

The Mechanism of Action of SAAP-148 Antimicrobial Peptide as Studied with NMR and Molecular Dynamics Simulations

 , and
, and 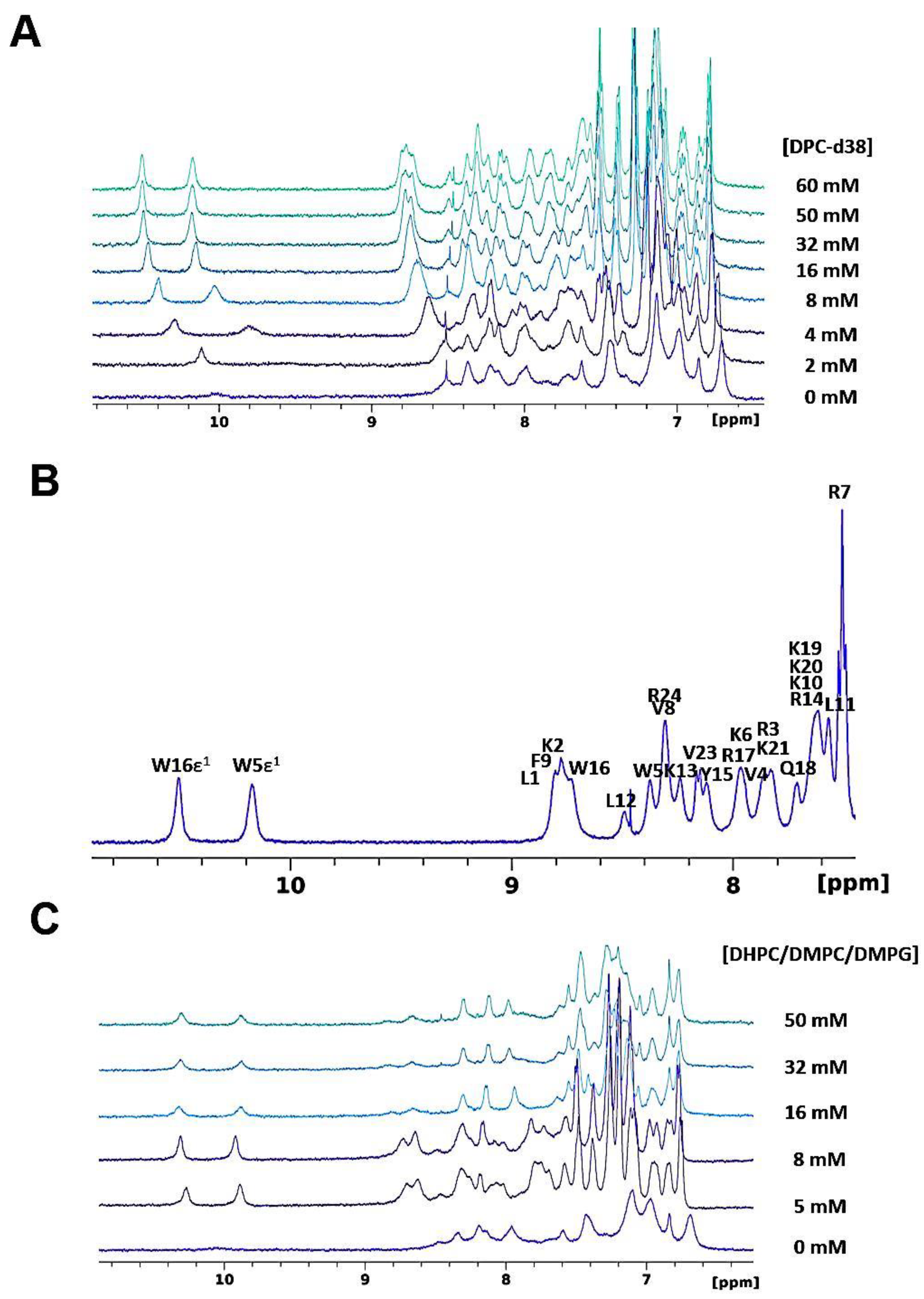{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptide Synthesis
2.2. Liquid-State NMR Spectroscopy and Sample Preparation
2.2.1. Titration of SAAP-148 with DPC Micelles
2.2.2. Paramagnetic NMR Experiment
2.2.3. Titration of SAAP-148 with Bicelles
2.2.4. NMR Acquisition and Processing
2.3. Solid-State NMR Spectroscopy and Sample Preparation
2.4. Circular Dichroism
2.5. Molecular Dynamics Simulations
3. Results and Discussion
3.1. Liquid-State NMR studies of SAAP-148 Interacting with Model Membranes
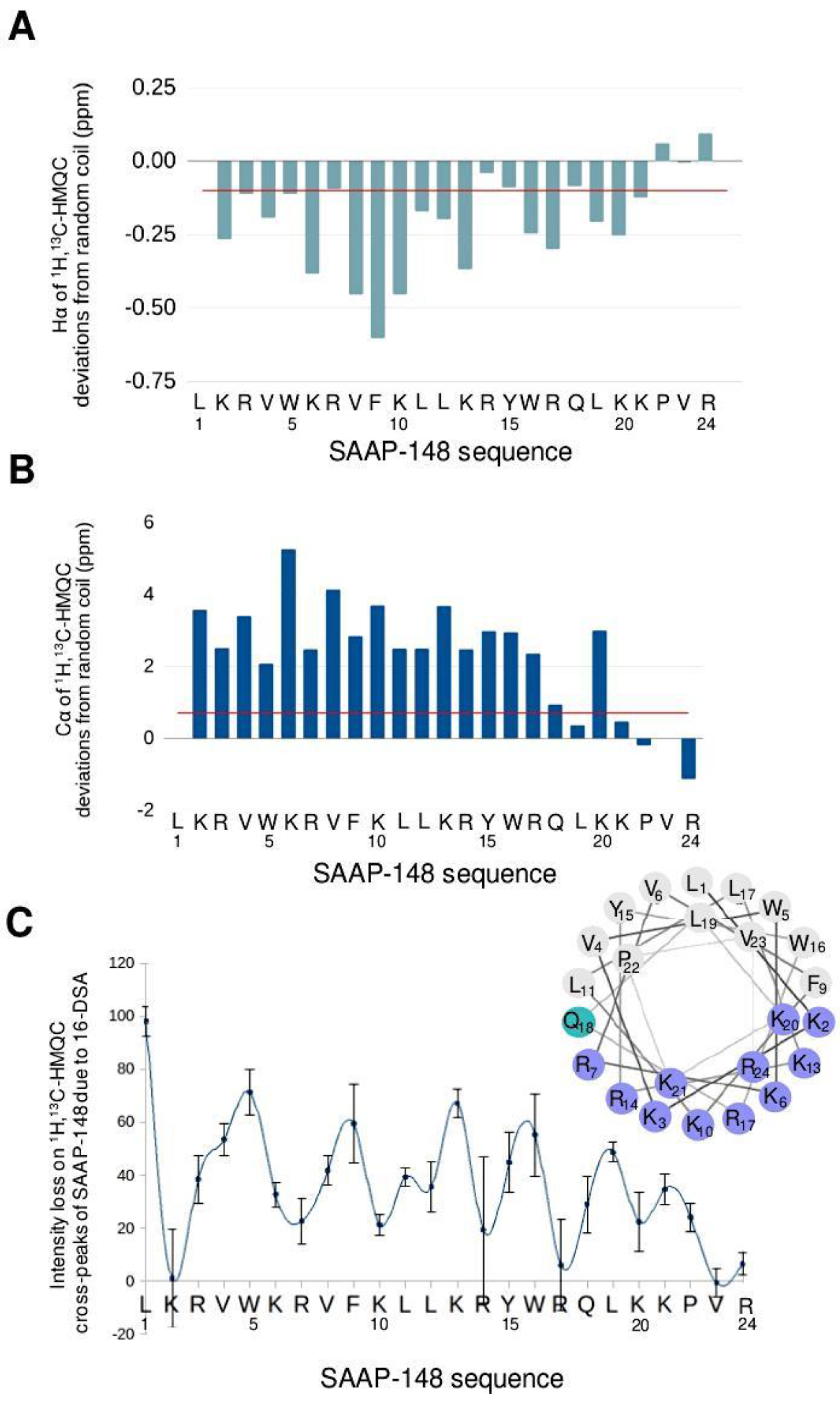
3.1.1. SAAP-148 in Aqueous Solution
3.1.2. SAAP-148 Interaction with Biomimetic Membrane Models
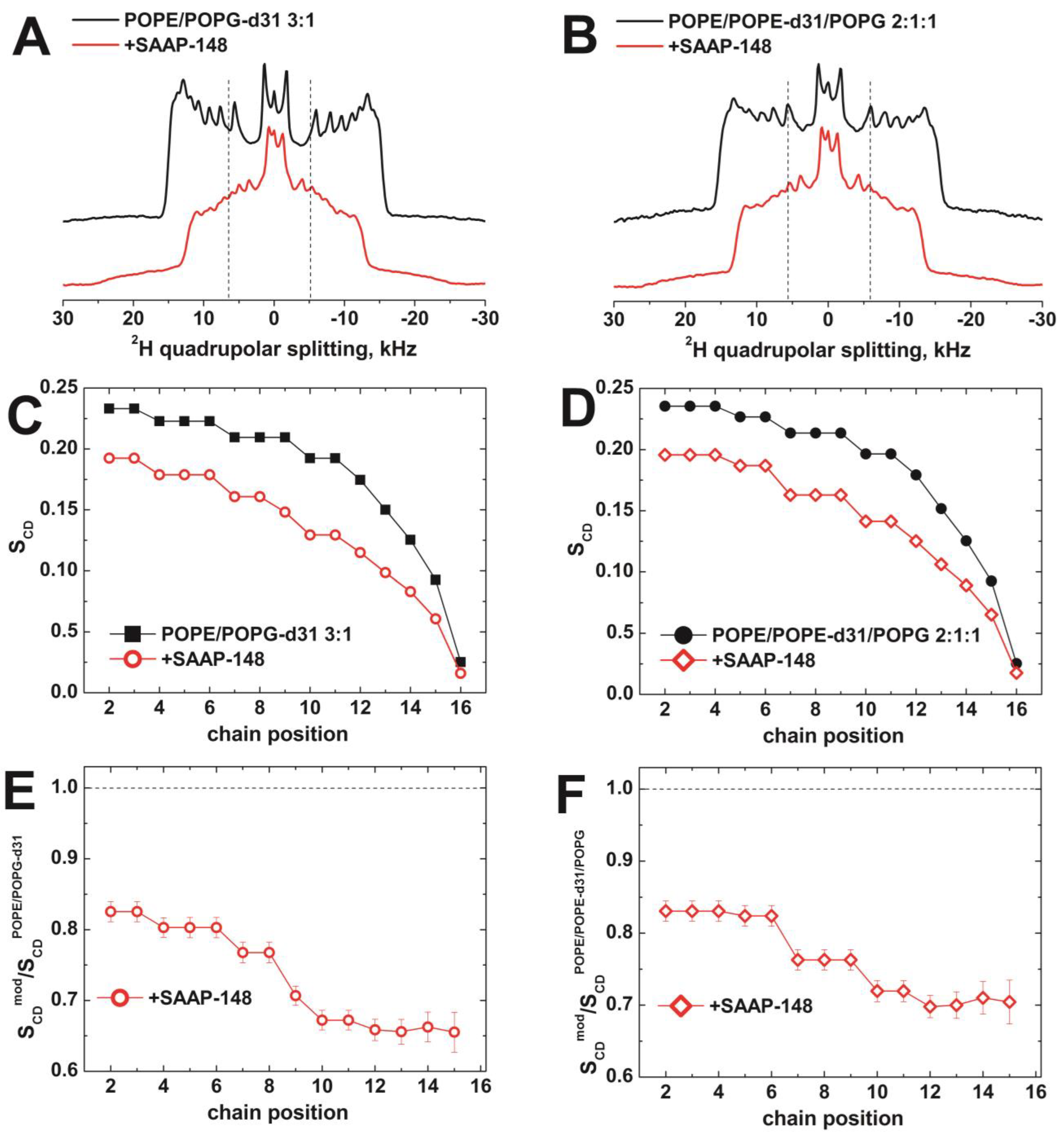
3.2. Solid-State NMR Studies on SAAP-148 Peptide Interacting with Model Membranes
3.2.1. The Orientation of SAAP-148 in Bacterial Biomimetic Membranes
3.2.2. The Effect of SAAP-148 on Biomimetic Membrane Dynamics
3.3. MD Simulations of SAAP-148 Interacting with Biomimetic Bilayers
3.3.1. Effect of SAAP-148 on Mammalian Model Membranes
3.3.2. Effect of SAAP-148 on Bacterial Model Membranes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical Relevance of the ESKAPE Pathogens. Expert Rev. Anti. Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K. Persister Cells and Infectious Disease; Springer: Berlin/Heidelberg, Germany, 2019; ISBN 9783030252410. [Google Scholar]
- Ramos-Vivas, J.; Chapartegui-González, I.; Fernández-Martínez, M.; González-Rico, C.; Fortún, J.; Escudero, R.; Marco, F.; Linares, L.; Montejo, M.; Aranzamendi, M.; et al. Biofilm Formation by Multidrug Resistant Enterobacteriaceae Strains Isolated from Solid Organ Transplant Recipients. Sci. Rep. 2019, 9, 8928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. The Potential of Antifungal Peptide Sesquin as Natural Food Preservative. Biochimie 2022, 203, 51–64. [Google Scholar] [CrossRef]
- Rizzetto, G.; Gambini, D.; Maurizi, A.; Candelora, M.; Molinelli, E.; Cirioni, O.; Brescini, L.; Giacometti, A.; Offidani, A.; Simonetti, O. Our Experience over 20 Years: Antimicrobial Peptides against Gram Positives, Gram Negatives, and Fungi. Pharmaceutics 2022, 15, 40. [Google Scholar] [CrossRef]
- Zhu, S.; Sani, M.-A.; Separovic, F. Interaction of Cationic Antimicrobial Peptides from Australian Frogs with Lipid Membranes. Pept. Sci. 2018, 110, e24061. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- van Gent, M.E.; van der Reijden, T.J.K.; Lennard, P.R.; de Visser, A.W.; Schonkeren-Ravensbergen, B.; Dolezal, N.; Cordfunke, R.A.; Drijfhout, J.W.; Nibbering, P.H. Synergism between the Synthetic Antibacterial and Antibiofilm Peptide (SAAP)-148 and Halicin. Antibiotics 2022, 11, 673. [Google Scholar] [CrossRef]
- Corbett, D.; Wise, A.; Langley, T.; Skinner, K.; Trimby, E.; Birchall, S.; Dorali, A.; Sandiford, S.; Williams, J.; Warn, P.; et al. Potentiation of Antibiotic Activity by a Novel Cationic Peptide: Potency and Spectrum of Activity of SPR741. Antimicrob. Agents Chemother. 2017, 61, e00200-17. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Martín, F.; D’Amelio, N. Molecular Basis of the Anticancer and Antibacterial Properties of CecropinXJ Peptide: An In Silico Study. Int. J. Mol. Sci. 2021, 22, 691. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; D’Amelio, N. Molecular Basis of the Anticancer, Apoptotic and Antibacterial Activities of Bombyx Mori Cecropin A. Arch. Biochem. Biophys. 2022, 715, 109095. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martín, F.; Herrera-León, C.; D’Amelio, N. Bombyx Mori Cecropin D Could Trigger Cancer Cell Apoptosis by Interacting with Mitochondrial Cardiolipin. Biochim. Biophys. Acta Biomembr. 2022, 1864, 184003. [Google Scholar] [CrossRef] [PubMed]
- Langel, Ü. CPP, Cell-Penetrating Peptides; Springer: Berlin/Heidelberg, Germany, 2019; ISBN 9789811387470. [Google Scholar]
- Lee, H.; Lim, S.I.; Shin, S.-H.; Lim, Y.; Koh, J.W.; Yang, S. Conjugation of Cell-Penetrating Peptides to Antimicrobial Peptides Enhances Antibacterial Activity. ACS Omega 2019, 4, 15694–15701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial Strategies of Resistance to Antimicrobial Peptides. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Baeder, D.Y.; Regoes, R.R.; Rolff, J. Predicting Drug Resistance Evolution: Insights from Antimicrobial Peptides and Antibiotics. Proc. Biol. Sci. 2018, 285, 20172687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, A.; Scieuzo, C.; Petrone, A.M.; Salvia, R.; Manniello, M.D.; Franco, A.; Lucchetti, D.; Vassallo, A.; Vogel, H.; Sgambato, A.; et al. Antimicrobial Peptides: A New Hope in Biomedical and Pharmaceutical Fields. Front. Cell. Infect. Microbiol. 2021, 11, 668632. [Google Scholar] [CrossRef]
- Rodríguez-Rojas, A.; Moreno-Morales, J.; James Mason, A.; Rolff, J. Cationic Antimicrobial Peptides Do Not Change Recombination Frequency in Escherichia Coli. Biol. Lett. 2018, 14, 20180006. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.W.; Charron, N.E. Understanding Membrane-Active Antimicrobial Peptides. Q. Rev. Biophys. 2017, 50, e10. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Hofferek, V.; Separovic, F.; Reid, G.E.; Aguilar, M.-I. The Role of Bacterial Lipid Diversity and Membrane Properties in Modulating Antimicrobial Peptide Activity and Drug Resistance. Curr. Opin. Chem. Biol. 2019, 52, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms: My Perspective. Adv. Exp. Med. Biol. 2019, 1117, 3–6. [Google Scholar] [PubMed]
- Vrancianu, C.O.; Gheorghe, I.; Czobor, I.B.; Chifiriuc, M.C. Antibiotic Resistance Profiles, Molecular Mechanisms and Innovative Treatment Strategies of. Microorganisms 2020, 8, 935. [Google Scholar] [CrossRef] [PubMed]
- Matthyssen, T.; Li, W.; Holden, J.A.; Lenzo, J.C.; Hadjigol, S.; O’Brien-Simpson, N.M. The Potential of Modified and Multimeric Antimicrobial Peptide Materials as Superbug Killers. Front. Chem. 2021, 9, 795433. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, H.; Fang, L.; Liu, D.; Liu, J.; Su, M.; Fang, Z.; Ren, W.; Jiao, H. The Modification and Design of Antimicrobial Peptide. Curr. Pharm. Des. 2018, 24, 904–910. [Google Scholar] [CrossRef]
- Sandín, D.; Valle, J.; Chaves-Arquero, B.; Prats-Ejarque, G.; Larrosa, M.N.; González-López, J.J.; Jiménez, M.Á.; Boix, E.; Andreu, D.; Torrent, M. Rationally Modified Antimicrobial Peptides from the N-Terminal Domain of Human RNase 3 Show Exceptional Serum Stability. J. Med. Chem. 2021, 64, 11472–11482. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, M.; Lai, R.; Zhang, Z. Chemical Modifications to Increase the Therapeutic Potential of Antimicrobial Peptides. Peptides 2021, 146, 170666. [Google Scholar] [CrossRef]
- de Breij, A.; Riool, M.; Cordfunke, R.A.; Malanovic, N.; de Boer, L.; Koning, R.I.; Ravensbergen, E.; Franken, M.; van der Heijde, T.; Boekema, B.K.; et al. The Antimicrobial Peptide SAAP-148 Combats Drug-Resistant Bacteria and Biofilms. Sci. Transl. Med. 2018, 10, eaan4044. [Google Scholar] [CrossRef] [Green Version]
- Scheper, H.; Wubbolts, J.M.; Verhagen, J.A.M.; de Visser, A.W.; van der Wal, R.J.P.; Visser, L.G.; de Boer, M.G.J.; Nibbering, P.H. SAAP-148 Eradicates MRSA Persisters Within Mature Biofilm Models Simulating Prosthetic Joint Infection. Front. Microbiol. 2021, 12, 625952. [Google Scholar] [CrossRef]
- Piller, P.; Wolinski, H.; Cordfunke, R.A.; Drijfhout, J.W.; Keller, S.; Lohner, K.; Malanovic, N. Membrane Activity of LL-37 Derived Antimicrobial Peptides against: Superiority of SAAP-148 over OP-145. Biomolecules 2022, 12, 523. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The SMART Model: Soft Membranes Adapt and Respond, Also Transiently, in the Presence of Antimicrobial Peptides. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, K.; Ryadnov, M.G.; Hoogenboom, B.W. Atomic Force Microscopy to Elucidate How Peptides Disrupt Membranes. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183447. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Henderson, J.M.; Waring, A.J.; Separovic, F.; Lee, K.Y.C. Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J. 2016, 111, 2176–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Spano, J.; Park, E.-K.; Wi, S. Evidence of Pores and Thinned Lipid Bilayers Induced in Oriented Lipid Membranes Interacting with the Antimicrobial Peptides, Magainin-2 and Aurein-3.3. Biochim. Biophys. Acta 2009, 1788, 1482–1496. [Google Scholar] [CrossRef] [Green Version]
- Zeth, K.; Sancho-Vaello, E. The Human Antimicrobial Peptides Dermcidin and LL-37 Show Novel Distinct Pathways in Membrane Interactions. Front. Chem. 2017, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Rakowska, P.D.; Jiang, H.; Ray, S.; Pyne, A.; Lamarre, B.; Carr, M.; Judge, P.J.; Ravi, J.; Gerling, U.I.M.; Koksch, B.; et al. Nanoscale Imaging Reveals Laterally Expanding Antimicrobial Pores in Lipid Bilayers. Proc. Natl. Acad. Sci. USA 2013, 110, 8918–8923. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshini, D.; Ivica, J.; Separovic, F.; de Planque, M.R.R. Characterisation of Cell Membrane Interaction Mechanisms of Antimicrobial Peptides by Electrical Bilayer Recording. Biophys. Chem. 2022, 281, 106721. [Google Scholar] [CrossRef]
- Huang, Y.-T.; Kumar, S.R.; Chan, H.-C.; Jhan, Z.-H.; Chen, D.W.; Lue, S.J. Efficacy of Antimicrobial Peptides (AMPs) against Escherichia Coli and Bacteria Morphology Change after AMP Exposure. J. Taiwan Inst. Chem. Eng. 2021, 126, 307–312. [Google Scholar] [CrossRef]
- Lee, C.-C.; Sun, Y.; Qian, S.; Huang, H.W. Transmembrane Pores Formed by Human Antimicrobial Peptide LL-37. Biophys. J. 2011, 100, 1688–1696. [Google Scholar] [CrossRef] [Green Version]
- Savini, F.; Loffredo, M.R.; Troiano, C.; Bobone, S.; Malanovic, N.; Eichmann, T.O.; Caprio, L.; Canale, V.C.; Park, Y.; Mangoni, M.L.; et al. Binding of an Antimicrobial Peptide to Bacterial Cells: Interaction with Different Species, Strains and Cellular Components. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183291. [Google Scholar] [CrossRef] [PubMed]
- Saigo, N.; Izumi, K.; Kawano, R. Electrophysiological Analysis of Antimicrobial Peptides in Diverse Species. ACS Omega 2019, 4, 13124–13130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordi, M.; Borzouyi, Z.; Chitsaz, S.; Asmaei, M.H.; Salami, R.; Tabarzad, M. Antimicrobial Peptides with Anticancer Activity: Today Status, Trends and Their Computational Design. Arch. Biochem. Biophys. 2023, 733, 109484. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Marx, L.; Blondelle, S.E.; Pabst, G.; Semeraro, E.F. Experimental Concepts for Linking the Biological Activities of Antimicrobial Peptides to Their Molecular Modes of Action. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183275. [Google Scholar] [CrossRef]
- Huang, H.W. DAPTOMYCIN, Its Membrane-Active Mechanism vs. that of Other Antimicrobial Peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183395. [Google Scholar] [CrossRef]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Aisenbrey, C.; Marquette, A.; Bechinger, B. The Mechanisms of Action of Cationic Antimicrobial Peptides Refined by Novel Concepts from Biophysical Investigations. Adv. Exp. Med. Biol. 2019, 1117, 33–64. [Google Scholar]
- Nielsen, J.T.; Mulder, F.A.A. POTENCI: Prediction of Temperature, Neighbor and pH-Corrected Chemical Shifts for Intrinsically Disordered Proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef]
- Aisenbrey, C.; Bertani, P.; Bechinger, B. Solid-State NMR Investigations of Membrane-Associated Antimicrobial Peptides. Methods Mol. Biol. 2010, 618, 209–233. [Google Scholar]
- Bechinger, B.; Opella, S.J. Flat-Coil Probe for NMR Spectroscopy of Oriented Membrane Samples. J. Magn. Reson. 1991, 95, 585–588. [Google Scholar] [CrossRef]
- Rance, M.; Byrd, R.A. Obtaining High-Fidelity Powder Spectra in Anisotropic Media: Phase-Cycled Hahn Echo Spectroscopy. J. Magn. Reson. 1983, 52, 221–240. [Google Scholar] [CrossRef]
- Pines, A.; Gibby, M.G.; Waugh, J.S. Proton-enhanced NMR of Dilute Spins in Solids. J. Chem. Phys. 1973, 59, 569–590. [Google Scholar] [CrossRef]
- Bertani, P.; Raya, J.; Bechinger, B. 15N Chemical Shift Referencing in Solid State NMR. Solid State Nucl. Magn. Reson. 2014, 61-62, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H.; Jeffrey, K.R.; Bloom, M.; Valic, M.I.; Higgs, T.P. Quadrupolar Echo Deuteron Magnetic Resonance Spectroscopy in Ordered Hydrocarbon Chains. Chem. Phys. Lett. 1976, 42, 390–394. [Google Scholar] [CrossRef]
- Batchelder, L.S.; Niu, C.H.; Torchia, D.A. Methyl Reorientation in Polycrystalline Amino Acids and Peptides: A Deuteron NMR Spin-Lattice Relaxation Study. J. Am. Chem. Soc. 1983, 105, 2228–2231. [Google Scholar] [CrossRef]
- Michalek, M.; Salnikov, E.S.; Bechinger, B. Structure and Topology of the Huntingtin 1–17 Membrane Anchor by a Combined Solution and Solid-State NMR Approach. Biophys. J. 2013, 105, 699–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1-2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces, Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry, Jerusalem, Israel, 13–16 April 1981; Springer: Dordrecht, The Netherlands; pp. 331–342. [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein Structure and Function Prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A Gen. Phys. 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2, e880. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Kelley, C.; Merritt, E.A.; Bersch, C.; Bröker, H.B.; Campbell, J.; Cunningham, R.; Denholm, D.; Elber, E.; Fearick, R. Gnuplot 5.4. 4: An Interactive Plotting Program; 2022. Available online: http://www.gnuplot.info/documentation.html (accessed on 1 May 2022).
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A Program for Display and Analysis of Macromolecular Structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Han, B.; Liu, Y.; Ginzinger, S.W.; Wishart, D.S. SHIFTX2: Significantly Improved Protein Chemical Shift Prediction. J. Biomol. NMR 2011, 50, 43–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, F.; Ramamoorthy, A.; Barany, G.; Veglia, G. On the Role of NMR Spectroscopy for Characterization of Antimicrobial Peptides. Membr. Proteins 2013, 159–180. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, I.; Auger, M. Bicelles as Model Membranes for Solid-and Solution-State NMR Studies of Membrane Peptides and Proteins. Concepts Magn. Reson. Part A Educ. J. 2005, 24, 17–37. [Google Scholar] [CrossRef]
- Randle, C.L.; Albro, P.W.; Dittmer, J.C. The Phosphoglyceride Composition of Gram-Negative Bacteria and the Changes in Composition during Growth. Biochim. Biophys. Acta 1969, 187, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martín, F.; D’Amelio, N. Biomembrane Lipids: When Physics and Chemistry Join to Shape Biological Activity. Biochimie 2022, 203, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jardetzky, O. Probability-Based Protein Secondary Structure Identification Using Combined NMR Chemical-Shift Data. Protein Sci. 2002, 11, 852–861. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. The Chemical Shift Index: A Fast and Simple Method for the Assignment of Protein Secondary Structure through NMR Spectroscopy. Biochemistry 1992, 31, 1647–1651. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13 C Chemical-Shift Index: A Simple Method for the Identification of Protein Secondary Structure Using 13 C Chemical-Shift Data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef]
- Wishart, D.S. Interpreting Protein Chemical Shift Data. Prog. Nucl. Magn. Reson. Spectrosc. 2011, 58, 62–87. [Google Scholar] [CrossRef]
- Hilty, C.; Wider, G.; Fernández, C.; Wüthrich, K. Membrane Protein-Lipid Interactions in Mixed Micelles Studied by NMR Spectroscopy with the Use of Paramagnetic Reagents. Chembiochem 2004, 5, 467–473. [Google Scholar] [CrossRef]
- Grisham, C.M. Paramagnetic Probes in NMR and EPR Studies of Membrane Enzymes. J. Biochem. Biophys. Methods 1980, 3, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Sommer, L.A.M.; Joel Janke, J.; Drew Bennett, W.F.; Bürck, J.; Ulrich, A.S.; Peter Tieleman, D.; Dames, S.A. Characterization of the Immersion Properties of the Peripheral Membrane Anchor of the FATC Domain of the Kinase “Target of Rapamycin” by NMR, Oriented CD Spectroscopy, and MD Simulations. J. Phys. Chem. B 2014, 118, 4817–4831. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals 2020, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Mól, A.R.; Castro, M.S.; Fontes, W. NetWheels: A Web Application to Create High Quality Peptide Helical Wheel and Net Projections. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B.; Salnikov, E.S. The Membrane Interactions of Antimicrobial Peptides Revealed by Solid-State NMR Spectroscopy. Chem. Phys. Lipids 2012, 165, 282–301. [Google Scholar] [CrossRef]
- Bechinger, B.; Sizun, C. Alignment and Structural Analysis of Membrane Polypeptides by15N and31P Solid-State NMR Spectroscopy. Concepts Magn. Reson. 2003, 18A, 130–145. [Google Scholar] [CrossRef]
- Salnikov, E.S.; James Mason, A.; Bechinger, B. Membrane Order Perturbation in the Presence of Antimicrobial Peptides by 2H Solid-State NMR Spectroscopy. Biochimie 2009, 91, 734–743. [Google Scholar] [CrossRef]
- Davis, J.H. The Description of Membrane Lipid Conformation, Order and Dynamics by 2H-NMR. Biochim. Biophys. Acta 1983, 737, 117–171. [Google Scholar] [CrossRef]
- Harmouche, N.; Bechinger, B. Lipid-Mediated Interactions between the Antimicrobial Peptides Magainin 2 and PGLa in Bilayers. Biophys. J. 2018, 115, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Herrera-León, C.; Ramos-Martín, F.; Antonietti, V.; Sonnet, P.; D’Amelio, N. The Impact of Phosphatidylserine Exposure on Cancer Cell Membranes on the Activity of the Anticancer Peptide HB43. FEBS J. 2022, 289, 1984–2003. [Google Scholar] [CrossRef]
- Herrera-León, C.; Ramos-Martín, F.; El Btaouri, H.; Antonietti, V.; Sonnet, P.; Martiny, L.; Zevolini, F.; Falciani, C.; Sarazin, C.; D’Amelio, N. The Influence of Short Motifs on the Anticancer Activity of HB43 Peptide. Pharmaceutics 2022, 14, 1089. [Google Scholar] [CrossRef] [PubMed]
- Vahedi, A.; Bigdelou, P.; Farnoud, A.M. Quantitative Analysis of Red Blood Cell Membrane Phospholipids and Modulation of Cell-Macrophage Interactions Using Cyclodextrins. Sci. Rep. 2020, 10, 15111. [Google Scholar] [CrossRef]
- Li, G.; Kim, J.; Huang, Z.; St Clair, J.R.; Brown, D.A.; London, E. Efficient Replacement of Plasma Membrane Outer Leaflet Phospholipids and Sphingolipids in Cells with Exogenous Lipids. Proc. Natl. Acad. Sci. USA 2016, 113, 14025–14030. [Google Scholar] [CrossRef] [Green Version]
- Luchini, A.; Vitiello, G. Mimicking the Mammalian Plasma Membrane: An Overview of Lipid Membrane Models for Biophysical Studies. Biomimetics 2020, 6, 3. [Google Scholar] [CrossRef]
- Zachowski, A. Phospholipids in Animal Eukaryotic Membranes: Transverse Asymmetry and Movement. Biochem. J. 1993, 294, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jalali, S.; Nilsson, B.L.; Dias, C.L. Binding Mechanisms of Amyloid-like Peptides to Lipid Bilayers and Effects of Divalent Cations. ACS Chem. Neurosci. 2021, 12, 2027–2035. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Monaco, I.; La Rosa, C.; Milardi, D. The Active Role of Ca Ions in Aβ-Mediated Membrane Damage. Chem. Commun. 2018, 54, 3629–3631. [Google Scholar] [CrossRef]
- Liu, Y.; Ren, B.; Zhang, Y.; Sun, Y.; Chang, Y.; Liang, G.; Xu, L.; Zheng, J. Molecular Simulation Aspects of Amyloid Peptides at Membrane Interface. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1906–1916. [Google Scholar] [CrossRef]
- Lockhart, C.; Klimov, D.K. Calcium Enhances Binding of Aβ Monomer to DMPC Lipid Bilayer. Biophys. J. 2015, 108, 1807–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkul, J.A.; Bevan, D.R. Perturbation of Membranes by the Amyloid β-Peptide—A Molecular Dynamics Study. FEBS J. 2009, 276, 3060–3075. [Google Scholar] [CrossRef] [PubMed]
- Khelashvili, G.; Plante, A.; Doktorova, M.; Weinstein, H. Ca2-Dependent Mechanism of Membrane Insertion and Destabilization by the SARS-CoV-2 Fusion Peptide. Biophys. J. 2021, 120, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.E.; Davidson, D.J.; Elaine Lau, Y.; Lee, K.; Scott, M.G.; Hancock, R.E.W. Impact of LL-37 on Anti-Infective Immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufourc, E.J.; Smith, I.C.; Dufourcq, J. Molecular Details of Melittin-Induced Lysis of Phospholipid Membranes as Revealed by Deuterium and Phosphorus NMR. Biochemistry 1986, 25, 6448–6455. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Martinez, G.V.; Brown, M.F.; Ramamoorthy, A. Perturbation of the Hydrophobic Core of Lipid Bilayers by the Human Antimicrobial Peptide LL-37. Biochemistry 2004, 43, 8459–8469. [Google Scholar] [CrossRef]
- Hénin, J.; Lelièvre, T.; Shirts, M.R.; Valsson, O.; Delemotte, L. Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1.0]. Living J. Comp. Mol. Sci. 2022, 4, 1583. [Google Scholar] [CrossRef]
- Pearlstein, R.A.; Dickson, C.J.; Hornak, V. Contributions of the Membrane Dipole Potential to the Function of Voltage-Gated Cation Channels and Modulation by Small Molecule Potentiators. Biochim. Et Biophys. Acta (BBA) Biomembr. 2017, 1859, 177–194. [Google Scholar] [CrossRef]
- Dreyer, J.; Zhang, C.; Ippoliti, E.; Carloni, P. Role of the Membrane Dipole Potential for Proton Transport in Gramicidin A Embedded in a DMPC Bilayer. J. Chem. Theory Comput. 2013, 9, 3826–3831. [Google Scholar] [CrossRef]
- Bechinger, B.; Juhl, D.W.; Glattard, E.; Aisenbrey, C. Revealing the Mechanisms of Synergistic Action of Two Magainin Antimicrobial Peptides. Front. Med. Technol. 2020, 2, 615494. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adélaïde, M.; Salnikov, E.; Ramos-Martín, F.; Aisenbrey, C.; Sarazin, C.; Bechinger, B.; D’Amelio, N. The Mechanism of Action of SAAP-148 Antimicrobial Peptide as Studied with NMR and Molecular Dynamics Simulations. Pharmaceutics 2023, 15, 761. https://doi.org/10.3390/pharmaceutics15030761
Adélaïde M, Salnikov E, Ramos-Martín F, Aisenbrey C, Sarazin C, Bechinger B, D’Amelio N. The Mechanism of Action of SAAP-148 Antimicrobial Peptide as Studied with NMR and Molecular Dynamics Simulations. Pharmaceutics. 2023; 15(3):761. https://doi.org/10.3390/pharmaceutics15030761
Chicago/Turabian StyleAdélaïde, Morgane, Evgeniy Salnikov, Francisco Ramos-Martín, Christopher Aisenbrey, Catherine Sarazin, Burkhard Bechinger, and Nicola D’Amelio. 2023. "The Mechanism of Action of SAAP-148 Antimicrobial Peptide as Studied with NMR and Molecular Dynamics Simulations" Pharmaceutics 15, no. 3: 761. https://doi.org/10.3390/pharmaceutics15030761
APA StyleAdélaïde, M., Salnikov, E., Ramos-Martín, F., Aisenbrey, C., Sarazin, C., Bechinger, B., & D’Amelio, N. (2023). The Mechanism of Action of SAAP-148 Antimicrobial Peptide as Studied with NMR and Molecular Dynamics Simulations. Pharmaceutics, 15(3), 761. https://doi.org/10.3390/pharmaceutics15030761