1. Introduction
Roughly 500,000 cases of leukemia are diagnosed worldwide every year, with about 300,000 patients succumbing to the disease [
1]. Leukemia is a general group of blood cancers derived from malignant bone marrow cells. Historically, treatments for leukemia have not been able to truly cure the disease due to the cancer’s persistence in other systems within the body, including the lymphatic system and lymph nodes, to which cancer drugs may have limited access [
2]. Molecularly targeted small molecule and antibody-based drugs have been developed to eliminate malignant cells. However, their cellular and cancer-laden tissue distribution and retention can be limited due to high clearance and tissue barriers [
3]. Only a fraction of orally administered small molecule drugs are able to reach the leukemic cell target as significant percentages of such drugs are subject to metabolic or excretory elimination.
Chemotherapeutic agents, including chlorambucil (alkylating agent), fludarabine (purine analogue), and cyclophosphamide (alkylating agent), are effective treatments for leukemia. However, at therapeutic doses these chemotherapeutic agents may carry significant side effects that can limit their application, particularly in weaker or older patients [
4]. With the introduction of molecularly targeted agents for specific druggable proteins that are overexpressed in leukemia, an additional safety margin is added. Newer clinical treatments for B-cell leukemia targeted to molecular checkpoints can be divided into three groups of compounds that inhibit the uncontrolled growth of B cells: (1) Bcl-2, a mitochondrial antiapoptotic protein, (2) Bruton’s tyrosine kinase (BTK) inhibitors (TKI’s), (3) monoclonal antibodies, several of which are targeted to CD20, a surface antigen on B cells [
5]. As molecularly targeted agents, these three compounds reduce off-target toxicities compared to earlier drugs, making them a preferred treatment option for patients from a wide demographic range [
6]. Biologic drugs have been largely successful, though their inherent structure limits them to only binding a single target per drug molecule. Resistance events to molecularly targeted single agent treatments are well documented in chronic use. Therefore, combination regimens (with multiple drugs targeted to varying mechanisms of action) are often used to reduce the risk of single-drug resistance [
7]. Combination regimens may provide synergy derived from two or more drug substances that both inhibit multiple pathways and improve potency [
8]. While the newer molecularly targeted drugs are typically approved as monotherapies, combination therapies with these newer drugs are also being considered for treatment durability.
Zanubrutinib is a second-generation TKI of Bruton’s Tyrosine Kinase (BTK) that has been recently introduced and approved by the FDA for several B cell-based blood cancers, including mantle cell lymphoma (MCL). The BTK inhibitor zanubrutinib is currently administered daily in an oral dosage form (considered an attractive treatment for patients). Due to a short plasma half-life of 2–4 h, oral zanubrutinib is administered twice daily to maintain adequate plasma concentrations of the drug [
9]. As chronic twice-daily oral dosing may cause pill fatigue, patient compliance is often an issue due to the physically taxing nature of such chemotherapeutics [
10]. Zanubrutinib is currently approved for patients with refracted MCL or similar diseases only as a monotherapy treatment, but combination therapy regimens consisting partly of zanubrutinib have been explored in a prominent phase 3 clinical trial, the SEQUOIA study [
11]. This study, currently in progress, is focused on chronic lymphocytic leukemia (CLL) treatment; it has so far reported improved progression-free survival in patients receiving zanubrutinib monotherapy compared to bendamustine-rituximab, a commonly used treatment targeting CD20-positive cells. An additional arm of the study is exploring patient tolerance of zanubrutinib in conjunction with venetoclax, a small-molecule inhibitor of Bcl-2; results have been positive, with 50/51 patients responding to treatment, but the study is ongoing [
12]. Studies have also indicated that the zanubrutinib-venetoclax combination can be used on leukemias beyond CLL.
As previously noted, even if oral venetoclax and zanubrutinib can be administered together, the asynchronized peak and time course of the two drugs will not provide consistent, sustained intracellular levels to maximally suppress leukemic cell growth. The percentage of oral drugs absorbed into blood from the gut mucosa is typically lower than that of intravenous injections. Drug metabolic enzymes found in the gut and liver may also reduce the percentage of active drug in the blood, leading to limited drug bioavailability, and, in some cases, sub-therapeutic plasma and intracellular dug levels. As a result, these events may lead to an increased risk of inducing drug resistant cells in tumor sites [
13]. In addition, daily (or more frequent) dosing, typically necessary for oral dosage form, can be cumbersome for patients, as high local concentration in the gut after oral dosing may lead to gastrointestinal injury. Additionally, zanubrutinib is typically administered twice daily, which over time can lead to pill fatigue in patients, further limiting the treatment due to missed doses. To address these limitations, we have evaluated the feasibility of a drug delivery system in which lower but sustained therapeutic levels persist in the blood for an extended period of time through the development of a combination of drugs that are targeted to multiple proteins in the leukemic cells. Drug combinations composed of molecularly targeted drug substances could greatly improve both the potency and patient tolerance of the combination drug product.
We have previously demonstrated that DcNP can stabilize combinations of hydrophobic (lopinavir and ritonavir, LogP = 5.9 and 6, respectively) and hydrophilic (tenofovir and emtricitabine, LogP = −1.6 and −0.6, respectively) HIV drugs with amphipathic lipid excipients [
14]. When given subcutaneously to nonhuman primates, DcNP both extends the plasma time course (long-acting behavior) of all three HIV drugs and leads to higher drug levels in lymphocytes than in plasma (demonstrating preferential cell targeting effects) [
15].
In this study, we evaluated whether a venetoclax and zanubrutinib drug combination could be assembled into a similar DcNP lipid nanoparticle to provide both long-acting plasma exposure and adequate drug concentrations; in addition, we evaluated if enhanced, synchronized cell uptake could be achieved. We have found that a nanoparticle formulation can greatly extend the half-lives of certain drugs when administered subcutaneously in mice compared to equivalent free drugs or DcNPs administered intravenously. As their protein targets are expressed across multiple forms of leukemia, both venetoclax and zanubrutinib have shown efficacy against different types of leukemia in phase III clinical trials [
11,
12,
13]. Thus, a DcNP approach for the venetoclax-zanubrutinib combination manifests broad treatment potential for several different types of leukemia beyond CLL.
2. Materials and Methods
2.1. Reagents
N-(carbonylmethoxypolyethyleneglycol-2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine, sodium salt (DSPE-mPEG2000), and 1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) (GMP grade) were purchased from Corden Pharma (Liestal, Switzerland). The two drug substances, zanubrutinib (BGB 3111) and venetoclax (ABT 199) were supplied by MedChemExpress (Monmouth Junction, NJ, USA). All other chemicals and solutions were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted.
2.2. Preparation and Characterization of Drug Combination Nanoparticles
To prepare the drug combination containing venetoclax and zanubrutinib, 9.6 mg venetoclax and 9.6 mg zanubrutinib, plus 33.6 mg DSPE-mPEG2000 and 85.4 mg DSPC, were dissolved together in 1 mL organic solvent in a glass tube. For the first production attempt, the chemical components were dissolved in an organic solution of ethanol with 5% ammonia, and were then subjected to rotary evaporation, followed by reconstitution in 0.9% NaCl and 20 mM NaHCO3 buffer using a stir bar. An Avanti Polar Lipids sonicator (Avanti, Alabaster, AL, USA) was used to reduce particle size after reconstitution in aqueous solvent. Sonication was performed at 40–45 °C for 5 min, followed by 5 min rest, followed by a final 5 min of sonication. The suspended drug combination was stabilized by lipid excipients and referred to as the drug combination nanoparticle, or VZ-DcNP. This suspension was diluted to defined concentrations with buffer solution. The second production method used tert-butyl alcohol (TBA) as an organic solvent, which was then removed via rotary evaporation and an additional 4 h of lyophilization to remove residual TBA solvent. The diameter of particles in suspension was estimated with a NICOMP 380 ZLS (NICOMP, Chicago, IL, USA). The final production method, consisting of solvent rotary evaporation, lyophilization, and sonication, was selected for assessment both in vitro and in vivo.
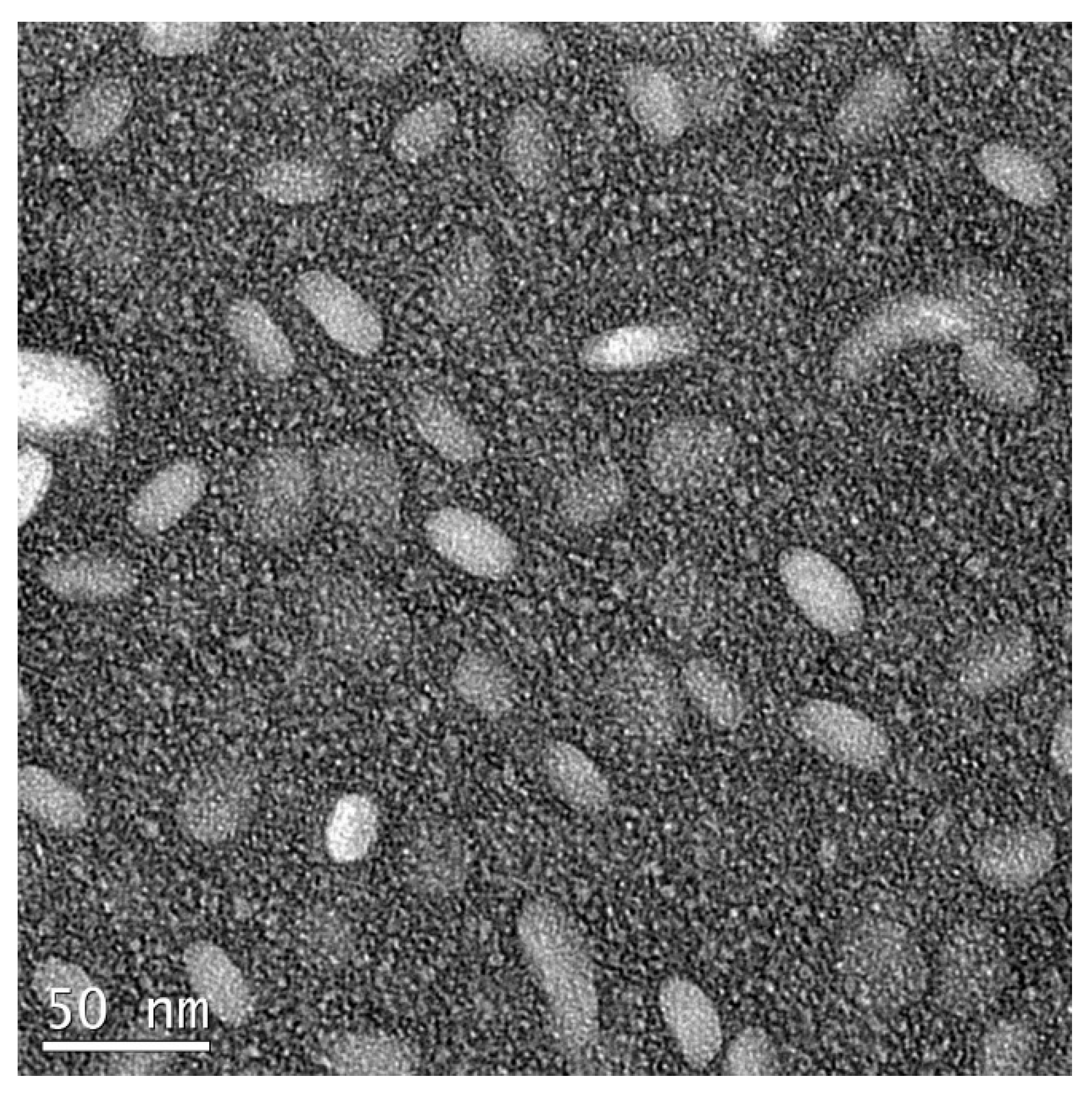
To estimate the percentage of venetoclax and zanubrutinib association to DcNP, the VZ-DcNP in suspension was first dialyzed (6–8 kDa molecular weight cutoff) in buffer under sink conditions. The sink conditions were generated by dialyzing 200 µL of VZ-DcNP suspension in 200 mL buffer solution (1000-fold volume change) for 4 h. The VZ-DcNP drug association efficiency (AE%) was determined by comparing the pre- and post-dialysis drug concentration ratios of each drug (V and Z). To determine VZ either in the dialysate or retentate, the samples were first extracted with organic solvent and analyzed with an LC-MS/MS assay as described below. Nanoparticles were also visually assessed for consistency using transmission electron microscopy with negative staining; sample suspensions containing the VZ-DcNPs were placed onto a TEM grid (copper, 300-mesh, coated with carbon and Formvar film), allowed to settle for 5 min, and then stained with 5% uranyl acetate as a negative stain. A Tecnai G2 F20 electron microscope (FEI, Hillsboro, OR, USA) was used at 200 kV.
2.3. Drug Extraction from VZ-DcNPs and LC-MS/MS Analysis
An extraction protocol was established to quantify concentrations of venetoclax and zanubrutinib in both nanoparticle-bound and free forms. Briefly, the drugs were solubilized by diluting the sample with ethyl acetate, which extracted them from either the DcNP complex, mouse plasma, or both. Following centrifugation, the supernatants were dried with nitrogen gas and then reconstituted in acetonitrile. Extracted drug solutions were then loaded onto a Shimadzu HPLC system coupled to a 3200 QTRAP mass spectrometer (Applied Biosystems, Grand Island, NY, USA). The HPLC system consisted of two Shimadzu LC-20A pumps, a DGU-20A5 degasser, and a Shimadzu SIL-20 AC HT autosampler. A Synergi Polar-RP column (100 × 2.0 mm) with a C8 guard column (4.0 × 2.0 mm) (Phenomenex, Torrance, CA, USA) was used for separations. Mobile phase A used water with 20 mM ammonium acetate and B used acetonitrile. The separations were done at room temperature with a flow rate of 0.55 mL/min. The mass spectrometer was equipped with an electrospray ionization (ESI) TurboIonSpray source, and the system was operated using Analyst software, version 1.5.2 (ABSciex, Framingham, MA, USA). Drug concentrations in various samples were calculated with standard curves prepared from normal mouse plasma containing known drug concentrations.
2.4. Drug Potency against Cancer Cells
K-562, a human chronic myelogenous leukemia cell line, was purchased from ATCC (Manassas, VA, USA). Human leukemic cell line MOLT-4 (of acute lymphoblastic leukemia origin), and acute promyelocytic leukemia HL-60 cells were obtained from Carrie Cummings at Fred Hutchinson Cancer Research Center (Seattle, WA, USA). They were grown in RPMI medium 1640, which contained 1% 100× Antibiotic-Antimycotic (Thermo Fisher Scientific, Waltham, MA, USA) and 10% fetal bovine serum. These were selected for evaluation due to their different degree of drug target expression, specifically Bruton’s tyrosine kinase (BTK) and B-cell lymphoma 2 (Bcl-2). HL-60 cells express both BTK [
16] and Bcl-2 [
17], while K-562 [
18,
19] cells only express BTK and MOLT-4 [
20,
21] cells express only Bcl-2.
Each cell line was first allowed to grow in black 96-well assay plates (Corning, NY, USA). After 1 h, 200 µL of varying concentrations of each drug (venetoclax or zanubrutinib), a combination of both free drugs (w/w 1:1), the same combination in DCNPs (VZ-DCNPs), or no drug (medium control) were added in RPMI medium to the cells. On day 5, drug treatment effects were estimated using an AlamarBlue Cell Viability Assay (Thermo Fisher Scientific, Waltham, MA, USA). The viable cells that produced positive fluorescence signals were quantified with a PerkinElmer 1420 Multilabel Counter plate reader. AlamarBlue was diluted 10-fold with cell media; the existing cell culture media in the plates was replaced with the 10% AlamarBlue media and allowed to incubate for 4 h. The cell culture media was then assessed for fluorescence, (λex = 570 nm, λem = 585 nm). Employing GraphPad software (Version 7.05) and using an Emax model, the mid-point of the inhibitory concentration curve (or IC50) was determined.
2.5. Effect of DcNP on Leukemic Cell Drug Uptake and Retention
One million HL-60 cells were aliquoted into several 1.5 mL Eppendorf tubes. To evaluate the effects of DcNP on VZ, we made a free-drug solution of venetoclax and zanubrutinib (1:1 w/w). A VZ-DcNP solution of identical drug concentrations was made and also added to each tube. Following drug exposure in a CO2 incubator, cells were removed at preselected timepoints (15 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, and 4 h), and were then washed twice with media to remove external drug and VZ-DCNPs. Cells were then lysed with acetonitrile. Drugs in the cells were quantified according to the aforementioned extraction protocol and LC-MS/MS methods.
2.6. Pharmacokinetic Analysis of VZ-DcNP versus Free Drugs
All animal studies were performed under a protocol approved by the University of Washington Institutional Animal Care and Use Committee. Female BALB/c mice were purchased from Charles River Laboratories (Wilmington, MA, USA). They were housed in a pathogen-free facility until use with a 12 h light/dark cycle. Three groups of three mice each were tested as follows: (group 1) an IV dose containing 30 mg/kg venetoclax and 30 mg/kg zanubrutinib in 0.9% NaCl, 20 mM NaHCO3 buffer with 5% DMSO and 5% Cremophor EL as solubilizing agents; (group 2) an intravenous dosing of venetoclax and zanubrutinib DCNPs equivalent in volume and drug molar concentration to that received by group 1; and (group 3) a subcutaneous injection of venetoclax and zanubrutinib DCNPs in the inner thigh of the right back leg. Plasma samples were collected at 5 min, 1 h, 3.5 h, 24 h, 48 h, 72 h, and 1 week through retro-orbital bleeding. The drugs in plasma samples were extracted and analyzed with an HPLC-MS/MS.
4. Discussion
Taking advantage of our ability to co-formulate venetoclax and zanubrutinib in a drug combination nanoparticle (DcNP), we have characterized the VZ-DcNP as stable with a high degree of drug association to DcNP, indicating that further purification is not necessary (
Table 2). In addition, the resulting VZ combination in DcNP is biologically active and shown to enhance the overall potency of VZ in fixed-dose combination at a 1:1 mole ratio (
Table 3). The overall enhanced potency against HL-60 leukemic cells appeared to enhance cell uptake by 42- and 5-fold for V and Z, respectively. We found that VZ-DCNPs are stable, scalable, and biocompatible, as the lipid excipients, DSPC and DPSE-mPEG
2000, provide a structural base to support the drug combination in nanoparticles appropriate for patient administration. These VZ-DcNPs are shown to extend plasma time-course and enhance the overall drug exposure of both venetoclax and zanubrutinib.
Unlike carriers that require drug encapsulation, it is noteworthy that both venetoclax and zanubrutinib were nearly completely associated to the DcNP lipid base structure in the VZ-DcNP. Thus, there is little or no drug loss during their synthesis, meaning that a final step to eliminate residual free drug is no longer necessary. In fact, our data indicate that the degree of drug association of both drugs to the VZ-DCNPs is 98% or more. Having a high degree of stable drug association may both reduce drug wastage in VZ-DcNP preparation and potentially minimize the risk of contamination while making the injectable VZ-DcNP product. In addition, we found that a lyophilized dosage form of VZ-DcNP could be produced, and that upon resuspension the product exhibited a mean diameter of ~30–40 nm, suitable for both intravenous and subcutaneous dosing. Coupled together, the high degree of association and the option of storing the drug product in lyophilized form (for producing a suspension on site) make VZ-DcNPs realistic for human testing.
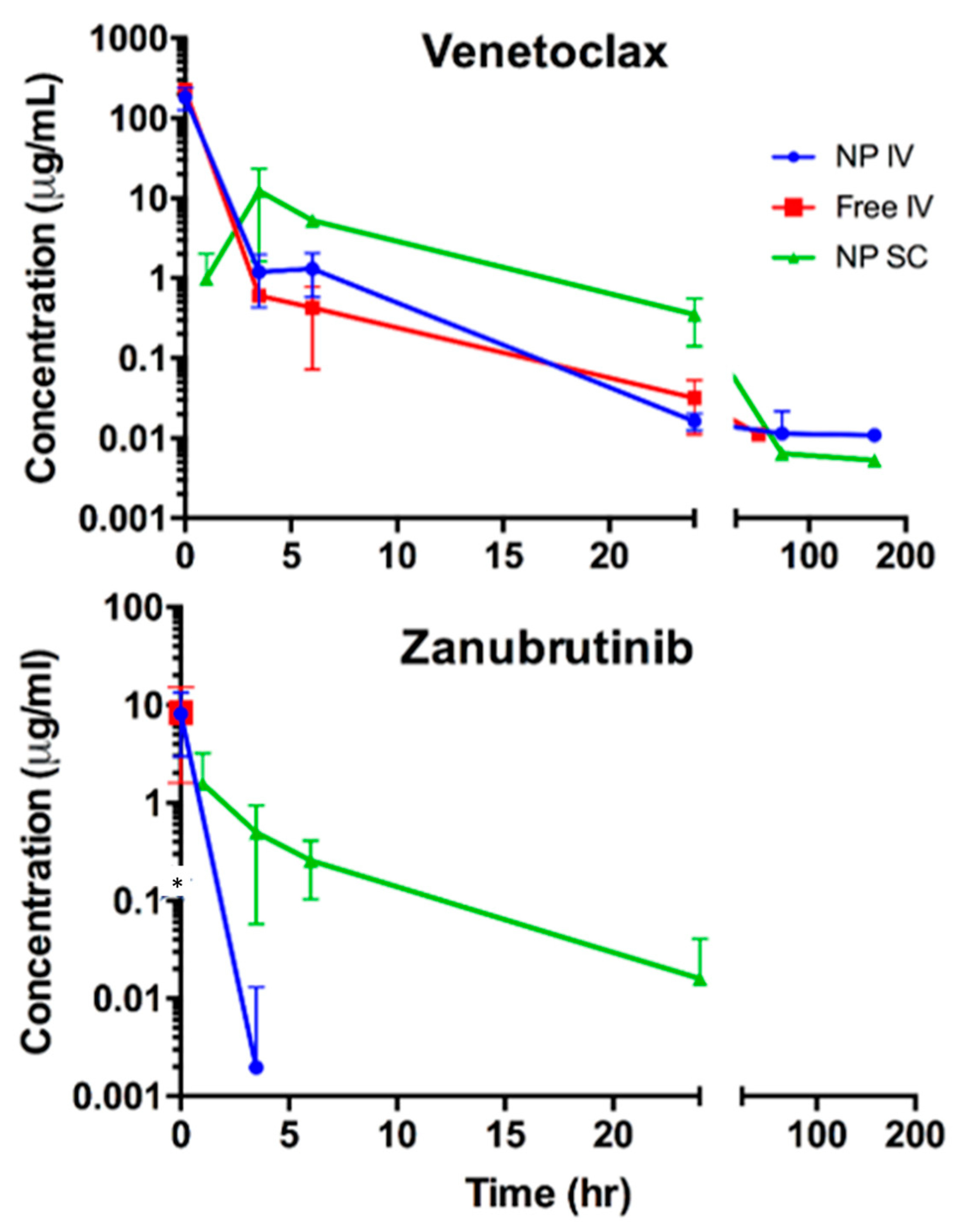
While the exact mechanisms responsible for the enhanced potency of VZ-DcNP compared to that of its free VZ counterpart are still unclear, it is likely that the small nanoparticles promote leukemic cell uptake and retention. The DcNP-associated drugs are taken up faster and are maintained at higher concentrations compared to equivalent soluble (free) VZ (
Figure 2). The enhanced uptake of VZ in DcNP parallels the improved potency of VZ in HL-60 leukemic cells expressing both VZ targets (Bcl-2 and BTK). This potency was also seen in K562 cells (BTK-expressing cell line) and MOLT-4 cells (Bcl-2-expressing cell line), indicating that both drugs were present and active. We found a roughly 82-fold enhanced potency for VZ-DcNP compared to the free formulation, and a 42- and 5-fold enhancement in cellular uptake for V and Z, respectively. Once again, the mechanisms leading to the disparity in the changes in cellular uptake are not clear, and they will be a subject of our future investigations. Regardless of the exact mechanisms, however, it is clear that both V and Z in DcNP are localized in the cells and are biologically active, leading to enhanced leukemic cell-growth suppression.
When delivered subcutaneously to BALB/c mice, VZ-DcNP was able to extend the presence of both drugs in plasma over an extended period of time, resulting in a significant extension of drug half-lives compared to those of free drugs given intravenously; a 42-fold increase for venetoclax and a 5-fold increase for zanubrutinib were observed. Intravenous VZ-DCNPs did not significantly alter the two drugs’ pharmacokinetics. VZ-DCNPs administered subcutaneously can greatly extend the plasma half-lives of associated drugs, demonstrating their ability to safely administer drugs over a longer period from a single injection, compared to the oral dosage forms that require more frequent dosing to achieve the same effect.
Currently, oral zanubrutinib (taken twice daily) and venetoclax (taken once daily) are in a phase 2 clinical study as a potential combination therapy for treating MCL (mantle cell lymphoma) and CLL (chronic lymphocytic leukemia) (NCT 05168930). Producing a long-acting and effective fixed-dose combination therapy intended to improve leukemic cell uptake and extend the time between doses may improve both patient uptake and acceptance.
New administration strategies for long-acting delivery of drugs that can overcome these limitations have been introduced. Long-acting cabotegravir with long-acting rilpivirine, explored through the CUSTOMIZE Hybrid III implementation-effectiveness study, is a novel formulation strategy that has been successfully implemented in HIV patient treatment, as the formulation can overcome the common problems with long-term drug treatment, namely patient adherence to the drug and maintaining adequate plasma drug levels. The VZ-DCNPs reported here can overcome these limitations imposed by daily oral dosing via subcutaneous administration of the nanoparticles: association with the biocompatible lipids safely retains the drugs in the subcutaneous space, protecting them from gastrointestinal and plasma metabolism while also slowly releasing the drugs over time into the plasma either through direct extravasation or through lymphatic uptake. These routes are likely responsible for the observed extended half-lives of the drugs, though more research is needed.
In leukemic cells, a fixed-dose combination of venetoclax and zanubrutinib exhibited good potency, with IC50′s in the low nanogram per milliliter range for HL-60 cells, which have high expression levels of both drugs’ targets. When formulated as a free drug with the same fixed-dose combination as VZ-DCNPs, the IC50 value for HL-60 cells was enhanced by about 1000-fold. The improvement in potency of the DCNPs over the free-drug combination is likely due to enhanced uptake and retention of the DcNP-bound drug as compared to the free drug. The pharmacokinetic study results indicate that in mice, subcutaneously administered VZ-DcNP was more favorable than both intravenous VZ-DcNP and intravenous free drug. Both drugs were detectable for a longer period in the plasma of subcutaneous VZ-DcNP-treated mice those that received free drug or DcNP-associated drug through an intravenous injection.







{kind=link}
{kind=link}
{kind=link}