
Design, Development, and Optimisation of Smart Linker Chemistry for Targeted Colonic Delivery—In Vitro Evaluation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthetic Procedures and Analytical Data
2.2. In Vitro Release Assays
3. Results and Discussion
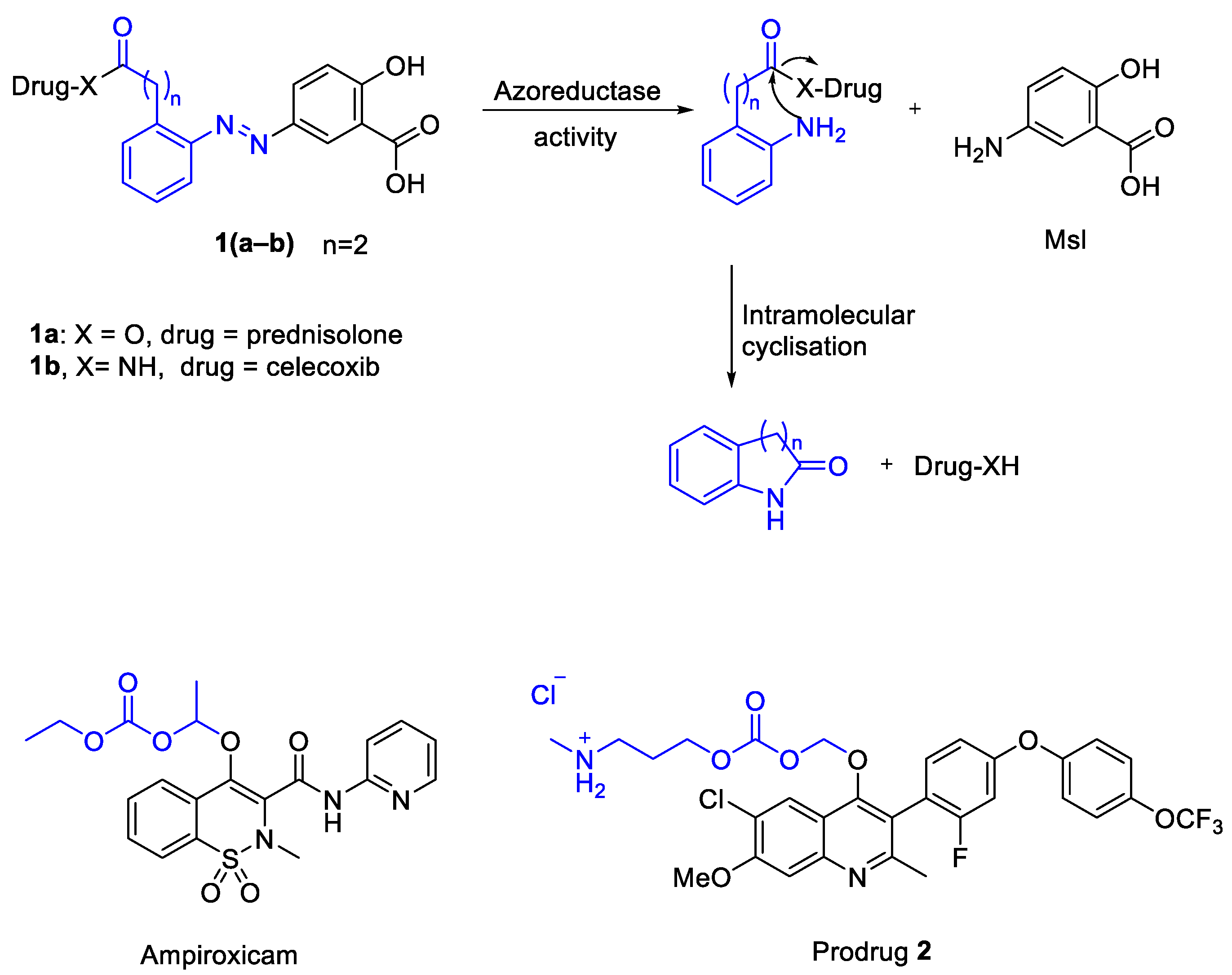
3.1. Proposed Release Mechanism
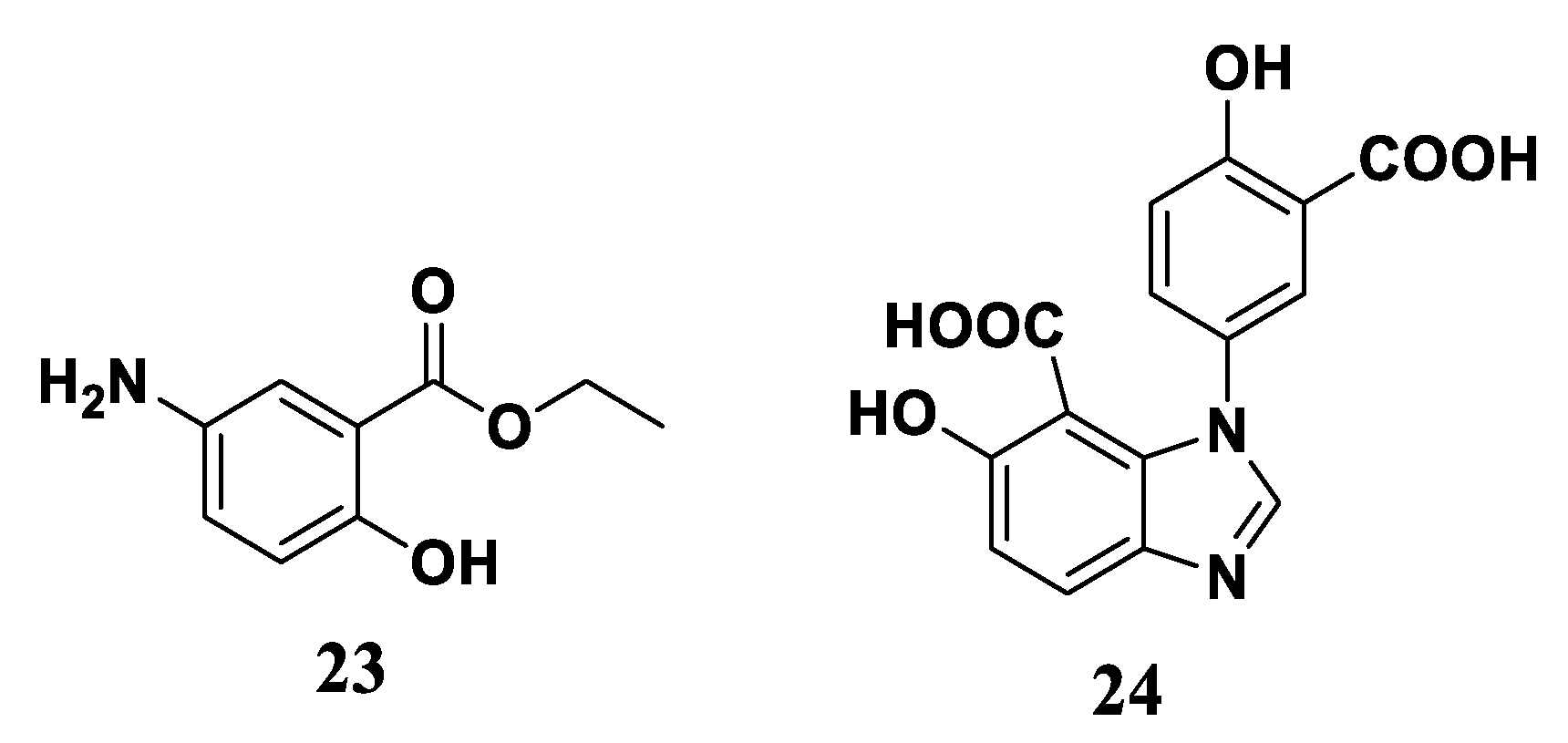
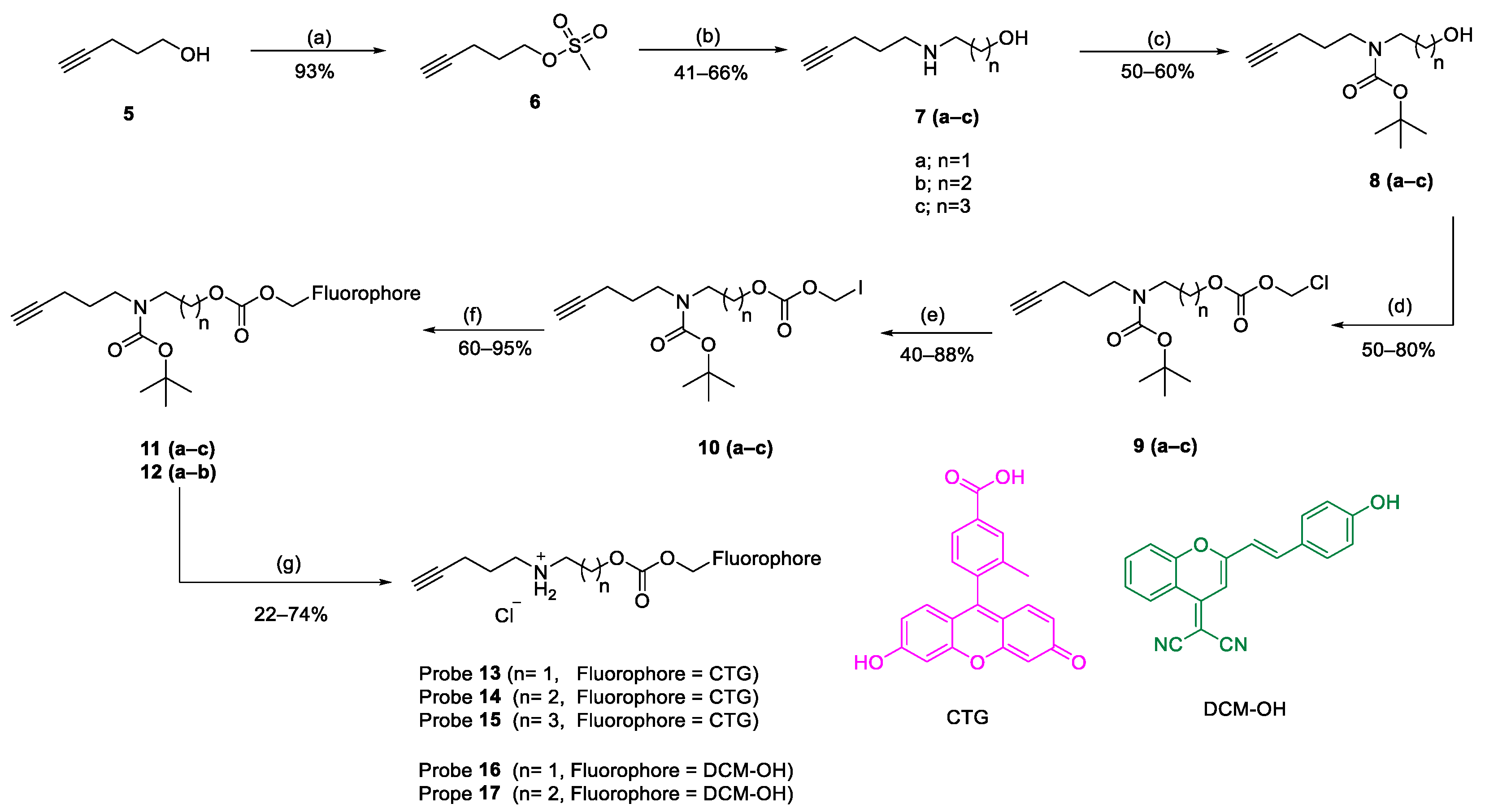
3.2. Synthesis
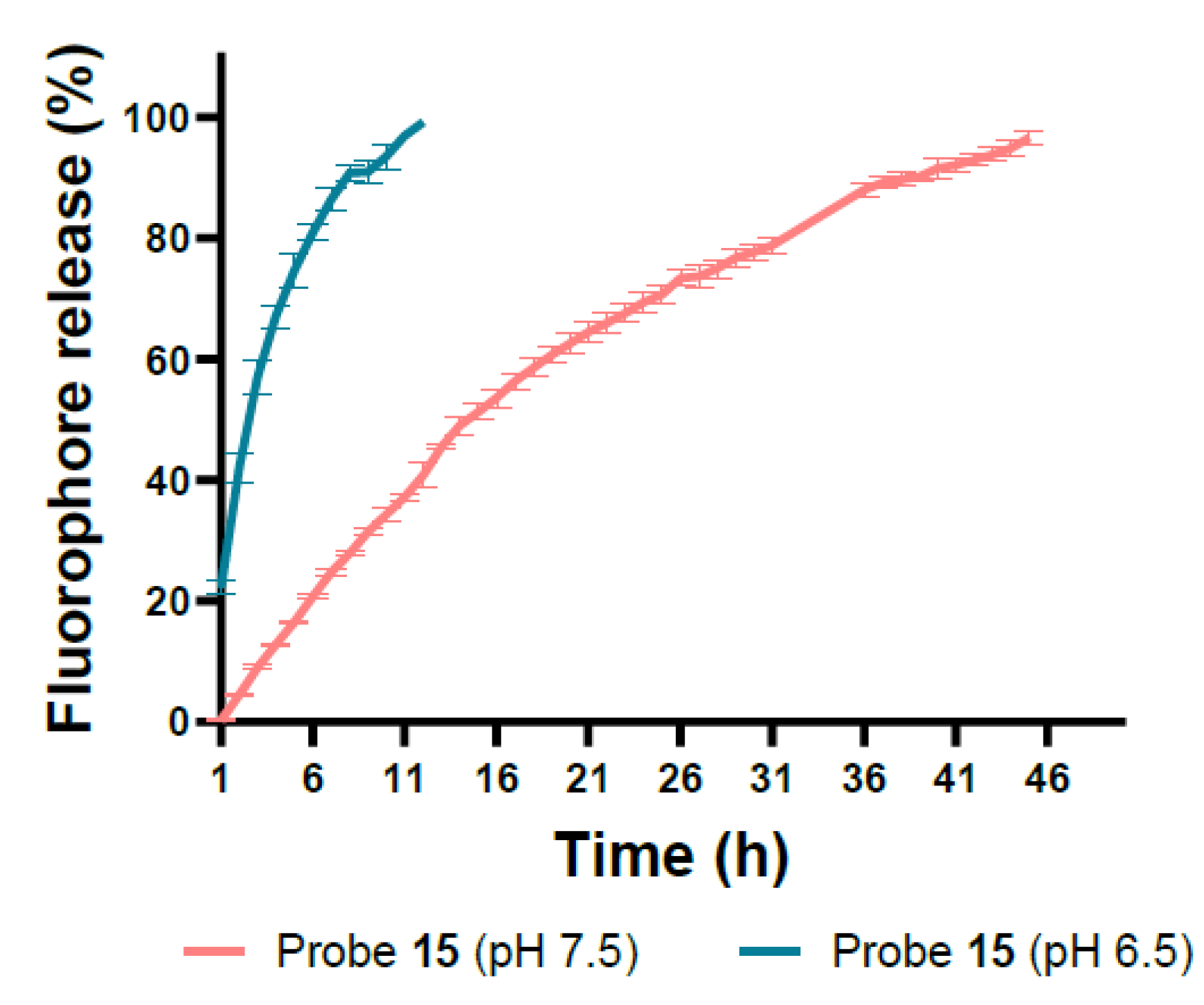
3.3. In Vitro Release of Probe Compounds
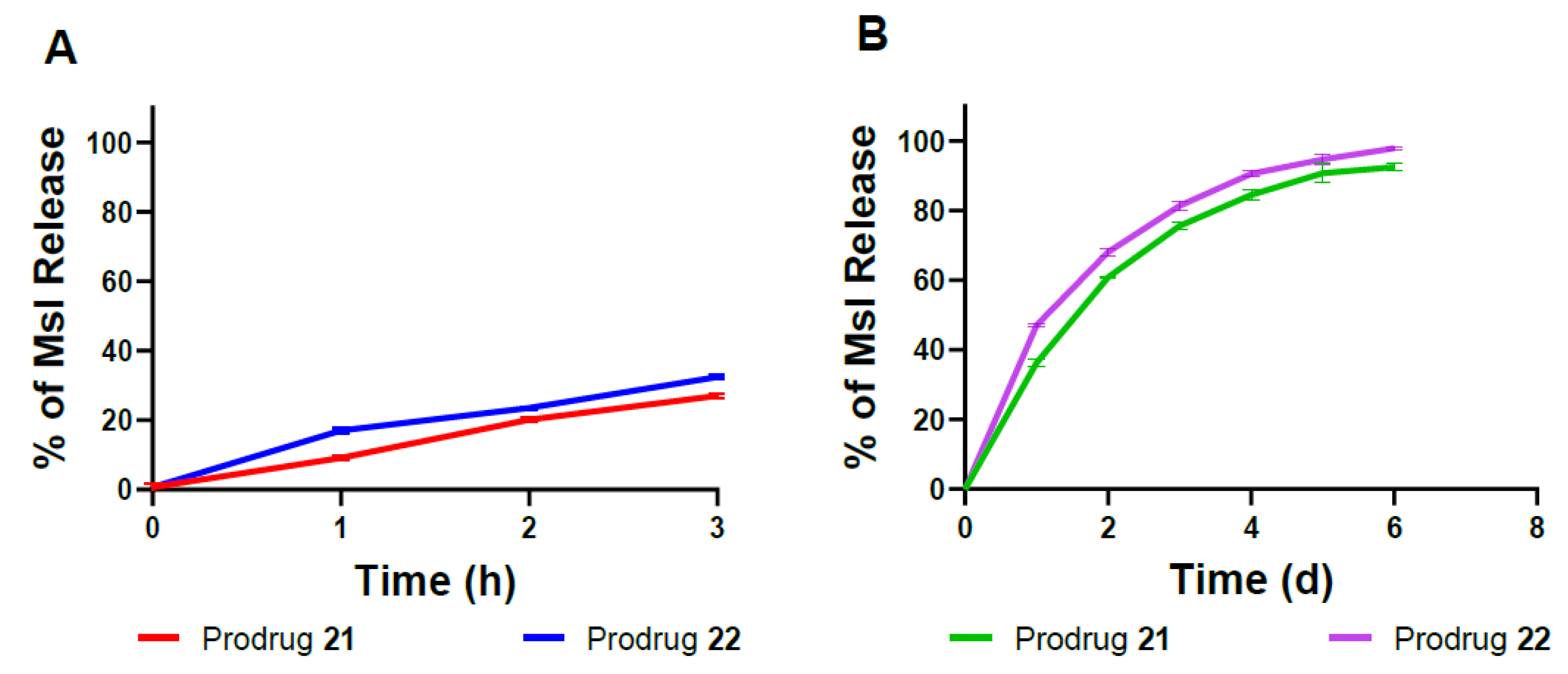
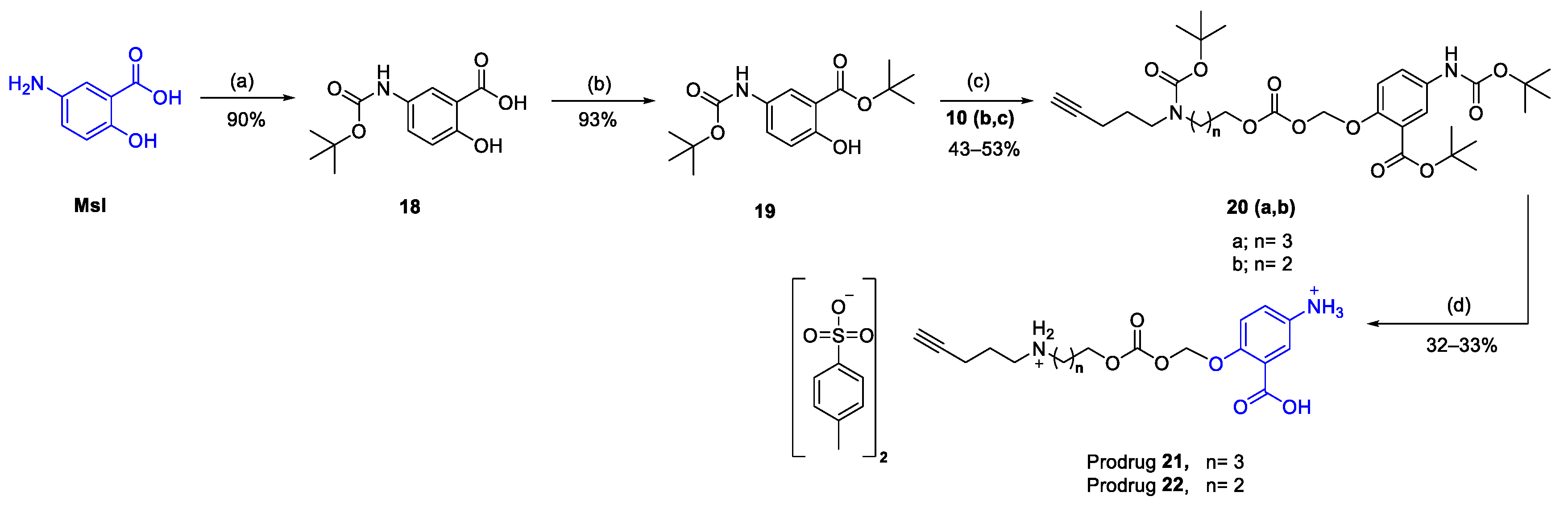
3.4. In Vitro Release Studies of Msl Prodrugs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Amino-AOCOM | Amino-alkoxycarbonyloxymethyl |
| GIT | Gastrointestinal tract |
| Msl | Mesalamine |
| CTG | 4-Carboxy-2-methyl Tokyo Green |
| DCM-OH | Dicyanomethylene-4H-pyran dye |
| Boc2O | Boc anhydride |
| CDI | 1,1′-Carbonyldiimidazole |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| Fe(II)–EDTA | Iron(II)ethylenediaminetetraacetic acid |
Appendix A
Appendix A.1. Preparation of Pent-4-yn-1-yl Methanesulfonate 6
Appendix A.2. General Procedure for the Synthesis of Compounds 7 (a–c)
Appendix A.3. General Procedure for the Synthesis of Compounds 8 (a–c) [49,50]
Appendix A.4. General Procedure for the Synthesis of Compounds 9 (a–c) [51]
Appendix A.5. General Procedure for the Synthesis of Compounds 10 (a–c) [21]
Appendix A.6. General Procedure for the Synthesis of Compounds 11 (a–c) and 12 (a–b)
Appendix A.7. General Procedure for the Synthesis of Probes 13–17
Appendix A.8. Procedure for the Synthesis of 5-((Tert-butoxycarbonyl)amino)-2-hydroxybenzoic Acid (18)
Appendix A.9. Procedure for the Synthesis of Tert-butyl 5-((tert-butoxycarbonyl)amino)-2-hydroxybenzoate (19)
Appendix A.10. General Procedure for the Synthesis of Compounds 20 (a–b)
Appendix A.11. General Procedure for the Synthesis of Msl Prodrugs 21 and 22
References
- Friend, D.R. New oral delivery systems for treatment of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2005, 57, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Philip, A.; Philip, B. Colon Targeted Drug Delivery Systems: A Review on Primary and Novel Approaches. Oman Med. J. 2010, 25, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Odeku, O.A.; Fell, J.T. In-vitro evaluation of khaya and albizia gums as compression coatings for drug targeting to the colon. J. Pharm. Pharmacol. 2005, 57, 163–168. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Wal, P.; Wal, A.; Rai, A.K. Primary and novel approaches for colon targeted drug delivery—A review. Int. J. Drug Deliv. 2010, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sang, Y.; Feng, J.; Li, Z.; Zhao, A. Polysaccharide-based micro/nanocarriers for oral colon-targeted drug delivery. J. Drug Target. 2016, 24, 579–589. [Google Scholar] [CrossRef]
- Zhu, Q.; Berzofsky, J.A. Oral vaccines: Directed safe passage to the front line of defense. Gut Microbes 2013, 4, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Sinha, V.R.; Singh, A.; Kumar, R.V.; Singh, S.; Kumria, R.; Bhinge, J.R. Oral colon-specific drug delivery of protein and peptide drugs. Crit. Rev. Ther. Drug Carr. Syst. 2007, 24, 63–92. [Google Scholar] [CrossRef]
- Patel, M.M. Getting into the colon: Approaches to target colorectal cancer. Expert Opin. Drug Deliv. 2014, 11, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Amidon, S.; Brown, J.E.; Dave, V.S. Colon-Targeted Oral Drug Delivery Systems: Design Trends and Approaches. AAPS PharmSciTech 2015, 16, 731–741. [Google Scholar] [CrossRef]
- Gazzaniga, A.; Maroni, A.; Sangalli, M.E.; Zema, L. Time-controlled oral delivery systems for colon targeting. Expert Opin. Drug Deliv. 2006, 3, 583–597. [Google Scholar] [CrossRef]
- Liu, L.; Yao, W.; Rao, Y.; Lu, X.; Gao, J. pH-Responsive carriers for oral drug delivery: Challenges and opportunities of current platforms. Drug Deliv. 2017, 24, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koev, T.T.; Harris, H.C.; Kiamehr, S.; Khimyak, Y.Z.; Warren, F.J. Starch hydrogels as targeted colonic drug delivery vehicles. Carbohydr. Polym. 2022, 289, 119413. [Google Scholar] [CrossRef] [PubMed]
- Musiał, W.; Kubis, A. Biodegradable polymers for colon-specific drug delivery. Polym. Med. 2005, 35, 51–61. [Google Scholar]
- Sinha, V.R.; Kumria, R. Colonic Drug Delivery: Prodrug Approach. Pharm. Res. 2001, 18, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.P.; Pope, D.J.; Gilbert, A.P.; Sacra, P.J.; Baron, J.H.; Lennard-Jones, J.E. Studies of two novel sulfasalazine analogs, ipsalazide and balsalazide. Dig. Dis. Sci. 1983, 28, 609–615. [Google Scholar] [CrossRef]
- Wadworth, A.N.; Fitton, A. Olsalazine: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in inflammatory bowel disease. Drugs 1991, 41, 647–664. [Google Scholar] [CrossRef]
- Wiggins, J.B.; Rajapakse, R. Balsalazide: A novel 5-aminosalicylate prodrug for the treatment of active ulcerative colitis. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1279–1284. [Google Scholar] [CrossRef]
- Ruiz, J.F.M.; Radics, G.; Windle, H.; Serra, H.O.; Simplício, A.L.; Kedziora, K.; Fallon, P.G.; Kelleher, D.P.; Gilmer, J.F. Design, Synthesis, and Pharmacological Effects of a Cyclization-Activated Steroid Prodrug for Colon Targeting in Inflammatory Bowel Disease. J. Med. Chem. 2009, 52, 3205–3211. [Google Scholar] [CrossRef]
- Ruiz, J.F.M.; Kedziora, K.; Keogh, B.; Maguire, J.; Reilly, M.; Windle, H.; Kelleher, D.P.; Gilmer, J.F. A double prodrug system for colon targeting of benzenesulfonamide COX-2 inhibitors. Bioorganic Med. Chem. Lett. 2011, 21, 6636–6640. [Google Scholar] [CrossRef]
- Ruiz, J.F.M.; Kedziora, K.; Pigott, M.; Keogh, B.; Windle, H.; Gavin, J.; Kelleher, D.P.; Gilmer, J.F. A nitrophenyl-based prodrug type for colorectal targeting of prednisolone, budesonide and celecoxib. Bioorganic Med. Chem. Lett. 2013, 23, 1693–1698. [Google Scholar] [CrossRef]
- Monastyrskyi, A.; Brockmeyer, F.; LaCrue, A.N.; Zhao, Y.; Maher, S.P.; Maignan, J.R.; Padin-Irizarry, V.; Sakhno, Y.I.; Parvatkar, P.T.; Asakawa, A.H.; et al. Aminoalkoxycarbonyloxymethyl Ether Prodrugs with a pH-Triggered Release Mechanism: A Case Study Improving the Solubility, Bioavailability, and Efficacy of Antimalarial 4(1H)-Quinolones with Single Dose Cures. J. Med. Chem. 2021, 64, 6581–6595. [Google Scholar] [CrossRef] [PubMed]
- Falkner, F.C.; Twomey, T.M.; Borger, A.P.; Garg, D.; Weidler, D.; Gerber, N.; Browder, I.W. Disposition of ampiroxicam, a prodrug of piroxicam, in man. Xenobiotica 1990, 20, 645–652. [Google Scholar] [CrossRef]
- Abinusawa, A.; Tenjarla, S.N. Release of 5-Aminosalicylic Acid (5-ASA) from Mesalamine Formulations at Various pH Levels. Adv. Ther. 2015, 32, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.F.; Pye, G.; Bramley, R.; Clark, A.G.; Dyson, T.J.; Hardcastle, J.D. Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 1988, 29, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Heck, H.; Casanovaschmitz, M.; Dodd, P.B.; Schachter, E.N.; Witek, T.J.; Tosun, T. Formaldehyde (CH2O) Concentrations in the Blood of Humans and Fischer-344 Rats Exposed to CH2O Under Controlled Conditions. Am. Ind. Hyg. Assoc. J. 1985, 46, 1–3. [Google Scholar] [CrossRef]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of pharmaceutical dosage forms through the small intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southwell, B.R.; Clarke, M.C.C.; Sutcliffe, J.; Hutson, J.M. Colonic transit studies: Normal values for adults and children with comparison of radiological and scintigraphic methods. Pediatr. Surg. Int. 2009, 25, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.R.; Rhee, P.-L. How to Interpret a Functional or Motility Test-Colon Transit Study. J. Neurogastroenterol. Motil. 2012, 18, 94–99. [Google Scholar] [CrossRef]
- Mineno, T.; Ueno, T.; Urano, Y.; Kojima, H.; Nagano, T. Creation of Superior Carboxyfluorescein Dyes by Blocking Donor-Excited Photoinduced Electron Transfer. Org. Lett. 2006, 8, 5963–5966. [Google Scholar] [CrossRef]
- Yamagishi, K.; Sawaki, K.; Murata, A.; Takeoka, S. A Cu-free clickable fluorescent probe for intracellular targeting of small biomolecules. Chem. Commun. 2015, 51, 7879–7882. [Google Scholar] [CrossRef]
- Gu, K.; Xu, Y.; Li, H.; Guo, Z.; Zhu, S.; Zhu, S.; Shi, P.; James, T.D.; Tian, H.; Zhu, W.-H. Real-Time Tracking and In Vivo Visualization of β-Galactosidase Activity in Colorectal Tumor with a Ratiometric Near-Infrared Fluorescent Probe. J. Am. Chem. Soc. 2016, 138, 5334–5340. [Google Scholar] [CrossRef] [PubMed]
- Saari, W.S.; Schwering, J.E.; Lyle, P.A.; Smith, S.J.; Engelhardt, E.L. Cyclization-activated prodrugs. Basic esters of 5-bromo-2’-deoxyuridine. J. Med. Chem. 1990, 33, 2590–2595. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, K.F.; Bundgaard, H. Cyclization-activated phenyl carbamate prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1993, 91, 39–49. [Google Scholar] [CrossRef]
- Matsumoto, H.; Sohma, Y.; Kimura, T.; Hayashi, Y.; Kiso, Y. Controlled drug release: New water-soluble prodrugs of an HIV protease inhibitor. Bioorganic Med. Chem. Lett. 2001, 11, 605–609. [Google Scholar] [CrossRef]
- Sharma, I.; Kaminski, G.A. Calculating pKavalues for substituted phenols and hydration energies for other compounds with the first-order fuzzy-border continuum solvation model. J. Comput. Chem. 2012, 33, 2388–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro, L.; Maisonneuve, S.; Retailleau, P.; Silvi, S.; Xie, J.; Métivier, R. Photophysical Properties of 4-Dicyanomethylene-2-methyl-6-(p-dimethylamino-styryl)-4H-pyran Revisited: Fluorescence versus Photoisomerization. Chem. A Eur. J. 2020, 26, 14341–14350. [Google Scholar] [CrossRef]
- Fredholt, K.; Mørk, N.; Begtrup, M. Hemiesters of aliphatic dicarboxylic acids as cyclization-activated prodrug forms for protecting phenols against first-pass metabolism. Int. J. Pharm. 1995, 123, 209–216. [Google Scholar] [CrossRef]
- Moharana, A.K.; Banerjee, M.; Sahoo, C.K.; Kanta, N. Development and validation of RP-HPLC method for mesalamine. Asian J. Pharm. Clin. Res. 2011, 4 (Suppl 2), 71–73. [Google Scholar]
- Sahoo, N.K.; Sahu, M.; Rao, P.S.; Ghosh, G. Validation of stability indicating RP-HPLC method for the estimation of mesalamine in bulk and tablet dosage form. Pharm. Methods 2013, 4, 56–61. [Google Scholar] [CrossRef]
- Jain, S.K.; Jain, N.; Khambete, H.; Rawal, A. Spectrophotometric method for simultaneous estimation of mesalamine and prednisolone in combined oral dosage form. Int. J. Pharm. Sci. Res. 2012, 3, 3707. [Google Scholar] [CrossRef]
- Moharana, A.K.; Banerjee, M.; Panda, S.S. Development and validation of UV spectrophotometric method for the deter-mination of mesalamine in bulk and tablet formulation. Int. J. Pharm. Pharm. Sci. 2011, 3, 19–21. [Google Scholar]
- Darak, V.; Karadi, A.B.; Arshad, M.; Ganure, A.L. Development and validation of HPLC method for determination of mesalamine in tablet dosage forms. Pharm. Sci. Monit. 2012, 3, 74–81. [Google Scholar]
- Kanubhai, T.R.; Mukesh, C.P.; Amit, R.K. Determination of Mesalamine related impurities from drug product by reversed phase validated UPLC method. E-J. Chem. 2011, 8, 131–148. [Google Scholar] [CrossRef]
- Nobilis, M.; Vybíralová, Z.; Sládková, K.; Lísa, M.; Holčapek, M.; Květina, J. High-performance liquid-chromatographic determination of 5-aminosalicylic acid and its metabolites in blood plasma. J. Chromatogr. A 2006, 1119, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Cornett, C.; Olsen, C.E.; Tjørnelund, J.; Hansen, S.H. Identification of major degradation products of 5-aminosalicylic acid formed in aqueous solutions and in pharmaceuticals. Int. J. Pharm. 1992, 88, 177–187. [Google Scholar] [CrossRef]
- Kobayashi, M.; Uneyama, K.; Hamada, N.; Kashino, S. Intramolecular cyclization of N1-(4-Oxo-2,5-cyclohexadien-1-ylidene)-N2-substituted-2,2,2-trifluoroethanimidamides (p-Benzoquinone imine derivatives): Syntheses of trifluoromethylated 6-hydroxybenzimidazoles and spiro dienone diazacarbocycles. J. Organ. Chem. 1995, 60, 6402–6407. [Google Scholar] [CrossRef]
- Rijk, M.C.M.; Van Schaik, A.; Van Tongeren, J.M. Disposition of 5-Aminosalicylic Acid by 5-Aminosalicylic Acid-Delivering Compounds. Scand. J. Gastroenterol. 1988, 23, 107–112. [Google Scholar] [CrossRef]
- O Nielsen, H.; Bondesen, S. Kinetics of 5-aminosalicylic acid after jejunal instillation in man. Br. J. Clin. Pharmacol. 1983, 16, 738–740. [Google Scholar] [CrossRef] [Green Version]
- Nani, R.R.; Gorka, A.P.; Nagaya, T.; Kobayashi, H.; Schnermann, M.J. Near-IR Light-Mediated Cleavage of Antibody-Drug Conjugates Using Cyanine Photocages. Angew. Chem. Int. Ed. 2015, 54, 13635–13638. [Google Scholar] [CrossRef]
- Schnermann, M.J.; Nani, R.R.; Gorka, A.P. Near-IR light-cleavable conjugates and conjugate precursors. WO/2018/031448, 2020. [Google Scholar]
- Manetsch, R.; Kyle, D.E.; Monastyrskyi, A.; LaCrue, A.N.; Maignan, J.R. Compounds and methods for their use in the treatment of malaria. WO2017127820A1, 2017. [Google Scholar]
- Taylor, S.J.; Kim, M.-J.; Nudel, K.; Briggs, T.F.; Yasuda, K.; Buckbinder, L.; Lanter, B.; Peck, S.C.; Snedeker, C.; She, A.; et al. Preparation of acylated active agents and methods of their use for the treatment of autoimmune disorders. WO2019236772, 2019. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | pH 5.0 | pH 6.5 | pH 7.5 |
|---|---|---|---|
| 13 | 6.3 ± 0.42 min | ND 2 | ND 2 |
| 14 | 3.5 ± 0.03 h | 8.1 ± 2.12 min | 1.7 ± 0.04 min |
| 15 | NR 1 | 13.2 ± 0.54 h | 2.4 ± 0.37 h |
| 16 | 20.6 ± 0.21 min | ND 2 | ND 2 |
| 17 | 16.4 ± 1.52 h | 28.0 ± 1.54 min | 6.8 ± 0.72 min |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd-Ellah, H.S.; Mudududdla, R.; Carter, G.P.; Baell, J.B. Design, Development, and Optimisation of Smart Linker Chemistry for Targeted Colonic Delivery—In Vitro Evaluation. Pharmaceutics 2023, 15, 303. https://doi.org/10.3390/pharmaceutics15010303
Abd-Ellah HS, Mudududdla R, Carter GP, Baell JB. Design, Development, and Optimisation of Smart Linker Chemistry for Targeted Colonic Delivery—In Vitro Evaluation. Pharmaceutics. 2023; 15(1):303. https://doi.org/10.3390/pharmaceutics15010303
Chicago/Turabian StyleAbd-Ellah, Heba S., Ramesh Mudududdla, Glen P. Carter, and Jonathan B. Baell. 2023. "Design, Development, and Optimisation of Smart Linker Chemistry for Targeted Colonic Delivery—In Vitro Evaluation" Pharmaceutics 15, no. 1: 303. https://doi.org/10.3390/pharmaceutics15010303
APA StyleAbd-Ellah, H. S., Mudududdla, R., Carter, G. P., & Baell, J. B. (2023). Design, Development, and Optimisation of Smart Linker Chemistry for Targeted Colonic Delivery—In Vitro Evaluation. Pharmaceutics, 15(1), 303. https://doi.org/10.3390/pharmaceutics15010303