
What Can RNA-Based Therapy Do for Monogenic Diseases?


Abstract
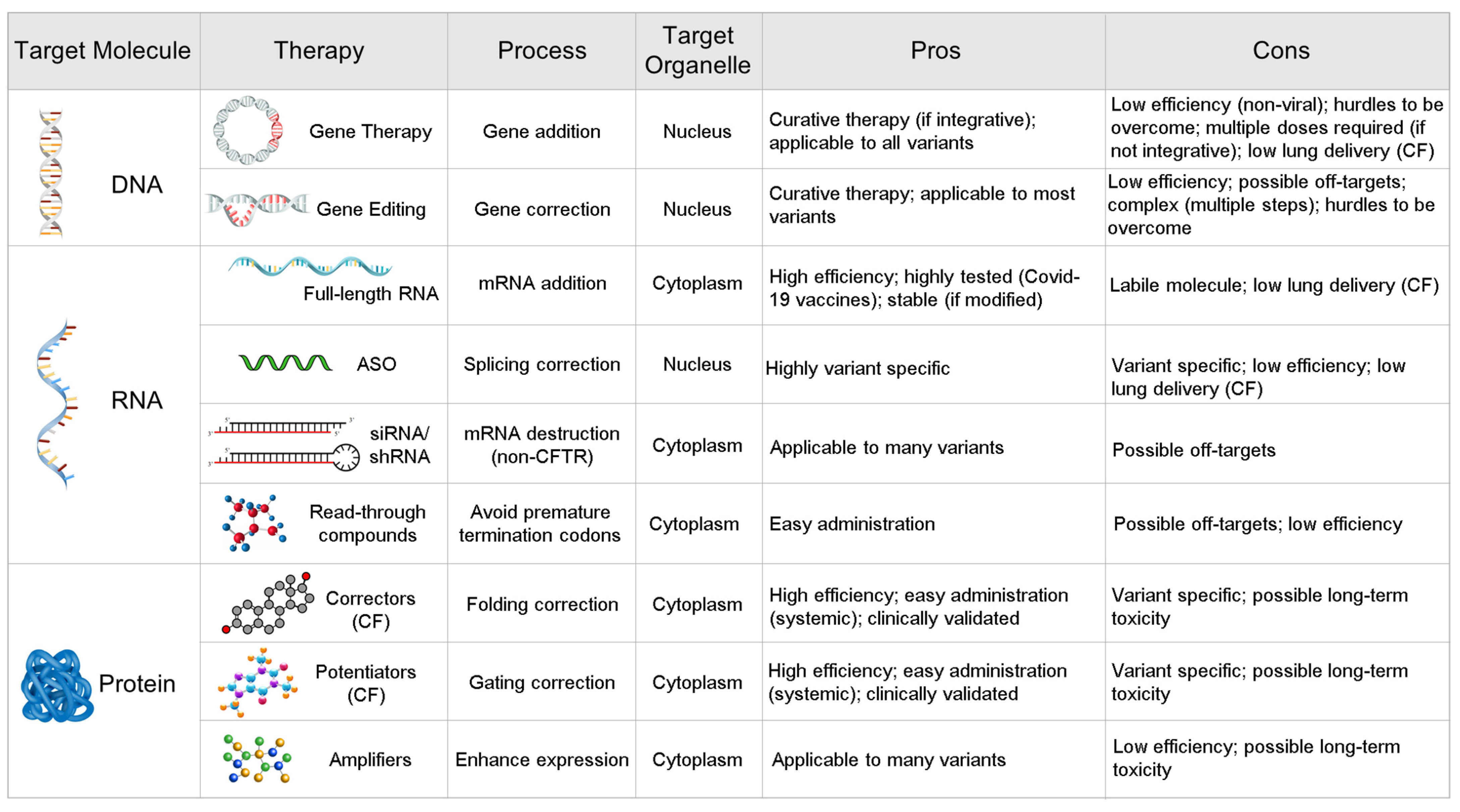
:1. Introduction
2. Mutation by Mutation: ASOs

{kind=link}
| Disease (Gene) | ASO Mechanism | Result | Model |
|---|---|---|---|
| CF (CFTR) | Block aberrant splice site (c.2657+5G>A variant) | Inclusion of skipped exon | Cells [25] |
| Block aberrant splice site (c.3718-2477C>T variant) | Exclusion of cryptic exon | Patient primary cells [26,27] | |
| Modulate splicing of exon containing p.Trp1282X variant | Skipping of variant containing exon | Patient primary cells [28,29,30] | |
| Block EJC deposition downstream of p.Trp1282X variant | Inhibition of NMD | Cells [32] | |
| SMA (SMN1) | Promote exon 7 inclusion in paralogous gene SMN2 | Enhanced compensatory SMN2 expression | Drug: Nusinersen [38] |
| DMD (DMD) | Skip variant contain exons without disrupting DMD open reading frame | Skipping of variant containing exons and production of edited but functional DMD | Drugs: Casimirsen [42], Eteplirsen [43], Golodirsen, Viltolarsen [44] |
3. Gene by Gene: mRNA Therapy
| Disease (Gene) | Method | Result | Model |
|---|---|---|---|
| CF (CFTR) | Transfection with optimized wt-CFTR mRNA | Increase in functional CFTR protein | Cells [50] |
| LNP-packaged modified CFTR mRNA | Increase in CFTR protein function | Patient-derived cells, mice [51] | |
| ReCode RTX00001 mRNA in SORT-LNP | Restoration of CFTR function | Primary hBE cells and mice [52] | |
| Arcturus LUNAR-LNP system | Significant in vivo functional restoration | Ferret and mice in [53] | |
| Nebulized MRT5005 mRNA | No adverse effects | First-in-human phases 1/2 trial [55,56] | |
| Haemophilia B (hEPO, hFIX) | LNPs and Lipid-like nanoparticle delivery of mRNAs | Production of a “depot” system of functional protein production in hepatocytes | Mice and non-human primates [58] |
| MMA (hMUT) | Intravenous administration of hMUT LNP-encapsulated mRNAs | Increased liver production of active MUT, survival and weight gain. | Mice [59,60] |
| Arginase 1 deficiency (ARG1) | Injection of LNP-encapsulated human codon-optimised mRNA | Increased liver hARG1 expression and activity, increased survival. | Mice [62,63] |
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Nopoulos, P.C. Huntington Disease: A Single-Gene Degenerative Disorder of the Striatum. Dialogues Clin. Neurosci. 2022, 18, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N.; Arteaga-Solis, E.; Baldock, C.; Collod-Béroud, G.; Booms, P.; De Paepe, A.; Dietz, H.C.; Guo, G.; Handford, P.A.; Judge, D.P.; et al. The molecular genetics of Marfan syndrome and related disorders. J. Med. Genet 2006, 43, 769–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef]
- Origa, R. β-Thalassemia. Genet Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal muscular atrophy. Orphanet. J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Wein, N.; Alfano, L.; Flanigan, K.M. Genetics and emerging treatments for Duchenne and Becker muscular dystrophy. Pediatr. Clin. N. Am. 2015, 62, 723–742. [Google Scholar] [CrossRef]
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef]
- Prakash, V.; Moore, M.; Yáñez-Muñoz, R.J. Current Progress in Therapeutic Gene Editing for Monogenic Diseases. Mol. Ther. 2016, 24, 465–474. [Google Scholar] [CrossRef]
- Apgar, T.L.; Sanders, C.R. Compendium of causative genes and their encoded proteins for common monogenic disorders. Prot Sci. 2022, 31, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Rodwell, C.; Aymé, S. (Eds.) 2014 Report on the State of the Art of Rare Disease Activities in Europe Part II: Key Developments in the Field of Rare Diseases in Europe in 2013. 2014, pp. 1–90. Available online: http://www.rd-action.eu/eucerd/SoA_main/2014ReportStateofArtRDActivitiesII.pdf (accessed on 16 December 2022).
- Cystic Fibrosis Mutation Database. Available online: http://genet.sickkids.on.ca/StatisticsPage.html (accessed on 15 December 2022).
- Fanen, P.; Wohlhuter-Haddad, A.; Hinzpeter, A. Genetics of cystic fibrosis: CFTR mutation classifications toward genotype-based CF therapies. Int. J. Biochem. Cell Biol. 2014, 52, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boeck, K.; Amaral, M.D. Progress in therapies for cystic fibrosis. Lancet. Respir. Med. 2016, 4, 662–674. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Garratt, A.; Hill, A. Worldwide rates of diagnosis and effective treatment for cystic fibrosis. J. Cyst. Fibros. 2022, 21, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, J.; Zhang, J.; Fortunak, J.; Hill, A. Current prices versus minimum costs of production for CFTR modulators. J. Cyst. Fibros. 2022, 21, 866–872. [Google Scholar] [CrossRef]
- Weatherall, D.J. Pharmacological treatment of monogenic disease. Pharmacogenom. J. 2003, 3, 264–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug. Dis. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic. Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Igreja, S.; Clarke, L.A.; Botelho, H.M.; Marques, L.; Amaral, M.D. Correction of a Cystic Fibrosis Splicing Mutation by Antisense Oligonucleotides. Hum. Mutat. 2016, 37, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Michaels, W.E.; Bridges, R.J.; Hastings, M.L. Antisense oligonucleotide-mediated correction of CFTR splicing improves chloride secretion in cystic fibrosis patient-derived bronchial epithelial cells. Nucl. Acids Res. 2020, 48, 7454–7467. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Irony-Tur Sinai, M.; Golec, A.; Barchad-Avitzur, O.; Mutyam, V.; Li, Y.; Hong, J.; Ozeri-Galai, E.; Hatton, A.; Leibson, C.; et al. Antisense oligonucleotide-based drug development for Cystic Fibrosis patients carrying the 3849+10 kb C-to-T splicing mutation. J. Cyst. Fibros. 2021, 20, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Avizur-Barchad, O.; Ozeri-Galai, E.; Elgrabli, R.; Schirelman, M.R.; Blinder, T.; Stampfer, C.D.; Ordan, M.; Laselva, O.; Cohen-Cymberknoh, M.; et al. Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J. Cyst. Fibros. 2022, 21, 630–636. [Google Scholar] [CrossRef]
- Michaels, W.E.; Pena-Rasgado, C.; Kotaria, R.; Bridges, R.J.; Hastings, M.L. Open reading frame correction using splice-switching antisense oligonucleotides for the treatment of cystic fibrosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2114886119. [Google Scholar] [CrossRef]
- Kim, Y.J.; Sivetz, N.; Layne, J.; Voss, D.M.; Yang, L.; Zhang, Q.; Krainer, A.R. Exon-skipping antisense oligonucleotides for cystic fibrosis therapy. Proc. Natl. Acad. Sci. USA 2022, 119, e2114858118. [Google Scholar] [CrossRef]
- Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Xu, H.; Avramescu, R.G.; Perdomo, D.; Zlock, L.; Nielson, D.W.; Finkbeiner, W.E.; Lukacs, G.L.; et al. Correctors and Potentiators Rescue Function of the Truncated W1282X-Cystic Fibrosis Transmembrane Regulator (CFTR) Translation Product. J. Biol. Chem. 2017, 292, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Nomakuchi, T.; Papaleonidopoulou, F.; Yang, L.; Zhang, Q.; Krainer, A.R. Gene-specific nonsense-mediated mRNA decay targeting for cystic fibrosis therapy. Nat. Commun. 2022, 13, 2978. [Google Scholar] [CrossRef]
- Thomas, G.S.; Cromwell, W.C.; Ali, S.; Chin, W.; Flaim, J.D.; Davidson, M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2013, 62, 2178–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeskamp, L.F.; Kastelein, J.J.P.; Moriarty, P.M.; Duell, P.B.; Catapano, A.L.; Santos, R.D.; Ballantyne, C.M. Safety and efficacy of mipomersen in patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2019, 280, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 22–31. [Google Scholar] [CrossRef]
- Brannagan, T.H.; Coelho, T.; Wang, A.K.; Polydefkis, M.J.; Dyck, P.J.; Berk, J.L.; Drachman, B.; Gorevic, P.; Whelan, C.; Conceição, I.; et al. Open-Label Extension Investigators. Long-term efficacy and safety of inotersen for hereditary transthyretin amyloidosis: NEURO-TTR open-label extension 3-year update. J. Neurol. 2022, 269, 6416–6427. [Google Scholar] [CrossRef]
- Brzustowicz, L.M.; Lehner, T.; Castilla, L.H.; Penchaszadeh, G.K.; Wilhelmsen, K.C.; Daniels, R.; Davies, K.E.; Leppert, M.; Ziter, F.; Wood, D. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3. Nature 1990, 344, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torroba, B.; Macabuag, N.; Haisma, E.M.; O’Neill, A.; Herva, M.E.; Redis, R.S.; Templin, M.V.; Black, L.E.; Fischer, D.F. RNA-based drug discovery for spinal muscular atrophy: A story of small molecules and antisense oligonucleotides. Exp. Opin. Drug. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.; Wood, M.J.A.; Roberts, T.C. Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing. RNA Biol. 2021, 18, 1048–1062. [Google Scholar] [CrossRef]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef]
- Syed, Y.Y. Eteplirsen: First Global Approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, M.; Frey, J.; Shohan, M.J.; Malek, S.; Mousa, S.A. Current and emerging therapies for Duchenne muscular dystrophy and spinal muscular atrophy. Pharmacol. Ther. 2021, 220, 107719. [Google Scholar] [CrossRef]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.B.; Manautou, J.E. Adverse Drug Reactions and Toxicity of the Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. 2022, 50, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release 2021, 333, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.M.; Lagerström-Fermér, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.C.; Rudvik, A.; Kjaer, M.; Collén, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef] [Green Version]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef] [PubMed]
- Bangel-Ruland, N.; Tomczak, K.; Fernández Fernández, E.; Leier, G.; Leciejewski, B.; Rudolph, C.; Rosenecker, J.; Weber, W.M. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: A novel alternative for cystic fibrosis gene therapy. J. Gene Med. 2013, 15, 414–426. [Google Scholar] [CrossRef]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, D.; Boudko, D.; Meleshkevitch, E.A.; Sidhu, M.S.; Poniatowski, J.R.; Gao, P.; Molla, T.; Comini, S.; Lister, H.; Coquelin, M.; et al. Functional Rescue of CFTR by Aerosolized Delivery of Optimized CFTR mRNA Using ReCode LNPs in Primary Human Bronchial Epithelial Cells Derived from Patients with Cystic Fibrosis. In Proceedings of the American Thoracic Society (ATS) International Conference, San Francisco, CA, USA, 18 May 2022. [Google Scholar]
- Harrison, P.T. CFTR RNA- and DNA-based therapies. Curr. Opin. Pharmacol. 2022, 65, 102247. [Google Scholar] [CrossRef]
- Pipeline of Arcturus-Owned mRNA Therapeutic Candidates. Available online: https://arcturusrx.com/mrna-medicines-pipeline/ (accessed on 16 December 2022).
- Zuckerman, J.; McCoy, K.; Schechter, M.S.; Dorgan, D.; Jain, M.; MacDonald, K.; Rowe, S. Safety and tolerability of a single dose of MRT5005, an inhaled CFTR mRNA therapeutic, in adult CF patients. Pediatr. Pulmonol. 2019, 54, S350. [Google Scholar]
- Study to Evaluate the Safety & Tolerability of MRT5005 Administered by Nebulization in Adults with Cystic Fibrosis (RESTORE-CF). Available online: https://clinicaltrials.gov/ct2/show/study/NCT03375047 (accessed on 16 December 2022).
- Lorenzer, C.; Dirin, M.; Winkler, A.M.; Baumann, V.; Winkler, J. Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 2015, 203, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRosa, F.; Guild, B.; Karve, S.; Smith, L.; Love, K.; Dorkin, J.R.; Kauffman, K.J.; Zhang, J.; Yahalom, B.; Anderson, D.G.; et al. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016, 23, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helman, G.; Pacheco-Colón, I.; Gropman, A.L. The urea cycle disorders. Semin. Neurol. 2014, 34, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asrani, K.H.; Cheng, L.; Cheng, C.J.; Subramanian, R.R. Arginase I mRNA therapy–a novel approach to rescue arginase 1 enzyme deficiency. RNA Biol. 2018, 15, 914–922. [Google Scholar] [CrossRef] [Green Version]
- Truong, B.; Allegri, G.; Liu, X.B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- NIH Clinical Trials Website. Available online: https://clinicaltrials.gov/ (accessed on 4 January 2023).
- Vavilis, T.; Stamoula, E.; Ainatzoglou, A.; Sachinidis, A.; Lamprinou, M.; Dardalas, I.; Vizirianakis, I.S. mRNA in the Context of Protein Replacement Therapy. Pharmaceutics 2023, 15, 166. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Dis. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Crooke, S.T.; Wang, S.; Vickers, T.A.; Shen, W.; Liang, X.H. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Dis. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, L.A.; Amaral, M.D. What Can RNA-Based Therapy Do for Monogenic Diseases? Pharmaceutics 2023, 15, 260. https://doi.org/10.3390/pharmaceutics15010260
Clarke LA, Amaral MD. What Can RNA-Based Therapy Do for Monogenic Diseases? Pharmaceutics. 2023; 15(1):260. https://doi.org/10.3390/pharmaceutics15010260
Chicago/Turabian StyleClarke, Luka A., and Margarida D. Amaral. 2023. "What Can RNA-Based Therapy Do for Monogenic Diseases?" Pharmaceutics 15, no. 1: 260. https://doi.org/10.3390/pharmaceutics15010260
APA StyleClarke, L. A., & Amaral, M. D. (2023). What Can RNA-Based Therapy Do for Monogenic Diseases? Pharmaceutics, 15(1), 260. https://doi.org/10.3390/pharmaceutics15010260