3.1. Experimental and Theoretical Lipophilicity
The RP-TLC method was used to evaluate the experimental lipophilicity of compounds
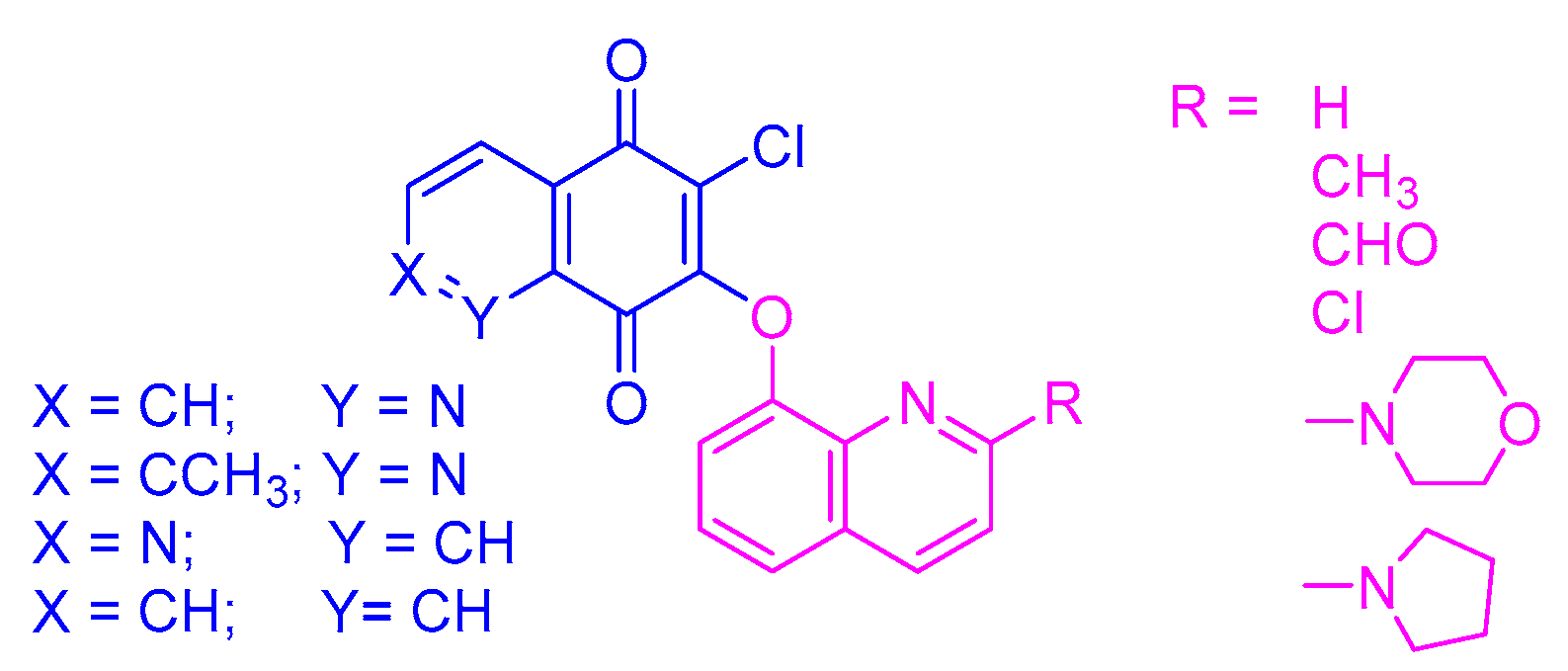
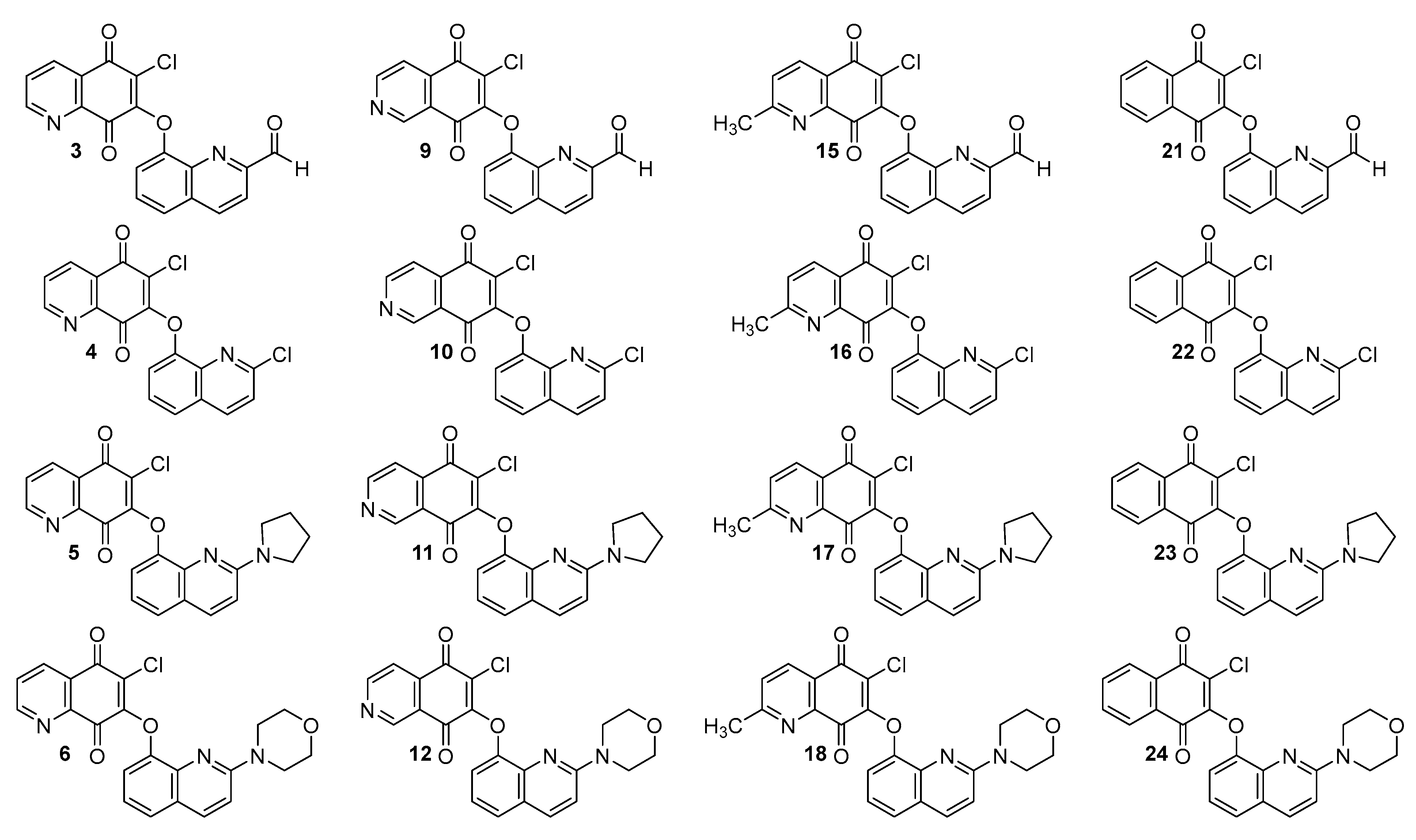
1–
24 (
Figure 2). The retardation parameter (R
f) was converted to the R
M parameter according to Equation (1):
The R
M parameter was calculated for every concentration of acetone and extrapolated to zero concentration of organic solvent in the mobile phase. The chromatographic parameter of lipophilicity (R
M0) was calculated using Equation (2):
where
C is the concentration of acetone in the mobile phase, while
b is the slope of the regression plot.
As seen in
Table 1, the correlation coefficient r covering the range of 0.968–0.999 shows a very good correlation between the concentration of acetone and the retardation factor (R
f).
In the next step, the relative lipophilicity parameter R
M0 was converted to the absolute lipophilicity parameter logP
TLC using the calibration curve. The obtained values of the R
M0 coefficient of the tested compounds were in the range of 1.51–4.51. The standard substances had to be selected in such a way that their literature values of log P
lit. were within a wider range than the range of tested compounds. As reference substances, benzamide (
A), acetanilide (
B), 4-bromoacetophenone (
C), benzophenone (
D), anthracene (
E), and dichlorodiphenyltrichloroethane (DDT) (
F) were used, for which the literature logP
lit values are in the range of 0.64–6.38 [
37,
38]. The R
M0 values for substances
A–
F were determined under the same conditions as for compounds
1–
24. The results are collated in
Table 2.
The calibration curve Equation (3) obtained by linear correlation between the literature value of logP
lit. and the experimental R
M0 parameter is as follows:
Equation (3) was used to obtain the logP
TLC parameter for all compounds
1–24 and the results are presented in
Table 3.
In general, the tested hybrids are characterized by rather low values of lipophilicity, varying in the range of 1.65–5.06. The highest values are seen for compounds
21–
24 (logP
TLC in the range 4.61–5.06) containing the 1,4-naphthoquinone moiety. Introduction of the nitrogen atom reduces the lipophilicity, while the changes in its position at the 5,8-quinolinedione moiety slightly affects the logP
TLC parameter. According to
Table 3, the trend of the values of logP
TLC is as follows: 5,8-quinolinedione (
1–
6) < 2-methyl-5,8-quinolinedione (
13–
18) < 5,8-isoquinolinedione (
7–
12). In the series of the 5,8-quinolinedione compounds (
1–
6), the lipophilicity depends on the type of substituent at the C-2 position of the quinone moiety with the order as follows: hydrogen atom (
1) < carbonyl group (
3) < methyl group (
2) < chloride atom (
4) < pyrrolidinyl ring (
5) < morpholinyl ring (
6). A similar correlation is observed for compounds with the 5,8-isoquinolinedione moiety (
7–
12). In the group of the 2-methyl-5,8-quinolinedione compounds (
13–
18), the lowest lipophilicity is observed for hybrid
14.
Lipophilicity correlates with hydrophobicity, which determines the solubility of the compound in water [
39,
40]. The hydrophobicity is described by the hydrophobicity index (φ
0), which can be calculated according to Equation (4).
If the value of the φ
0 index is in the range of 65.88–81.13, it means that the compounds show a moderate solubility in water (
Table 1). In the series of tested compounds, the hybrids with the 5,8-quinolinedione moiety (
1–
6) or 2-methyl-5,8-quinolinedione (
13–
18) possess a comparable solubility in water, varying in the range of 65.88–75.30. Compounds
1–
6 and
7–
12 differ in the position of the nitrogen atom on the 5,8-quinolinedione moiety. However, hybrids with the 5,8-quinolinedione moiety (
1–
6) show a lower value of φ
0 index than those with the 5,8-isoquinolinedione moiety (
7–
12), which means that the position of the nitrogen atom influences their solubility in water. The 1,4-naphthoquinone hybrids (
19–
24) possess the lowest water solubility.
The theoretical lipophilicity can be evaluated by the on-line available programs [
29,
30,
33]. The results of the theoretical approach are presented in
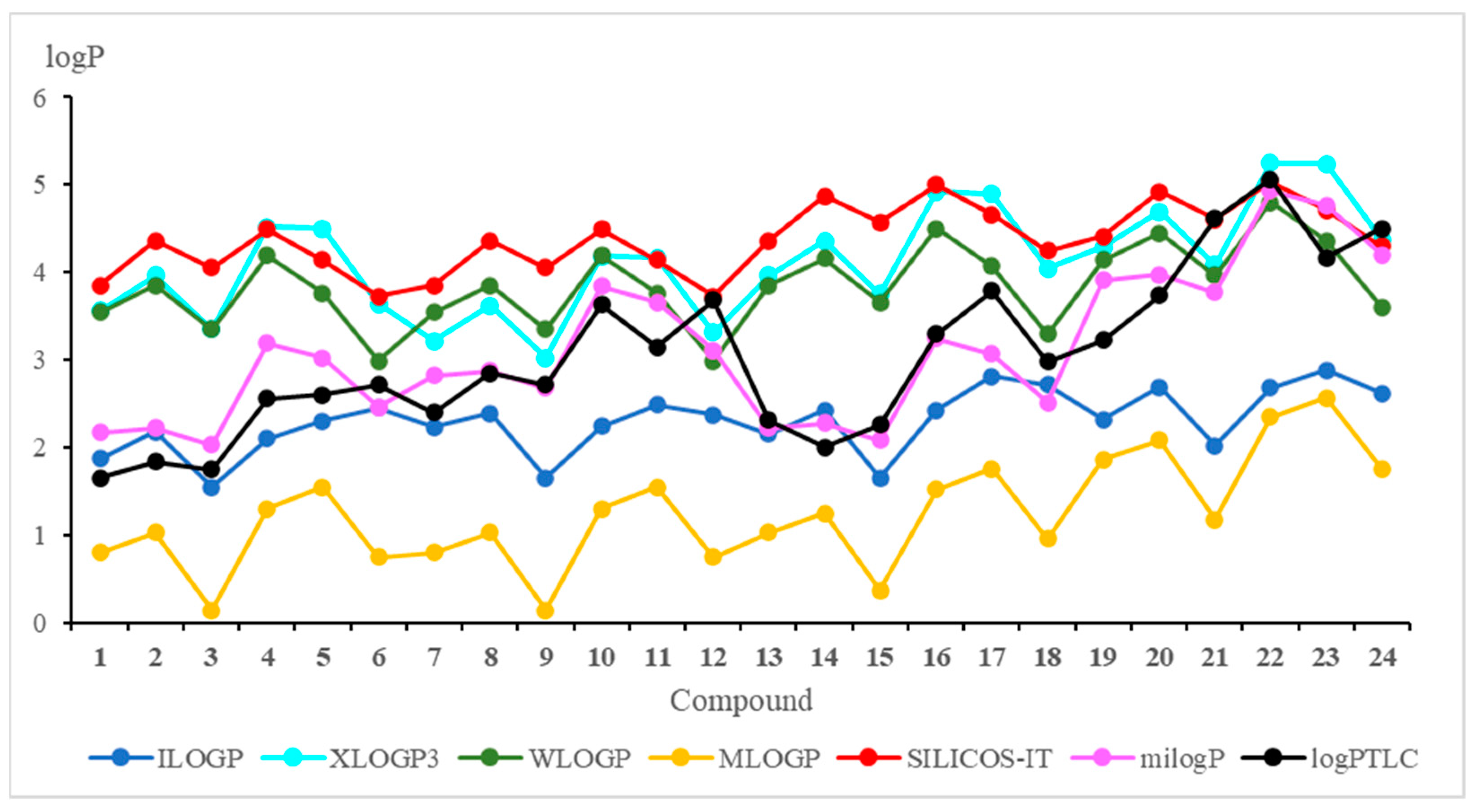
Figure 3 and
Table S1.
Figure 3 shows that the milogP program gives the logP values most similar to the experimental ones. Furthermore, it can be seen from the chemical structure of hybrids that logP depends on the substituent at the C-2 position of the quinoline moiety, and this relationship has the following order: morpholinyl ring (
6,
12,
18, and
24) > pyrrolidinyl ring (
5,
11,
17, and
23) > carbonyl group (
3,
9,
15, and
21) > hydrogen atom (
1,
7,
13, and
19) > methyl group (
2,
8,
14, and
20) > chloride atom (
4,
10,
16, and
22).
Comparison of the calculated logP values for compounds with the 5,8-quinolinedione (1–6) and 5,8-isoquinolinedione (7–12) moieties shows that the lipophilicity as determined by the WLOGP, MLOGP and SILICOS-IT programs has the same value for hybrids with the same quinoline moiety (1 and 7; 2 and 8; 3 and 9; 4 and 10; 5 and 11; 6 and 12) while the experimental lipophilicities (logPTLC) are different. It can be concluded that, for compounds containing the 5,8-quinolinedione moiety, these programs are not suitable for calculations of lipophilicity because they do not reproduce well the experimental values.
In
Table 4, the correlation equations between theoretical and experimental lipophilicity are presented. The highest correlation factor (r = 0.884) is observed for milogP program while the WLOGP program gives the worst correlation with the experiment (r = 0.417).
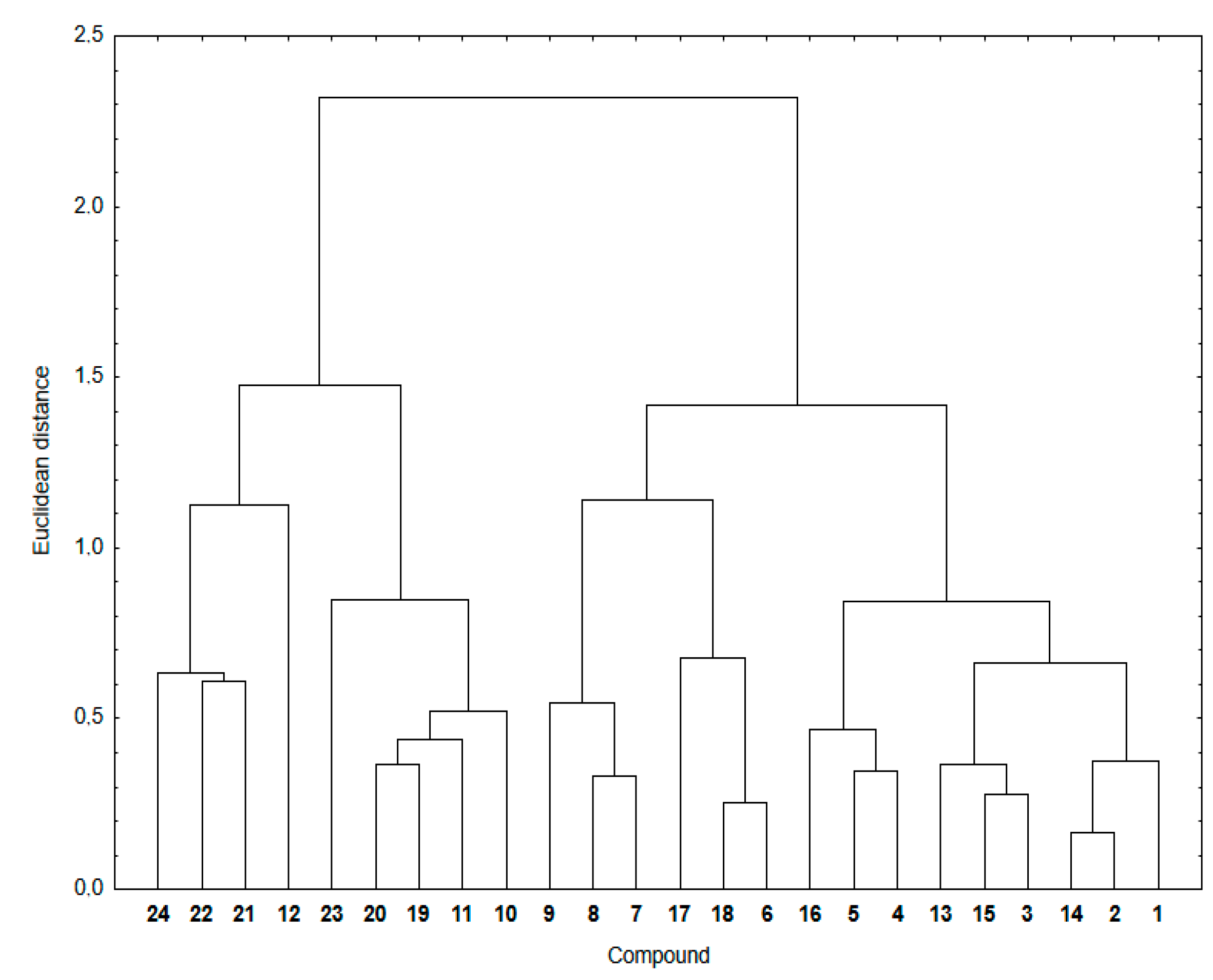
Figure 4 shows a dendrogram indicating the similarity relationship between experimental and calculated lipophilicity for compounds
1–
24. The theoretical lipophilicity data covers all used calculation methods.
As seen in
Figure 4, the hybrids
1–
24 are arranged in two main clusters. The first consists of the 1,4-naphthoquinone (
19–
24) and some 5,8-isoquinolinedione (
10–
12) hybrids. The second contain hybrids with the 5,8-quinolinedione (
1–
6 and
13–
18) and 5,8-isoquinolinedione (
7–
9) moieties.
The cluster presentation is based on the Euclidean distance (ED) values [
41,
42,
43]. The Euclidean distance is the distance in the Euclidean space of two objects whose similarity is examined by means of the similarity analysis. According to the principles of this analysis, the smaller the ED, the greater the similarity of two objects. Objects with a small ED from one another are located in the same region of the Euclidean space. To convert this distance metric to a similarity metric, we divided the object’s distance (ED) by the maximum distance in this set and then subtracted it from 1 to evaluate the similarity parameter between 0 and 1.
Table 5 presents the similarity parameters for experimental and calculated lipophilicity for compounds
1–
24.
According to the calculation method, hybrid 12 shows a similarity parameter equal to 0, which is the smallest possible. It can be seen that, for most compounds, the similarity parameter is not very high, varying in the range 0.67–0.85. Furthermore, for highly lipophilic hybrids (12, 21–24), the similarity parameters show the lowest values, covering the range of 0.00–0.46. Compounds with the same substituent at the C-2 position of the quinoline moiety have comparable ED distances, which means that the quinoline moiety affects the lipophilicity of hybrids.
3.2. ADMET Analysis
The lipophilicity is also related to other ADMET parameters such as molecular mass (MW), topological polar surface area (TPSA), number of rotatable bonds (RT), and number of acceptors (HA) and donors (HD) of the hydrogen bond. According to the rules of Lipinski and Veber, these parameters allow us to determine the bioavailability of the drug after oral administration [
5,
6,
11,
44].
As seen in
Table 6, the tested hybrids meet all Lipinski rules, meaning that the molecular mass is less than 500 g/mol, and the number of donors (HD) and acceptors (HA) of hydrogen bond are less than 5 and 10, respectively. Moreover, the experimental lipophilicity is less than 5 (
Table 4). The TPSA and RT of hybrids
1–
24 are in the range 56.26–86.22 and 2–3, respectively. According to Veber’s rule, these compounds should be well absorbed orally.
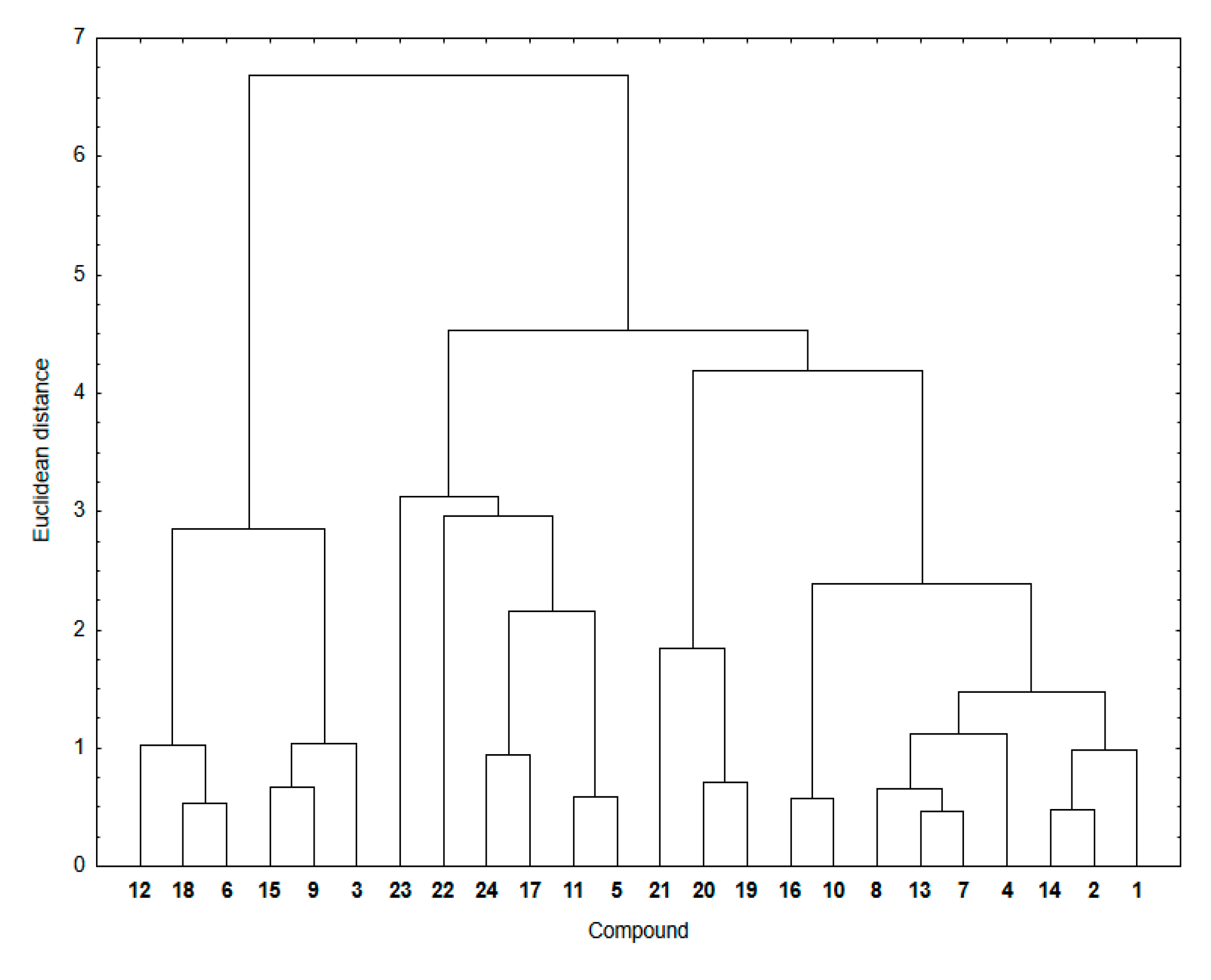
Similarity analysis was used to examine a relationship between the ADMET parameters mentioned above and experimental lipophilicity for hybrids
1–
24. In
Figure 5, the cluster analysis dendrogram showing similarities between these two sets of data is presented.
The dendrogram of the similarity analysis shows two main clusters (
Figure 5). The first includes the 5,8-quinolinedione hybrids with the morpholinyl ring (
6,
12 and
18) and carbonyl group (
3,
9 and
15) at the C-2 position of the quinoline ring. The second cluster is divided into three subclusters (
Figure 5). The first subcluster consists of compounds with the pyrrolidinyl ring (
5,
11,
17 and
23) at the C-2 position of the quinoline ring and compounds with the 1,4-naphthoquinone moiety (
22 and
24). The second includes the 1,4-naphthoquinone compounds (
19–
21). The third consists of compounds with the 5,8-quinolinedione (
1–
2,
4,
13–
14, and
16) and 5,8-isoquinolinedione (
7–
8, and
10) moiety. As before, the similarity parameters were calculated and collated in
Table 7.
It was found that, for most hybrids, the similarity parameters are high, ranging around 0.70–0.85. This means that there is a significant similarity between ADMET parameters—which can correlate with the descriptors of bioavailability—and lipophilicity of the hybrids. The exception are hybrids with high lipophilicity, for which the similarity parameters are very low, varying in the range 0.00–0.67.
In conclusion, it can be stated that, based on the similarity analysis, the relationship between ADMET parameters and experimental lipophilicity shows the lowest similarity for hybrids with the higher lipophilicity. Structural changes, such as varying the position of the nitrogen atom or substitution of the CH3 group, affect the lipophilicity of hybrids, and they also influence the similarity parameters in the similarity analysis. As the results so far have shown, lipophilicity can be determined experimentally or theoretically using appropriate computer programs.
The other method to determine lipophilicity (logP
calc) is the use of ADMET parameters (
Table 5). Using the Statistica program, the multilinear regression (MLR) Equation (5) has been determined, as shown below:
The lipophilicities calculated by this method for compounds
1–
24 are summarized in
Table S2. The absolute error varied in the range of 0.02–0.50. It can be noticed that there is good agreement between the lipophilicity determined in this way and the experimental one.
The bioavailability parameters influence the pharmacokinetic properties, which determine the absorption of the potential drug. Prediction of the oral and transdermal absorption was performed in silico using the Caco-2 permeability (logPapp), human intestinal absorption (HIA), and skin permeability (logKp) models. Moreover, the neurotoxicity of the compounds was designated by blood–brain barrier permeability (logBB) and central nervous system (logPS) penetration [
32,
44]. The pharmacokinetic parameters obtained in silico by the pkCSM software are presented in
Table 8.
Lipinski and Veber descriptors are associated with pharmacokinetic parameters such as the Caco-2 permeability (logPapp) and human intestinal absorption (HIA). The compound is well absorbed and transported across the intestinal mucosa if the logPapp and HIA value are higher than 0.9 and 30%, respectively [
32]. As seen in
Table 6, all hybrids could be well absorbed and transmitted by the intestinal mucosa. The Caco-2 permeability (logPapp) depends on the type of the 1,4-quinone moiety and the order is as follows: 5,8-quinolinedione (
1–
6) > 2-methylo-5,8-quinolinedione (
13–
18) > 5,8-isoquinolinedione (
7–
12) > 1,4-naphthoquinone (
19–
24). The HIA index depends slightly on the type of 1,4-quinone moiety. The tested hybrids show high skin permeability because the logKp values are lower than −2.5.
One of the most important properties of a potential drug is its neurotoxicity, which is characterized by the blood–brain barrier permeability (logBB) and central nervous system penetration (logPS). The logBB values for hybrids with the 5,8-quinolinedione (
1–
6 and
13–
18) and the 5,8-isoquinolinedione (
7–
12) moieties range from −0.671 to −1.009, which means that the compounds slowly pass through the blood–brain barrier [
32]. Moreover, the logPS for compounds
1–
18 varies from −2.045 to −2.966, which proves their poor penetration of the central nervous system [
32]. Replacing the nitrogen atom with a carbon atom (
19–
24) causes an increase in the logBB which allows the compound to penetrate the blood–brain barrier. Similar results were obtained for logPS. For these reasons, hybrids with the 1,4-naphtoquinone moiety can be neurotoxic.
3.3. Quantum Chemical Descriptors
Molecular parameters, such as energy of HOMO (E
HOMO) and LUMO (E
LUMO) orbitals allow us to determine the global reactivity descriptors, including the ionization potential (I), electron affinity (A), hardness (η), chemical potential (µ), electronegativity (χ) and electrophilicity index (ω) [
45,
46,
47]. These parameters can be useful for characterizing the ability of a tested compound to interact with the electrophilic and nucleophilic molecules. The energy of the HOMO and LUMO orbitals and the global descriptors are presented in
Table 9.
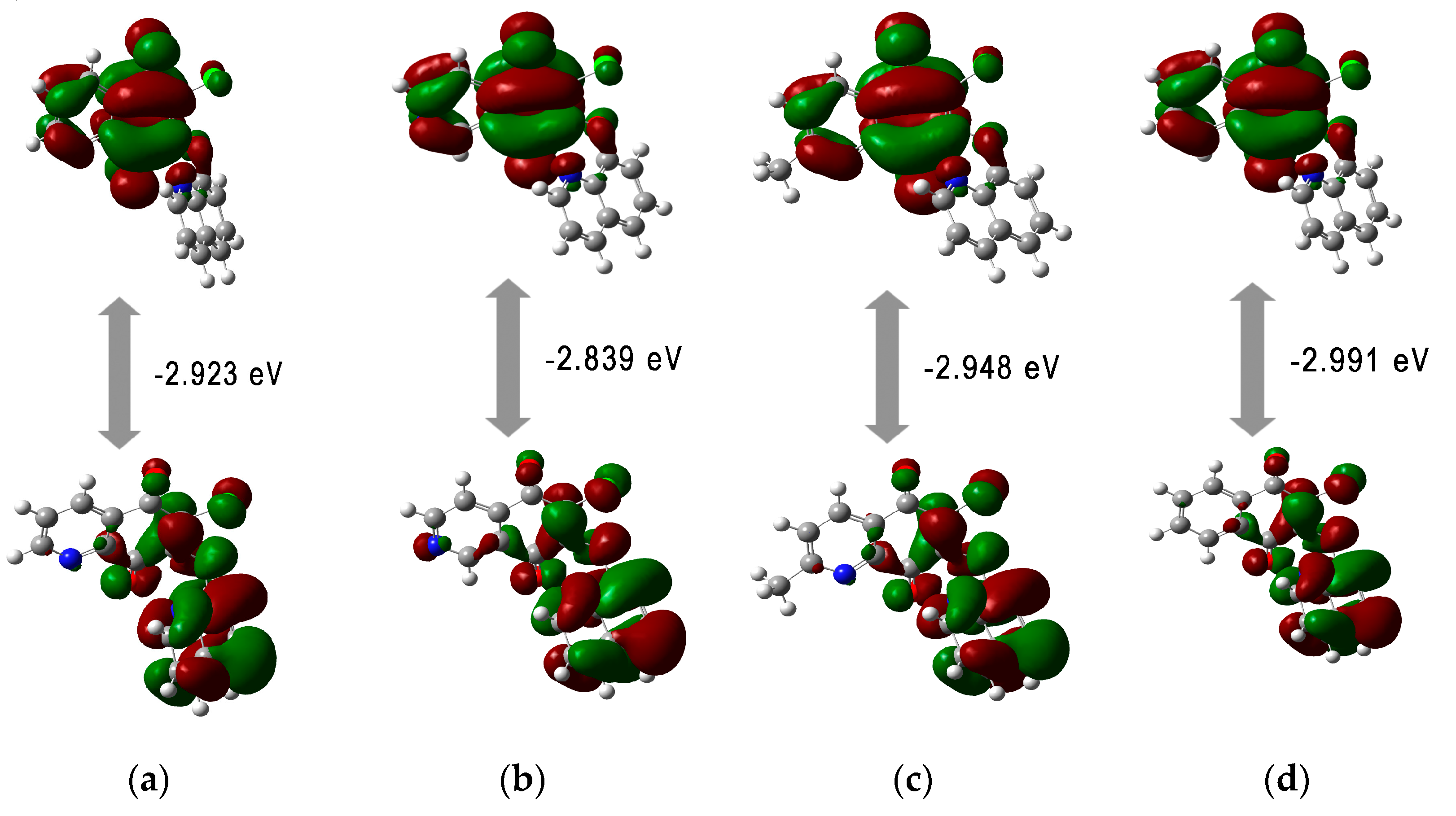
Upon analyzing the energy orbitals in relation to the molecular structure of hybrids, it can be seen that they depend on the type of the substituent at the C-2 position in the quinone moiety. Introduction of nucleophilic groups such as pyrrolidinyl (
5,
11,
17, and
23) and morpholinyl (
6,
12,
18, and
24) rings increases the energy of the HOMO orbital. However, the LUMO energy does not depend on the type of the 1,4-quinone scaffold. The HOMO orbitals are dispersed throughout the quinone scaffold and the carbonyl groups at the C-5 and C-8 positions of the 1,4-quinone moiety. The LUMO orbitals are localized at the 1,4-quinone moiety. The distribution of the HOMO and LUMO across the entire molecule indicates that the molecular system has good charge transfer capabilities (
Figure 6).
All tested compounds possess comparable HOMO-LUMO energy gaps (ΔE), indicating comparable chemical reactivity. The ΔE values range from −2.045 eV to −3.240 eV showing that the hybrids
1–
24 are characterized by high reactivity against biological targets [
48]. The calculated reactivity descriptors show that hybrids have high softness and flexibility in gaining electrons. The high softness value is useful because soft drugs interact easily with an enzyme target. Moreover, the soft drug can be better metabolized into non-toxic compounds [
49]. High value of electrophilicity index (ω) (7.733–11.502 eV) characterizes the tested molecules as strong electrophiles, according to the electrophilicity ranking of organic molecules [
47].
The multilinear regression (MLR) Equation (6) was used to determine the enzymatic conversion rate of NQO1 (logNQO1
calc) based on the quantum chemical properties, such as energy of LUMO (E
LUMO) orbital and electrophilicity index (ω).
In
Table S3, the enzymatic conversion rates of NQO1 (logNQO1
calc) for hybrids
1–
24 are collated. The absolute error varieds in the range of 0.01–0.30. The obtained results indicated that the quantum chemical descriptor could be used to determine the enzymatic conversion rate of NQO1.
3.4. Docking Study
According to our previous research, the tested hybrids
1–
24 induced the mitochondrial apoptotic pathway. Molecular mechanics studies showed that hybrids reduced the number of mRNA copies of gene encoding the BCL-2 protein [
22]. These results inspired the study of interactions between the ligand and the BCL-2 protein using the AutoDock Vina program [
35]. Venetoclax, an inhibitor of this protein, was used as the reference substance [
50].
As can be seen in
Table 10, the scoring values (ΔG) obtained for hybrids
1–
24 are lower than for venetoclax. It means that these compounds show a higher affinity for the BCL-2 protein than the reference substance. Comparing the scoring values across all compounds
1–
24 shows that the type of 1,4-quinone affects the affinity of the ligand for the active center of the protein, and the order is as follows: 2-methyl-5,8-quinolinedione (
13–
18) > 1,4-naphthoquinone (
19–
24) > 5,8-isoquinolinedione (
7–
12) > 5,8-quinolinedione (
1–
6). It wasw found that the type of substituent at the C-2 position of the quinoline moiety affects the score value. The lowest value of ΔG was obtained for compounds with the amine substituent (
5–
6,
11–
12,
17–
18, and
23–
24), while the highest was for compounds with the hydrogen atom (
1,
7,
13,
19) at the C-2 position of the quinone moiety (
Table 10).
Detailed analysis was performed for compounds with the 2-methyl-5,8-quinolinedione moiety (
13–
18). Its aim was to determine the influence of the type of quinoline substituent on the interaction with the BCL-2 protein. As can be seen in
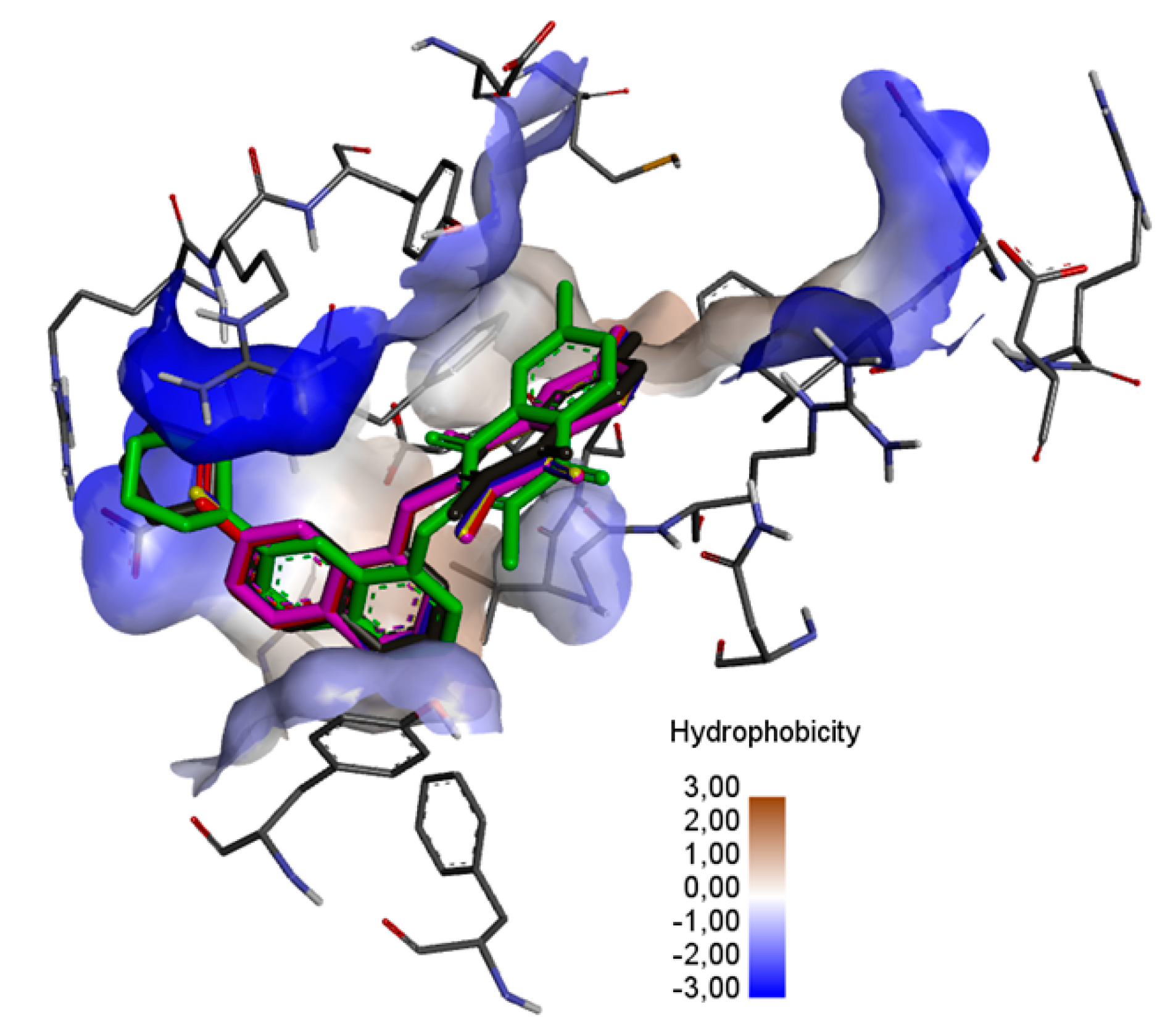
Figure 7, the ligands are localized deep within the hydrophobic matrix of the protein active center.
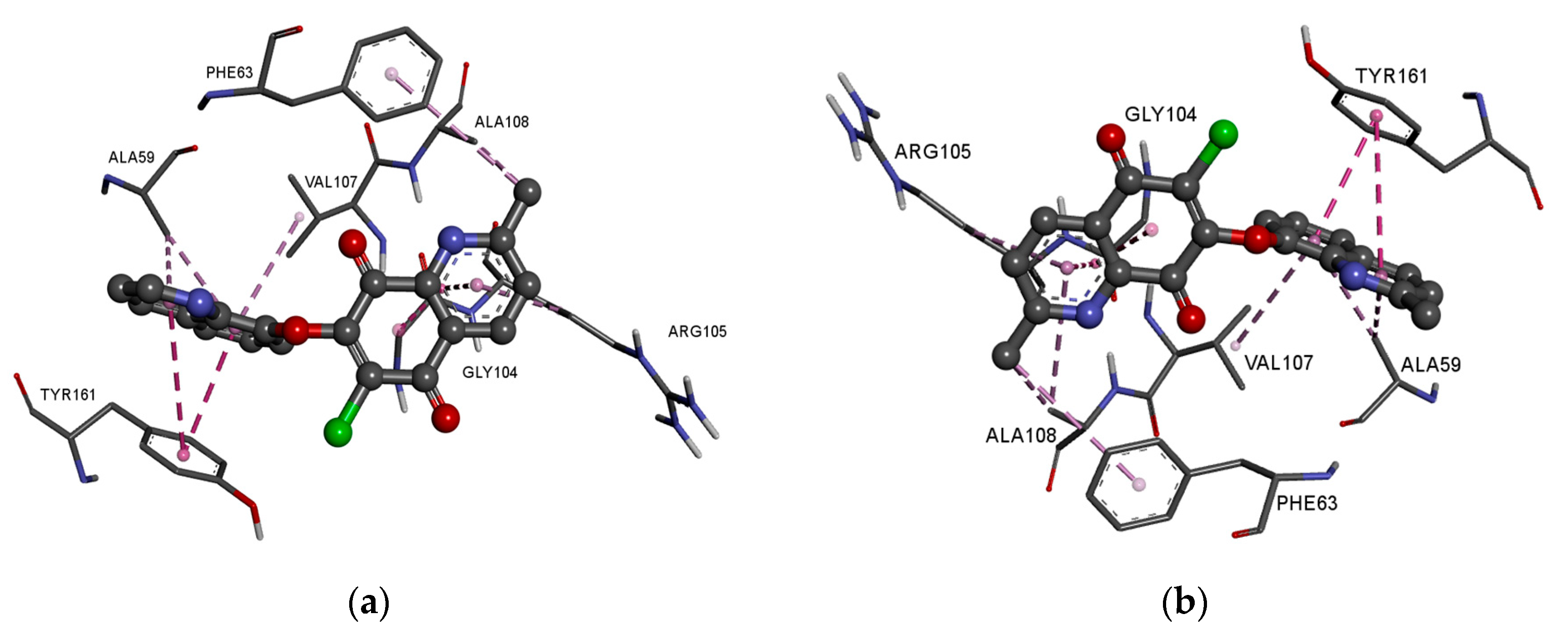
In the complex of ligands
13–
16 with the BCL2 protein, the 2-methyl-5,8-quinolinedione moiety creates the hydrophobic interaction with the glycine (GLY104), arginine (ARG105), alanine (ALA105) and phenylalanine (PHE63), while the quinoline substituent interacts with the alanine (ALA59), tyrosine (THY161) and valine (VAL107) (
Figure 8a–f,
Table S4).
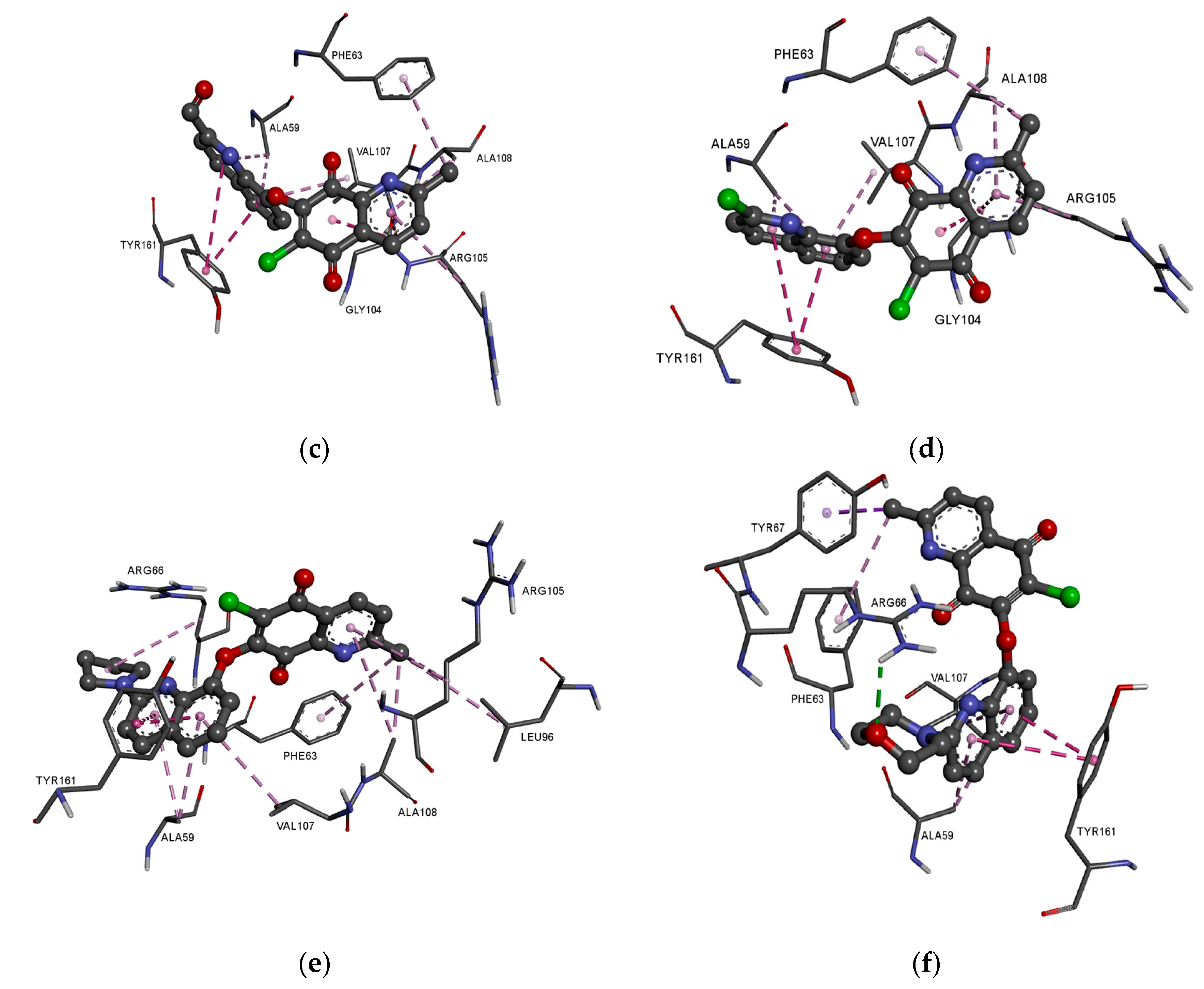
The presence of an additional amine ring (
17–
18) leads to a change in the arrangement of ligand in the active site of the protein (
Figure 8e,f). Comparing the arrangement of
13–
16 and
17 shows that the 2-methyl-5,8-quinolinedione and quinoline moiety create an additional hydrophobic interaction with leucine (LUE96) and arginine (ARG66), respectively (
Figure 8e,
Table S4). The arrangement of
18 in the active site of the protein is completely different from the others. In this case, the 2-methyl-5,8-quinolinedione interacts with phenylalanine (PHE63) and tyrosine (TYR67) via a hydrophobic interaction. The quinoline moiety interacts with alanine (ALA59), tyrosine (TYR161), and valine (VAL107). In contrast, the oxygen atom at the morpholine ring creates a hydrogen bond with arginine (ARG66) (
Figure 8f,
Table S4).
Comparing the arrangement of 2-methylo-5,8-quinolinedione (
13–
18) (
Figure 8) with Venetoclax (
Figure S2) shows that tested ligand interacts with similar amino acid residues in the active center of the BCL-2 protein. The hybrids
13–
18 and reference substance interact with phenylalanine (PHE63), tyrosine (TYR67) and glycine (GLY104).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}