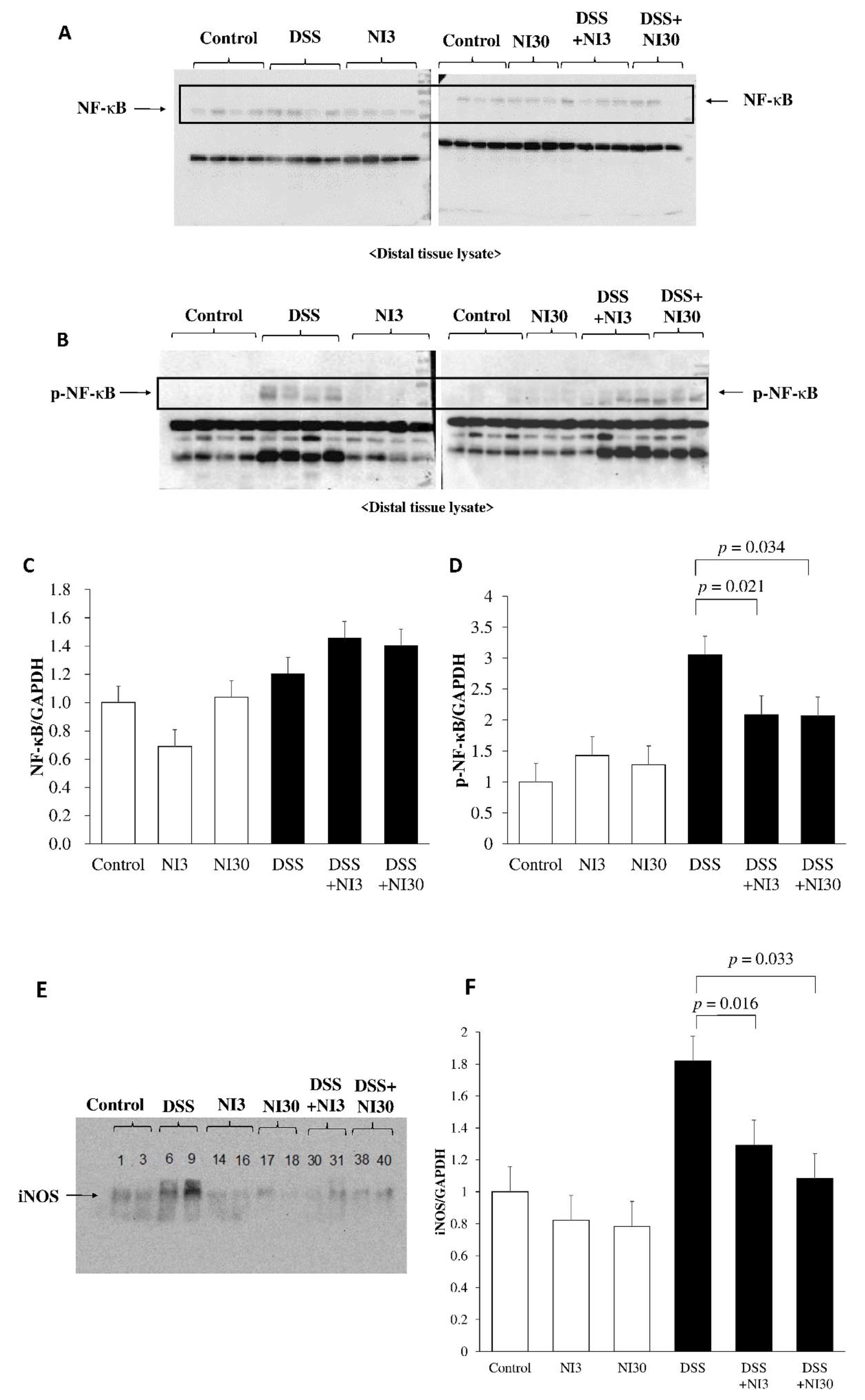
3.4. Western Blot Assay
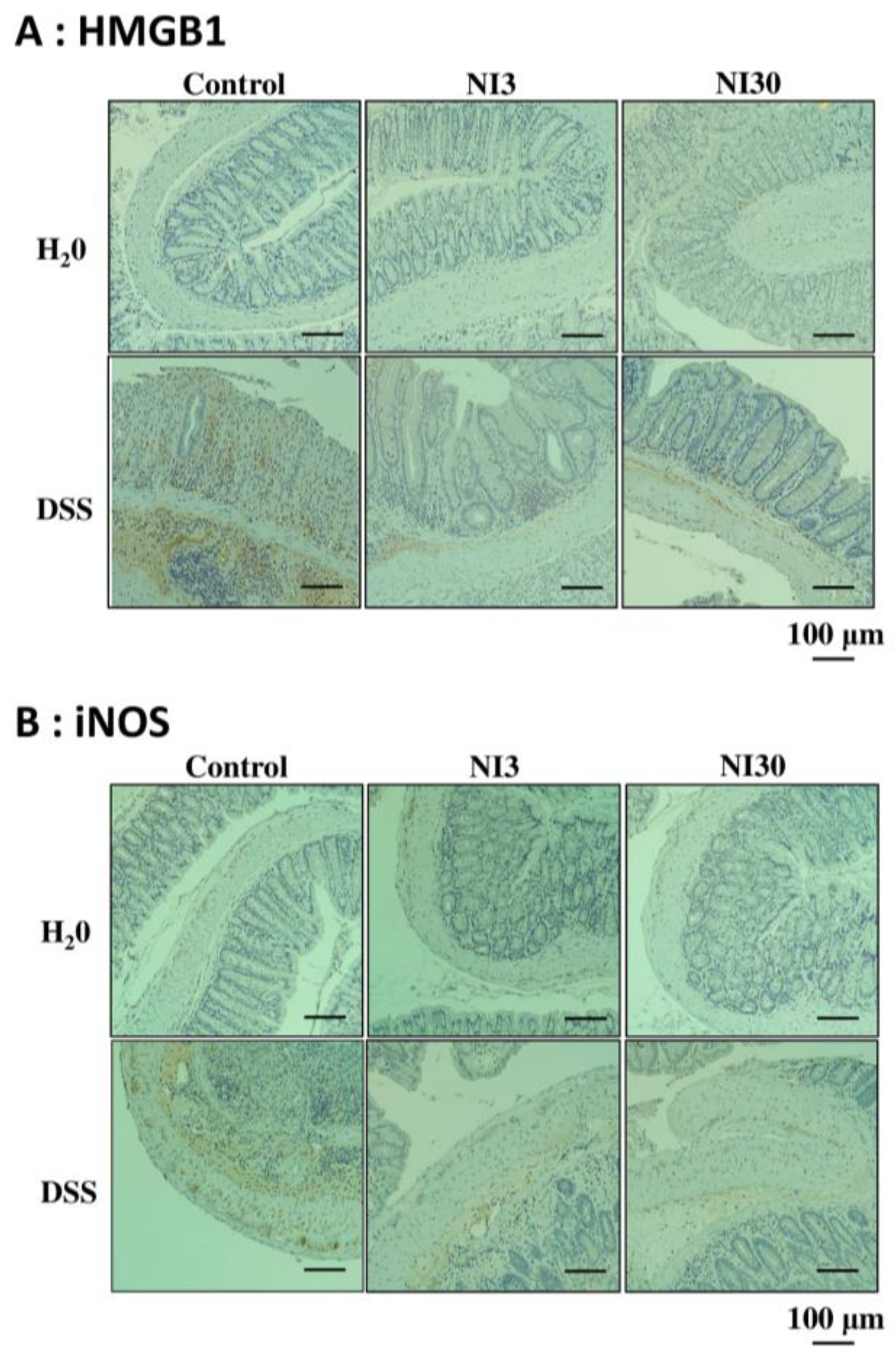
3.5. Immunohistochemical Staining
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogler, G. Resolution of inflammation in inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2017, 2, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Mowat, C.; Cole, A.; Windsor, A.; Ahmad, T.; Arnott, I.; Driscoll, R.; Mitton, S.; Orchard, T.; Rutter, M.; Younge, L.; et al. Guidelines for the management of inflammatory bowel disease in adults. Gut 2011, 60, 571–607. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, O.H.; Ainsworth, M.A. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 2013, 369, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W.; et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 376, 1723–1736. [Google Scholar] [CrossRef]
- Sands, B.E.; Peyrin-Biroulet, L.; Loftus, E.V., Jr.; Danese, S.; Colombel, J.F.; Törüner, M.; Jonaitis, L.; Abhyankar, B.; Chen, J.; Rogers, R.; et al. Vedolizumab versus Adalimumab for Moderate-to-Severe Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1215–1226. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Hagiwara, C.; Tanaka, M.; Kudo, H. Increase in colorectal epithelial apoptotic cells in patients with ulcerative colitis ultimately requiring surgery. J. Gastroenterol. Hepatol. 2002, 17, 758–764. [Google Scholar] [CrossRef]
- Souza, H.S.; Tortori, C.J.; Castelo-Branco, M.T.; Carvalho, A.T.; Margallo, V.S.; Delgado, C.F.; Dines, I.; Elia, C.C. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: Evidence of altered expression of FasL and perforin cytotoxic pathways. Int. J. Colorectal Dis. 2005, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Geng, H.; Tan, X.D. Cell death of intestinal epithelial cells in intestinal diseases. Sheng Li Xue Bao 2020, 72, 308–324. [Google Scholar] [PubMed]
- Watson, A.J. Necrosis and apoptosis in the gastrointestinal tract. Gut 1995, 37, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Koo, S.Y.; Ahn, B.H.; Park, O.; Park, D.H.; Seo, D.O.; Won, J.H.; Yim, H.J.; Kwak, H.S.; Park, H.S.; et al. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO− scavenger. Arch. Pharm. Res 2010, 33, 1813–1823. [Google Scholar] [CrossRef]
- Thu, V.T.; Kim, H.K.; Long, L.T.; Lee, S.R.; Hanh, T.M.; Ko, T.H.; Heo, H.J.; Kim, N.; Kim, S.H.; Ko, K.S.; et al. NecroX-5 prevents hypoxia/reoxygenation injury by inhibiting the mitochondrial calcium uniporter. Cardiovasc. Res. 2012, 94, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Im, K.I.; Kim, N.; Lim, J.Y.; Nam, Y.S.; Lee, E.S.; Kim, E.J.; Kim, H.J.; Kim, S.H.; Cho, S.G. The Free Radical Scavenger NecroX-7 Attenuates Acute Graft-versus-Host Disease via Reciprocal Regulation of Th1/Regulatory T Cells and Inhibition of HMGB1 Release. J. Immunol. 2015, 194, 5223–5232. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Park, E.; Ahn, B.H.; Kim, H.J.; Park, J.H.; Koo, S.Y.; Kwak, H.S.; Park, H.S.; Kim, D.W.; Song, M.; et al. NecroX-7 prevents oxidative stress-induced cardiomyopathy by inhibition of NADPH oxidase activity in rats. Toxicol. Appl. Pharmacol. 2012, 263, 1–6. [Google Scholar] [CrossRef]
- Jin, S.A.; Kim, S.K.; Seo, H.J.; Jeong, J.Y.; Ahn, K.T.; Kim, J.H.; Choi, D.E.; Park, J.H.; Lee, J.H.; Choi, S.W.; et al. Beneficial Effects of Necrosis Modulator, Indole Derivative NecroX-7, on Renal Ischemia-Reperfusion Injury in Rats. Transplant. Proc. 2016, 48, 199–204. [Google Scholar] [CrossRef]
- Kim, D.; Koo, J.S.; Park, J.H.; Hwang, S.H.; Lee, D.; Choe, J.W.; Hyun, J.J.; Jung, S.W.; Jeen, Y.T.; Lee, S.W. The protective effect of necrosis inhibition on acute murine colitis induced by dextran sulphate sodium. J. Crohn’s Colitis 2019, 13, S116. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15.25.11–15.25.14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.C.; Bériou, G.; Josien, R. Dextran Sulfate Sodium (DSS)-Induced Acute Colitis in the Rat. Methods Mol. Biol. 2016, 1371, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Nakagawa, S.; Yamaguchi, T.; Yoshida, N.; Minami, M.; Kita, M.; Imanishi, J.; et al. Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J. Gastroenterol. Hepatol. 2003, 18, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, H.K.; Kim, Y.K.; Park, J.H.; Ryu, M.J.; Chang, J.Y.; Hwang, J.H.; Lee, C.H.; Kim, S.H.; Kim, H.J.; Kweon, G.R.; et al. The indole derivative NecroX-7 improves nonalcoholic steatohepatitis in ob/ob mice through suppression of mitochondrial ROS/RNS and inflammation. Liver Int. 2015, 35, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; de Haar, C.; Chen, M.; Deuring, J.; Gerrits, M.M.; Smits, R.; Xia, B.; Kuipers, E.J.; van der Woude, C.J. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut 2010, 59, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011, 22, 83–89. [Google Scholar] [CrossRef]
- Kwon, D.J.; Bae, Y.S.; Ju, S.M.; Youn, G.S.; Choi, S.Y.; Park, J. Salicortin suppresses lipopolysaccharide-stimulated inflammatory responses via blockade of NF-κB and JNK activation in RAW 264.7 macrophages. BMB Rep. 2014, 47, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, Y.; Dong, H.; Wang, B.; Ji, H.; Liu, X. Trilobatin attenuates the LPS-mediated inflammatory response by suppressing the NF-κB signaling pathway. Food Chem. 2015, 166, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Andresen, L.; Jørgensen, V.L.; Perner, A.; Hansen, A.; Eugen-Olsen, J.; Rask-Madsen, J. Activation of nuclear factor kappaB in colonic mucosa from patients with collagenous and ulcerative colitis. Gut 2005, 54, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Im, K.I.; Nam, Y.S.; Kim, N.; Song, Y.; Lee, E.S.; Lim, J.Y.; Jeon, Y.W.; Cho, S.G. Regulation of HMGB1 release protects chemoradiotherapy-associated mucositis. Mucosal Immunol. 2019, 12, 1070–1081. [Google Scholar] [CrossRef] [Green Version]
- Antoine, D.J.; Harris, H.E.; Andersson, U.; Tracey, K.J.; Bianchi, M.E. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol. Med. 2014, 20, 135–137. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef]
- Degryse, B.; Bonaldi, T.; Scaffidi, P.; Müller, S.; Resnati, M.; Sanvito, F.; Arrigoni, G.; Bianchi, M.E. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol. 2001, 152, 1197–1206. [Google Scholar] [CrossRef]
- Müller, S.; Scaffidi, P.; Degryse, B.; Bonaldi, T.; Ronfani, L.; Agresti, A.; Beltrame, M.; Bianchi, M.E. New EMBO members’ review: The double life of HMGB1 chromatin protein: Architectural factor and extracellular signal. EMBO J. 2001, 20, 4337–4340. [Google Scholar] [CrossRef]
- Tirone, M.; Tran, N.L.; Ceriotti, C.; Gorzanelli, A.; Canepari, M.; Bottinelli, R.; Raucci, A.; Di Maggio, S.; Santiago, C.; Mellado, M.; et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J. Exp. Med. 2018, 215, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Di Maggio, S.; Scavello, F.; D’Ambrosio, A.; Bianchi, M.E.; Capogrossi, M.C. The Janus face of HMGB1 in heart disease: A necessary update. Cell. Mol. Life Sci. 2019, 76, 211–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Cross, R.K.; Wilson, K.T. Nitric oxide in inflammatory bowel disease. Inflamm. Bowel Dis. 2003, 9, 179–189. [Google Scholar] [CrossRef]
- Kimura, H.; Miura, S.; Shigematsu, T.; Ohkubo, N.; Tsuzuki, Y.; Kurose, I.; Higuchi, H.; Akiba, Y.; Hokari, R.; Hirokawa, M.; et al. Increased nitric oxide production and inducible nitric oxide synthase activity in colonic mucosa of patients with active ulcerative colitis and Crohn’s disease. Dig. Dis. Sci. 1997, 42, 1047–1054. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Feature | Score | Description |
|---|---|---|
| Weight loss (from baseline) | 0 | No weight loss or increase |
| 1 | Weight loss of 1–10% | |
| 2 | Weight loss of 11–15% | |
| 3 | Weight loss of 16–20% | |
| 4 | Weight loss > 20% | |
| Stool consistency | 0 | Well-formed pellets |
| 2 | Semi-formed stool (no anal adherence) | |
| 4 | Liquid stool (anal adherence) | |
| Bleeding | 0 | Absence |
| 2 | Blood-tinged stool | |
| 4 | Gross bleeding |
| Feature | Score | Description |
|---|---|---|
| Inflammation severity | 0 | None |
| 1 | Mild | |
| 2 | Moderate | |
| 3 | Severe | |
| Inflammation extent | 0 | None |
| 1 | Mucosa | |
| 2 | Mucosa and submucosa | |
| 3 | Transmural | |
| Crypt damage | 0 | None |
| 1 | Basal 1/3 damaged | |
| 2 | Basal 2/3 damaged | |
| 3 | Crypt loss and surface-epithelium retention | |
| 4 | Crypt and surface-epithelium loss | |
| Percent involvement | 0 | None |
| 1 | 1–25% | |
| 2 | 26–50% | |
| 3 | 51–75% | |
| 4 | 76–100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Koo, J.S.; Kim, S.H.; Park, Y.S.; Choe, J.W.; Kim, S.Y.; Hyun, J.J.; Jung, S.W.; Jung, Y.K.; Yim, H.J. The Effect of Necrosis Inhibitor on Dextran Sulfate Sodium Induced Chronic Colitis Model in Mice. Pharmaceutics 2023, 15, 222. https://doi.org/10.3390/pharmaceutics15010222
Kim D, Koo JS, Kim SH, Park YS, Choe JW, Kim SY, Hyun JJ, Jung SW, Jung YK, Yim HJ. The Effect of Necrosis Inhibitor on Dextran Sulfate Sodium Induced Chronic Colitis Model in Mice. Pharmaceutics. 2023; 15(1):222. https://doi.org/10.3390/pharmaceutics15010222
Chicago/Turabian StyleKim, Dongwoo, Ja Seol Koo, Soon Ha Kim, Yeong Seo Park, Jung Wan Choe, Seung Young Kim, Jong Jin Hyun, Sung Woo Jung, Young Kul Jung, and Hyung Joon Yim. 2023. "The Effect of Necrosis Inhibitor on Dextran Sulfate Sodium Induced Chronic Colitis Model in Mice" Pharmaceutics 15, no. 1: 222. https://doi.org/10.3390/pharmaceutics15010222
APA StyleKim, D., Koo, J. S., Kim, S. H., Park, Y. S., Choe, J. W., Kim, S. Y., Hyun, J. J., Jung, S. W., Jung, Y. K., & Yim, H. J. (2023). The Effect of Necrosis Inhibitor on Dextran Sulfate Sodium Induced Chronic Colitis Model in Mice. Pharmaceutics, 15(1), 222. https://doi.org/10.3390/pharmaceutics15010222