1. Introduction
Appropriate formulation of Biopharmaceutics Classification System (BCS) Class II drugs, characterised by low solubility and high permeability, is essential to maximising their oral bioavailability and thereby minimising waste. Many formulation approaches have been investigated in an attempt to increase the rate and extent of dissolution of BCS Class II drugs, with solid dispersion technology being especially well studied. Over the last decade, advanced fabrication technologies such as electrospinning and centrifugal spinning (melt and solution) have been used to prepare solid dispersions in the form of polymer- or sugar-based nano- and micro-fibres [
1,
2,
3,
4,
5,
6]. Typically, the formulation design is for the drug to be molecularly dispersed within the fibres, leading to enhanced in vitro dissolution and in vivo bioavailability. Several excellent reviews on this area of pharmaceutical formulation science have recently been published, providing an in-depth analysis of the current understanding of the subject [
7,
8,
9,
10,
11].
Despite the potential dissolution and oral bioavailability advantages of fibre-based formulations, they require further processing into a patient-friendly format before use. However, due to the particular morphological features of nano- or micro-fibres (high surface area to volume ratio, high porosity, and low density), the incorporation of these systems into a conventional oral dosage form capable of being manufactured reproducibly on a large scale may be challenging. In particular, content (dose) uniformity in the final product is critical and will be dependent on adequate mixing of the fibres and the additional formulation excipients, potentially requiring a milling stage with concomitant risks of both chemical and physical instability of the drug. Tabletting (compression) of “fluffy” material such as fibre mats to obtain a robust product may be mechanically difficult, carries a risk of inducing recrystallisation of an amorphous material, and may result in a decrease in the dissolution rate compared with the native uncompressed fibres due to effective surface area changes. Finally, if the dissolution and oral bioavailability advantages of the original fibres are dependent on the drug being present in the amorphous state, the risks of recrystallisation on storage, leading to deleterious changes in the dissolution profile over time, need to be evaluated and mitigated by judicious choice of packaging and storage conditions.
There have been relatively few studies on the downstream processing of electrospun fibres. Electrospun drug-loaded fibre mats have been manually folded and placed in gelatine capsule shells [
12], although this was more of a processing aid for dissolution studies than an attempt to develop a viable pharmaceutical product. Some studies [
13,
14,
15,
16] have shown that electrospun drug-loaded fibre mats can be manually compressed after direct loading of the fibre mat into the tablet die. Other authors have ground the fibre mats to effect size reduction and mixed them with large quantities of conventional tabletting excipients prior to compression, reflecting a more conventional oral tablet formulation [
17,
18,
19,
20,
21,
22]. Two recent studies have focussed on the mixing step in the conversion of electrospun drug-loaded fibre mats into tablets. Fülöp et al. [
23] showed that it is possible to prepare low-dose tablets with acceptable content uniformity from electrospun drug-loaded fibre mats after high-shear dry mixing with conventional tabletting excipients on a reasonable development lab scale (500 g) and compression on a rotary tablet press. Szabó et al. [
24] successfully applied continuous manufacturing processing techniques (mixing, compression) with inline analysis to prepare tablets from electrospun drug-loaded microfibre mats; in this case, the milled fibres were treated as a starting material. A detailed study on the mechanical and compression behaviour of electrospun drug-loaded nanofibre mats has recently been published [
25]. The presence of the drug, at 10%
w/
w loading, was shown to affect the mechanical properties of the nanofibre mat, in that the drug-loaded samples exhibited greater stiffness and lower ductility than the placebos. Interestingly, only the drug-loaded nanofibrous tablets showed clear fracture behaviour during diametral compression testing after preparation, with the placebo nanofibrous tablets and the tablets prepared from equivalent physical mixtures all showing continuous yielding and tablet shape change instead. The study by Démuth et al. [
26] illustrated the need for judicious choice of excipients in developing tablet formulations from amorphous solid dispersions. They studied electrospun nanofibres containing itraconazole, a poorly water-soluble drug. A total drug release of only 80% of the theoretical amount was observed from tablets containing the water-insoluble lubricant magnesium stearate, which was attributed to the amorphous itraconazole in the nanofibres crystallising on the magnesium stearate during the dissolution test. When a more water-soluble lubricant (sodium stearyl fumarate) was used instead, dissolution was both faster and more extensive, with drug release values of >95% of the theoretical value being achieved.
Much less attention has been paid to the technique of centrifugal spinning (sometimes described as rotary spinning), either in the solution mode or the melt mode, as a means of preparing micro- and nanofibres that can be further processed for pharmaceutical use. Two studies [
27,
28] used centrifugal solution spinning of aqueous solutions of drug and polymer to generate drug-loaded nanofibre mats, which were then micronised, mixed with standard tabletting excipients, and compressed into tablets. Centrifugal melt spinning was used in two studies [
29,
30] to prepare nanofibre mats of drug and sucrose/polyvinylpyrrolidone (PVP), which were then directly compressed into tablets with no further processing. In both cases, dissolution from the tablets was slightly slower than from the nanofibre mats, and in one case [
29], the tablets were observed to be sticky, indicating that, although the nanofibre mats could be compacted, further work is required to convert the nanofibres into a useable formulation.
In our previous work [
3,
4], we have used centrifugal melt spinning to successfully manufacture sucrose-based microfibres containing three clinically significant BCS Class II drugs (itraconazole, olanzapine, and piroxicam) at 10%
w/
w drug loading. Upon manufacture, both the drug and the carrier (sucrose) were observed to exist in an amorphous state in the microfibres, although small amounts of crystalline drug were detected in the piroxicam-loaded microfibres. Under both sink and non-sink conditions, all the fresh drug-loaded microfibres showed significantly superior dissolution behaviour to the raw drugs and to simple physical mixtures of equivalent formulation, with supersaturation being observed in the non-sink testing protocol. The amorphous nature of both the drug and the microfibre matrix, the solubilizing capability of sucrose, and the high surface area to volume ratio of the microfibres all contributed to these effects. Surprisingly, all three drug-loaded microfibres retained their dissolution and supersaturation advantages after exposure to 75% relative humidity (RH) at 25 °C for short periods (around 1 day), which resulted in the crystallisation of the sucrose carrier leading to the collapse of the microfibres to powder. No change in the dissolution profile was observed after 8 months of further exposure to 25 °C/75% RH. Interestingly, while olanzapine and piroxicam recrystallised along with the sucrose, itraconazole appeared to be present in an amorphous state in the humidity-treated samples. These results show that there is considerable potential for the use of centrifugally melt-spun microfibres in enhancing the dissolution profiles of poorly water-soluble drugs. Additionally, these studies indicate that the relationship between the physical form of the system and the drug dissolution behaviour may be more complex than anticipated, with recrystallisation on exposure to high humidity conditions not necessarily leading to the decrease in dissolution rate that may be commonly expected.
This then raises the question of whether it is possible to deliberately recrystallise freshly prepared drug-loaded microfibres by exposure to an environment with controlled high humidity, causing the collapse of the microfibres into powder, and use this powder as the drug source for further processing into conventional tablets for ease of administration to patients. Such an approach should have the dual advantages of retaining the dissolution benefit of the microfibres while using a source material (powder) that is more easily processed into tablets than the freshly prepared microfibres themselves. It is this question that is explored in the current study. Here we have investigated the feasibility of preparing tablet formulations using standard pharmaceutical excipients and processing methods that contain drug-loaded microfibres produced by centrifugal melt spinning and exposed to high humidity conditions. Our aim is to develop a tablet product with a fast dissolution profile that is both scalable and economically viable. Itraconazole was chosen as the model drug for this study, as there is a particular need for an improved oral formulation of itraconazole, given its relatively low oral bioavailability (55%), and the variability in plasma levels reported following oral administration under different feeding regimes [
31,
32]. However, as a low level of drug loading in the microfibres (10%
w/
w in our previous studies) will inevitably lead to a low drug content in the final tablets, the effects of increasing the itraconazole content of the microfibres have also been investigated. The flow properties of the drug-loaded microfibres, alone and in formulation mixtures, were studied, along with their mixing and segregation potentials. Selected formulations were compressed, and their pharmaceutical performance was assessed in terms of compliance with standard mandatory pharmacopoeial tests and their disintegration and dissolution profiles.
3. Results and Discussion
3.1. Preparation of Microfibres with Increasing Drug Loading and Characterisation of the Fresh Microfibres
Microfibres were successfully prepared by centrifugal melt spinning of physical mixtures of sucrose and itraconazole as described above. The calculated percentage yields (% of theoretical, mean ± SD, n = 6) were 94.6 ± 1.5, 93.4 ± 1.7, and 94.1 ± 1.4, respectively, for the 20, 30, and 50%
w/
w drug-loaded microfibres, similar to the value of 95.4 ± 2.1 observed for the 10%
w/
w sample [
4]. Drug content uniformity values (% of theoretical, mean ± SD, n = 6) of 98.6 ± 1.5 and 99.4 ± 1.3 for the 20 and 30%
w/
w samples, respectively, were again similar to that seen for the 10%
w/
w sample, 99.5 ± 1.1 [
4], indicating full incorporation and homogeneous distribution of the drug in the microfibre product. However, at 50%
w/
w itraconazole loading, a higher mean value and greater variation were observed, i.e., 117.1 ± 9.8, suggesting that the drug may not be homogeneously distributed within the sucrose matrix at high drug incorporation, possibly due to the limited loading capacity of the sucrose carrier. This is discussed in more detail below. Surprisingly, itraconazole was able to form microfibres alone, with no sucrose carrier. The optimum temperature for spinning pure itraconazole was found to be 183 °C, with a yield of 65.7 ± 4.3% of theoretical. At lower temperatures, no microfibres were formed even though the drug was molten, whereas at higher temperatures, the yields were lower as some of the drug stuck to the spinneret. The lower spinning temperature for pure itraconazole compared with the mixed itraconazole-sucrose systems is a consequence of itraconazole’s lower melting point of 166 °C [
33] compared with that of sucrose (186 °C) [
34]. We believe this is the first report of pure itraconazole microfibres being formed using any production technique.
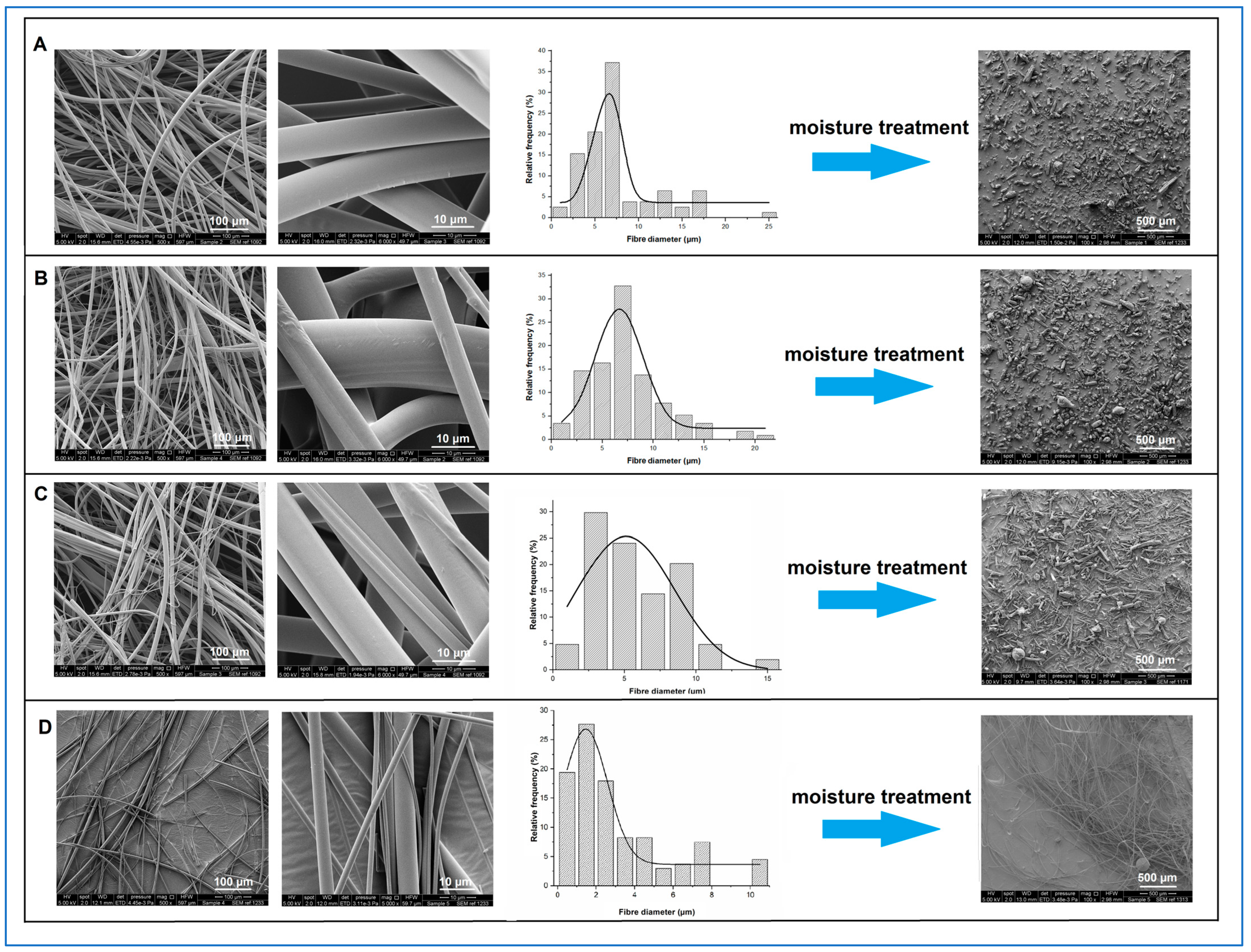
As shown in
Figure 1, under SEM, all freshly prepared itraconazole-loaded sucrose microfibres showed smooth surface morphology with no defects or evidence of surface or bulk drug crystallisation.
The mean microfibre diameter was independent of itraconazole content, with values of circa 7 µm being observed for all samples, as detailed in
Table 2. However, visual inspection of the 50%
w/
w itraconazole-loaded samples and closer analysis of their SEM images showed the presence of thin (1 to 5 µm diameter), grey-coloured microfibres with a visibly different texture from the bulk of the microfibres. These thinner microfibres were similar in appearance and diameter to the pure itraconazole microfibres (3.22 ± 2.54 µm, mean ± SD), suggesting that some of the thinner microfibres in the 50%
w/
w itraconazole-loaded sample were in fact pure itraconazole microfibres.
XRPD diffractograms of all fresh drug-loaded sucrose microfibres (20, 30, and 50%
w/
w) and the pure drug microfibres showed the typical broad halo pattern of an amorphous material, as shown in
Figure 2, as had previously been observed for the freshly prepared drug-free and 10%
w/
w drug-loaded sucrose systems.
Figure 3 shows the MTDSC reversing heat flow traces of the spun microfibres and the equivalent quench-cooled samples.
All samples showed glass transitional behaviour, indicating the generation of amorphous dispersions. Interestingly, the quench-cooled sucrose-itraconazole samples all showed two separate glass transitions assigned to the individual components, with that of sucrose occurring at 68 ± 4.3 °C, indicated with a red arrow, and that of itraconazole occurring at 60 ± 0.2 °C, indicated with a blue arrow. These data indicate phase separation, i.e., lack of miscibility, of the two components, as discussed previously [
35] for other systems. Further evidence of phase separation is offered by the presence of the two endothermic events occurring at around 74 and 90 °C, ascribed to the formation of a chiral nematic mesophase of pure itraconazole (90 °C) and rotational restriction of the molecules (74 °C) upon cooling from the melt, which are reversible upon re-heating, leading to the observed endotherms [
36]. Both of these endothermic transitions and the glass transition are clearly visible for the quench-cooled pure itraconazole sample here. Microfibres containing 20 and 30%
w/
w itraconazole showed a single mixed-phase glass transition, indicated with a green arrow, at 74.7 ± 1.1 °C and 73.2 ± 1.3 °C, respectively, similar to that observed for the 10%
w/
w drug-loaded sample at 74.1 ± 1.9 °C [
4]. Conversely, the thermal behaviour of the 50%
w/
w itraconazole-loaded microfibres was more similar to that of the corresponding quench-cooled sample, with both individual substance glass transitions and both itraconazole endothermic events being observed. It is interesting to note that the endothermic transitions are less pronounced for the microfibres, possibly indicating a lower degree of phase separation compared with the equivalent quench-cooled sample. The pure itraconazole microfibres showed the expected glass transition and endothermic transitions.
Taken together, these initial characterisation data suggest that the melt centrifugal process may increase the degree of mixing and miscibility of the drug (itraconazole) in the carrier (sucrose) compared with the simple melt quenching method, possibly due to the application of high centrifugal forces, with the true limit of miscibility being between 30 and 50% w/w of itraconazole. In the 50% w/w itraconazole-loaded samples, at least some of the excess drug appears to be ejected as pure drug microfibres rather than solidifying as conventional drug particles. This observation also provides an explanation for the more variable content uniformity data seen for the 50% w/w-loaded samples than those with lower drug content: each sample taken for analysis is likely to contain a different proportion of the various populations of microfibres, leading to greater variability in the results.
3.2. High Humidity Treatment of Fresh Microfibres
All high-drug-loaded microfibres showed sucrose recrystallisation after storage at 25 °C/75% RH in open containers, as demonstrated by the appearance of sucrose Bragg peaks in the XRPD diffractograms and collapse of the microfibre structure, as described previously for the 10%
w/
w system [
4]. Interestingly, the time required for the microfibres to collapse increases as a function of the itraconazole content. The 10%
w/
w sample was observed to collapse within 24 h, whereas microfibres containing higher amounts of itraconazole required significantly longer times to do so. Specifically, systems containing 20, 30, and 50%
w/
w itraconazole were seen to collapse in 4.2 ± 1.3, 7.4 ± 1.9, and 19.7 ± 2.4 days, respectively (n = 3 for each formulation). This is likely due to differences in the water uptake tendency of the microfibres with the higher concentrations of this highly lipophilic drug. Our previous studies [
4] on centrifugally spun sucrose microfibres containing 10%
w/
w of olanzapine (log P = 2.2), piroxicam (log P = 3.06), or itraconazole (log P = 5.66) showed that the more hydrophobic the drug, the lower the moisture uptake as determined by dynamic vapour sorption, and the slower the sucrose recrystallisation and microfibre collapse. This was attributed to the increased hydrophobicity of the microfibre surface in the presence of the drug. It is logical, therefore, to expect that increasing the quantity of itraconazole in the microfibres would further retard the diffusion of water through the hydrophilic sucrose matrix, slowing down the sucrose recrystallisation and the collapse of fibrous structure.
For all subsequent studies, all microfibre formulations were held at 25 °C/75% RH in open containers for 30 days in order to ensure that the sucrose had fully recrystallised in all samples and comparisons between formulations were valid.
3.3. Characterisation of the 30-Day Humidity-Treated Microfibres
After exposure of freshly prepared samples to high humidity conditions, microfibres with 20 and 30%
w/
w itraconazole collapsed into powders with similar average particle size and morphology to those observed for the previously investigated system containing 10%
w/
w itraconazole [
4]. At 50%
w/
w drug loading, there are more particles with a more elongated morphology compared with all other systems. However, in all cases, the moisture-treated itraconazole-sucrose systems collapsed into significantly smaller particles than the drug-free sucrose sample.
Figure 1 shows the SEM micrographs of the fresh microfibres with their corresponding diameter frequency diagrams and the SEM images of the collapsed powder after humidity treatment.
Table 2 summarises the size data for the samples studied here and the 10%
w/
w drug-loaded and pure sucrose samples for comparison. The pure itraconazole microfibres showed no change in appearance after exposure to the high humidity conditions for 30 days, with the measured diameter of 3.13 ± 2.50 µm (mean ± SD) showing no significant difference to that of the fresh samples (
p > 0.05). The different collapse behaviour of the microfibres (total collapse for the pure sucrose and 10, 20, and 30%
w/
w itraconazole-loaded microfibres; partial collapse of the 50%
w/
w itraconazole-loaded microfibres; no observable collapse for the pure itraconazole microfibres) suggest that the dominant factor in the collapse process is the sucrose. For the 50%
w/
w itraconazole-loaded samples, those microfibres containing sucrose will collapse after humidity treatment, whereas microfibres containing pure or almost pure drug will not, leading to the observation of a mixed population of particles and microfibres after treatment.
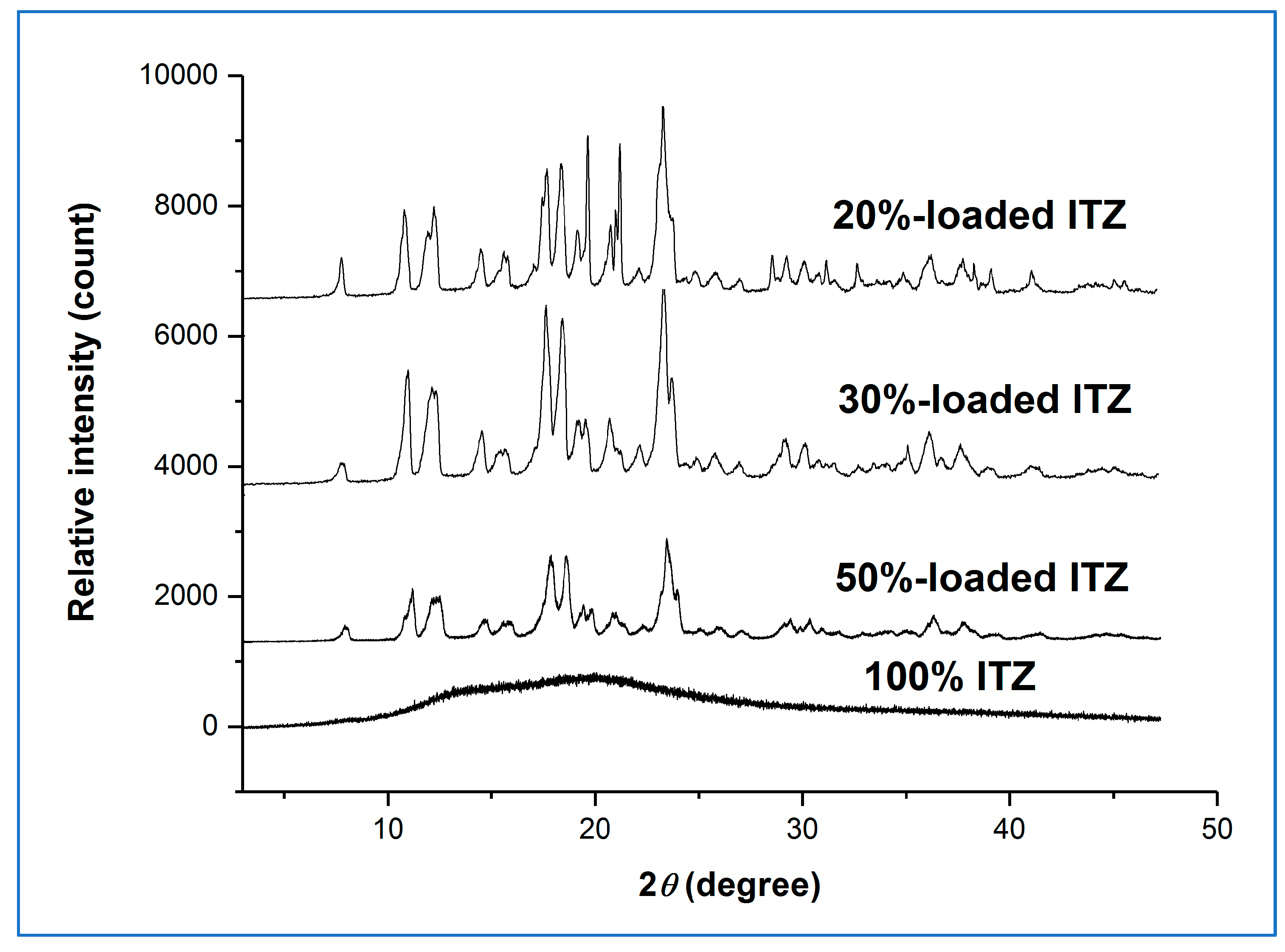
Figure 4 shows the XRPD diffractograms for the high humidity-treated microfibre formulations.
The presence of the characteristic Bragg peaks for crystalline sucrose and the absence of peaks corresponding to crystalline itraconazole confirmed that the sucrose had largely or fully recrystallised from all the aged sucrose-itraconazole systems, whereas it is inferred that the itraconazole remained in the amorphous state. We had previously observed this behaviour for the itraconazole-sucrose system containing 10%
w/
w drug [
4]. However, no recrystallisation of the pure itraconazole microfibre system was observed after 30 days, as indicated by the presence of a broad halo pattern and the absence of any Bragg peaks for crystalline itraconazole. The MTDSC reversing heat flow traces of the high humidity-treated microfibre formulations are shown in
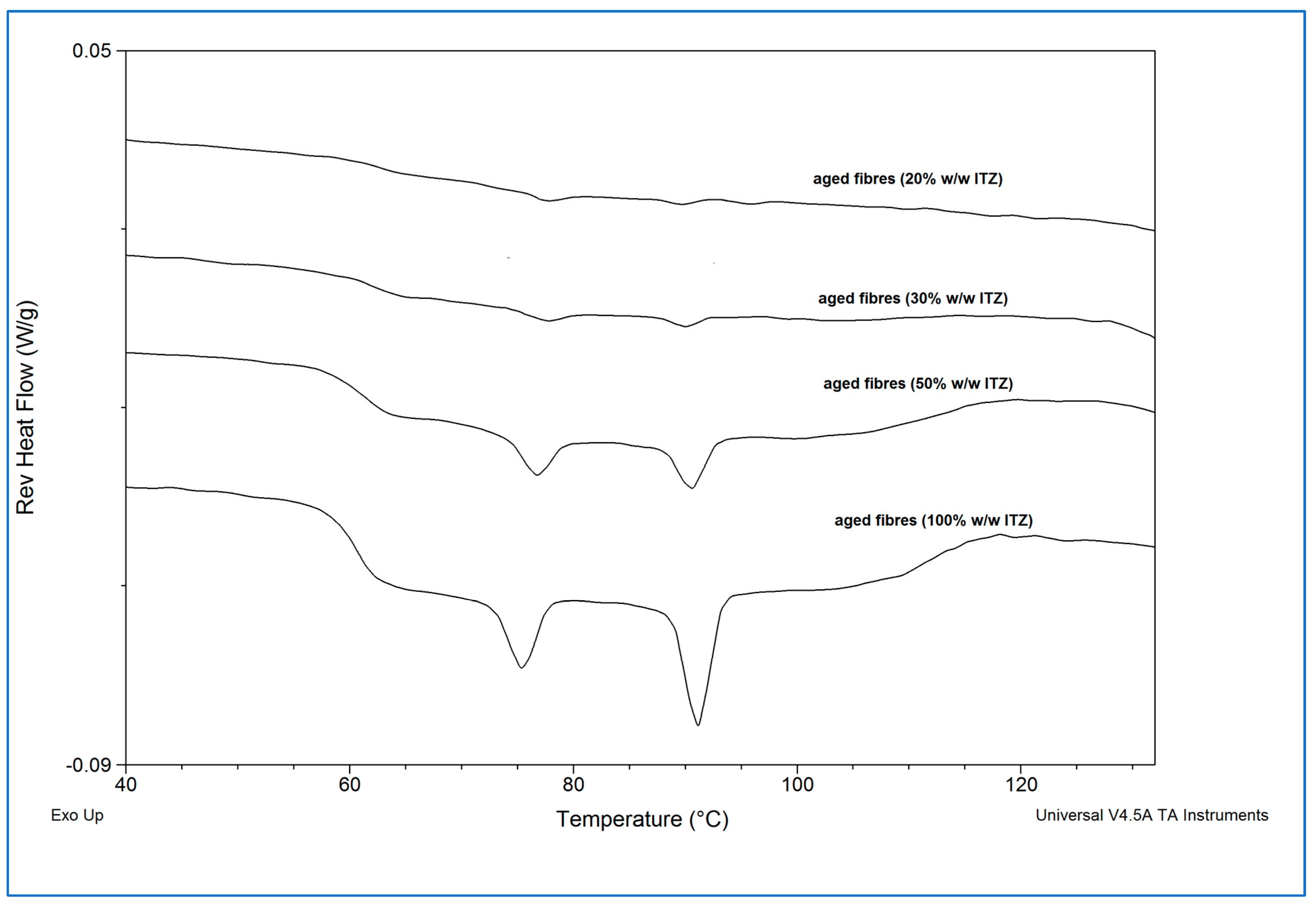
Figure 5.
The absence of the glass transition associated with sucrose (circa 68 °C) and the presence of the glass transition corresponding to itraconazole (circa 60 °C), followed by the two endothermic transitions associated with amorphous itraconazole (circa 74 °C and 90 °C) confirmed that sucrose fully recrystallised from all the itraconazole-sucrose systems, whereas itraconazole remained in the amorphous state.
Drug content uniformity was measured for the moisture-treated samples, with values (% of theoretical, mean ± SD, n = 6) of 99.1 ± 1.2, 97.9 ± 1.7 and 106.3 ± 11.2 for the 20, 30, and 50% w/w samples, respectively. These are essentially unchanged from the fresh samples, suggesting that the microfibre collapse did not adversely affect the drug distribution in the samples.
Overall, physical and chemical analysis of the humidity-treated microfibres shows that the collapse is due to recrystallisation of the sucrose carrier and that the drug remains in the amorphous form, possibly explained by its high hydrophobicity, which reduces its interaction with water during storage.
3.4. Non-Sink Dissolution Testing of Fresh and Aged Microfibres
In our previous study [
4], humidity-treated 10%
w/
w itraconazole-loaded sucrose microfibres exhibited an unexpected increase (circa 1.25-fold) in the solubility of itraconazole, observed under non-sink conditions, compared with the freshly prepared microfibres, which themselves showed very significant itraconazole solubility increases (circa 8-fold) compared with the pure crystalline drug or an equivalent physical mix. The solubility advantage of the treated samples was maintained even after 8 months of further exposure to high humidity conditions. Here, we have studied the dissolution and solubility behaviour of the samples with higher drug content, both fresh and aged for 30 days at 25 °C/75% RH, under the same non-sink test conditions and with the same total amount of drug in each experiment. The concentration–time profiles of the fresh and aged samples are shown in
Figure 6 and
Figure 7, respectively.
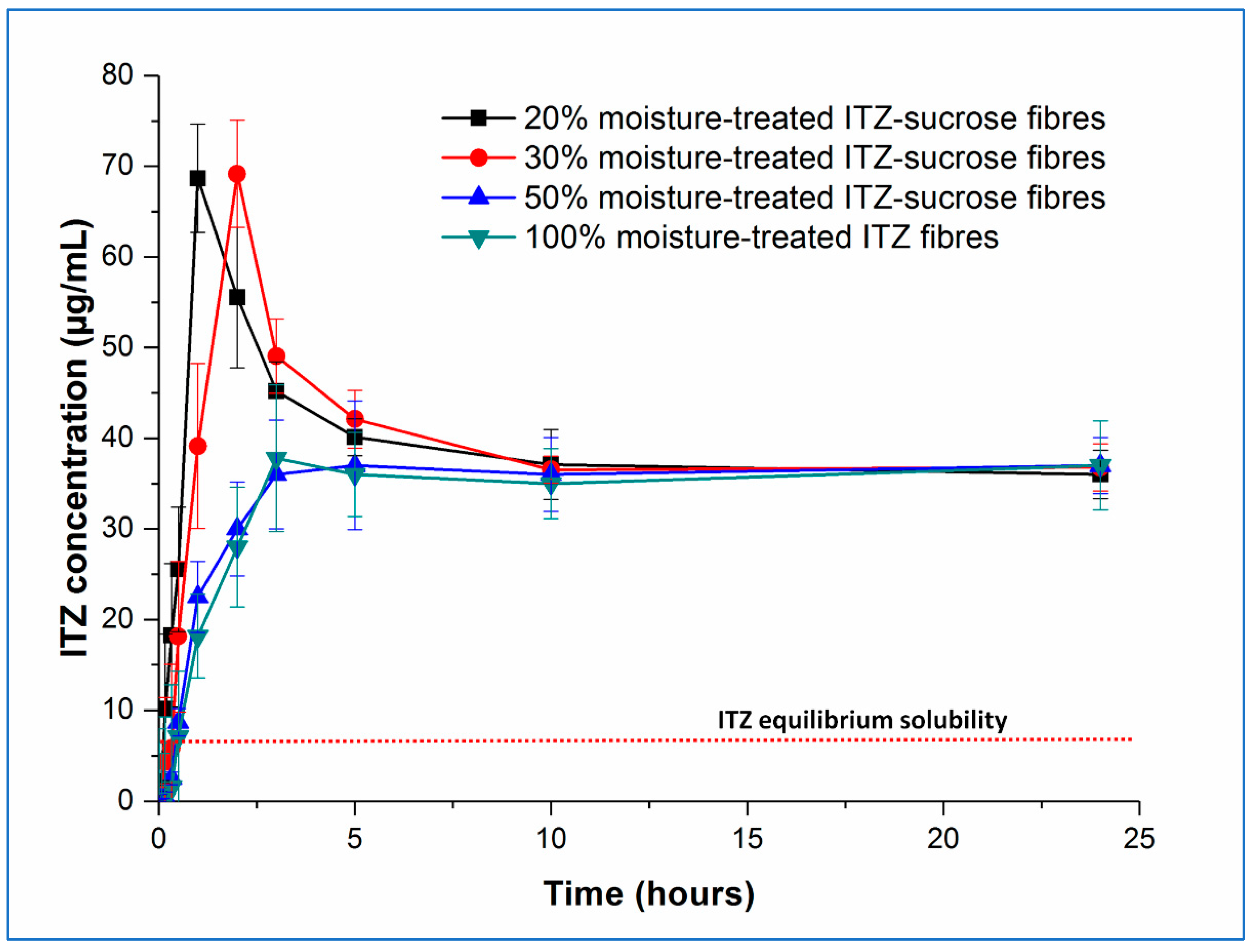
Examining the fresh samples first, in all cases, the initial drug dissolution process was relatively rapid, forming supersaturated solutions with itraconazole concentrations far exceeding the measured crystalline itraconazole equilibrium solubility (approximately 7 µg/mL). It is interesting to note the behaviour of the pure amorphous itraconazole microfibres, as this gives an indication of the amorphous itraconazole equilibrium solubility (approximately 40 µg/mL) as well as allowing interrogation of the role of sucrose in the dissolution and supersaturation processes.
Both the rate of supersaturation generation and the peak concentration of the itraconazole in solution are affected by the drug-carrier ratio. The time required (T
max) to reach the maximum level of drug supersaturation (C
max) increases with increasing drug loading, with T
max values being approximately 1, 2, and 3 h for the 20, 30, and 50%
w/
w itraconazole-loaded microfibres, respectively. Samples containing 20 and 30%
w/
w of itraconazole reached an equivalent maximum level of supersaturation (
p > 0.05) (C
max = 57.7 ± 5.9 and 59.2 ± 4.7 µg/mL, respectively), following which drug precipitation occurred, with the final observed dissolved concentrations stabilising at the level of the equilibrium solubility of amorphous itraconazole. In contrast, at 50%
w/
w drug loading, the C
max was significantly lower (
p < 0.05), reaching only 44.9 ± 3.9 µg/mL and rapidly decreasing down to the pure amorphous itraconazole solubility. The 10%
w/
w drug-loaded sample showed the same trend as the 20 and 30%
w/
w drug-loaded samples: T
max was shorter at approximately 30 min, and C
max was not significantly different (
p > 0.05) at 56.78 ± 5.9 µg/mL [
4].
This observed pattern of supersaturation behaviour can be explained by a combination of the effect of the dissolving hydrophilic sucrose and the solid-state characteristics of the microfibres. At 50% w/w drug loading, the sucrose-itraconazole system is at least partially phase-separated, and the actual amount of sucrose in the dissolving sample is low. It is reasonable to suggest that the rapid dissolution of this small amount of sucrose is able to affect only marginally the initial drug dissolution step, leaving the phase-separated amorphous drug to regulate the final dissolution step, hence the final drug concentration cannot substantially exceed the solubility of the equivalent pure amorphous drug. At higher sucrose ratios (lower drug content), where the drug and carrier are molecularly dispersed in the solid microfibres, the initial drug dissolution profiles are more controlled by the concomitant dissolution of the carrier and some of the amorphous drug. This will lead to a more consistent Cmax, as this will ultimately be dependent on the extent of the interactions between the two components and the maximum increase in solubility of itraconazole that may be effected by the presence of sucrose. The shorter Tmax with the lower drug contents is a reflection of the higher sucrose:drug ratio, leading to greater interaction between the two components initially and a faster rate of supersaturation generation. The decrease in measured drug concentration from the Cmax down to the level of equilibrium amorphous drug solubility is likely caused by the complete dissolution of the carrier, leaving the remaining undissolved amorphous drug to control the rest of the process. Even though the very high levels of supersaturation are maintained for a relatively short period (up to about 2 h), this may be sufficient to allow enhanced oral absorption, as considered in more detail below.
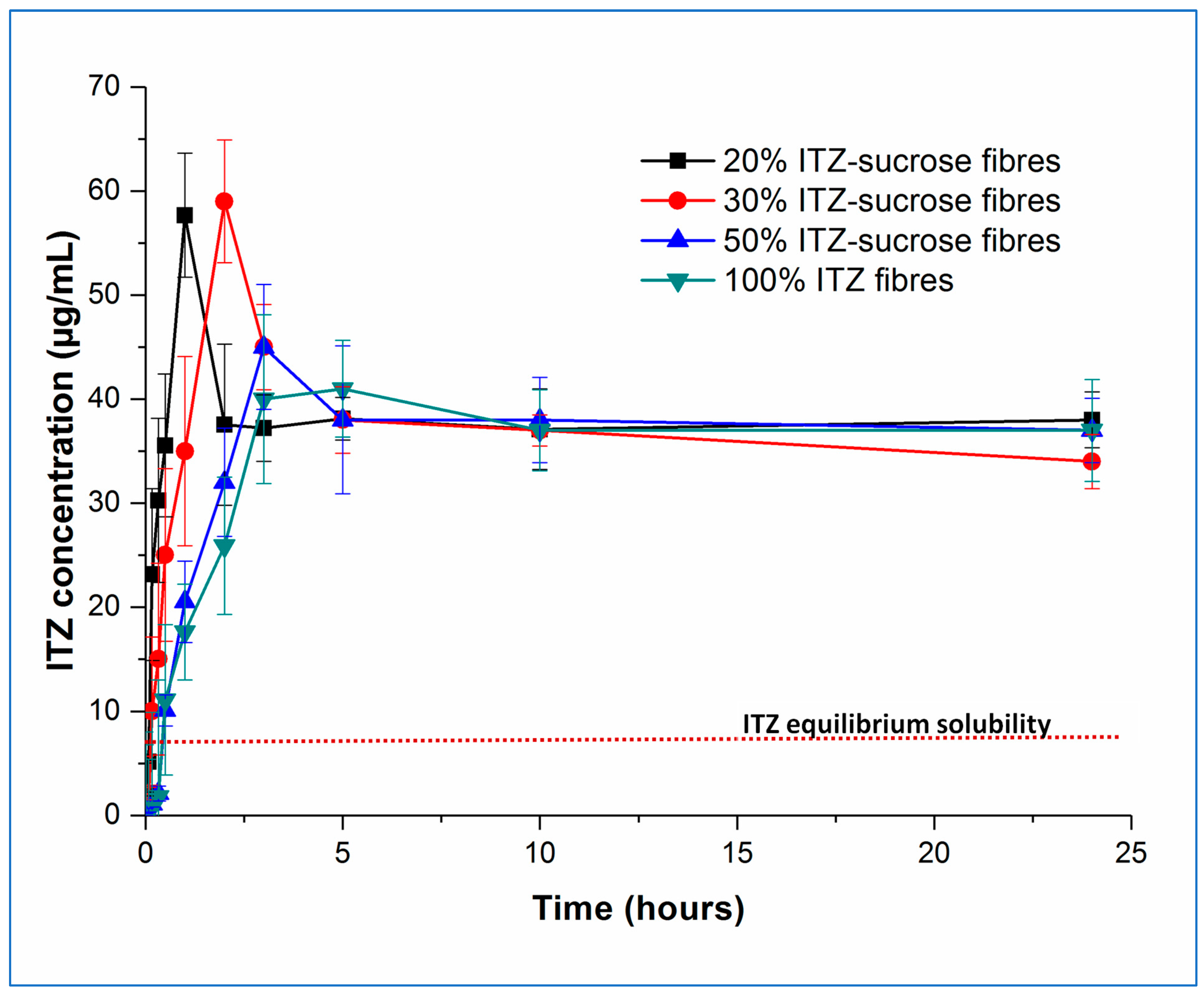
Examining now the dissolution–supersaturation profiles for the humidity-treated samples, at first sight, the profiles of the aged samples are very similar to those of the fresh samples. As expected based on the physical analysis described above, there was no change in the behaviour of the pure amorphous itraconazole microfibres, and the dissolution profile of the 50%
w/
w itraconazole sample has decreased to match that of the pure itraconazole microfibres, losing the initial solubility benefit seen in the fresh sample. In contrast, the initial dissolution rate of the 20 and 30%
w/
w drug-loaded samples decreased compared with the fresh samples, but the T
max values remained the same and the supersaturation (C
max) levels were increased, to 68.15 ± 4.12 and 69.17 ± 7.45 µg/mL, respectively, a similar profile to the 10%
w/
w-loaded samples previously described [
4]. The C
max values for the aged 20 and 30%
w/
w drug-loaded samples were not significantly different from each other but were significantly different (
p < 0.05) from the corresponding fresh samples. This may be explained by the slower dissolution of crystalline sucrose in the aged samples compared with the amorphous sucrose in the fresh samples, leading to slower initial dissolution of the drug but preventing a too-rapid buildup of supersaturation, which would then lead to rapid precipitation back to the equilibrium amorphous itraconazole solubility levels. These observations are in agreement with the theoretical model proposed by Han and Lee [
37], in which rapid supersaturation generation above a critical value is followed by rapid desaturation based on the decreasing energy barrier to nucleation and precipitation seen at high supersaturation levels.
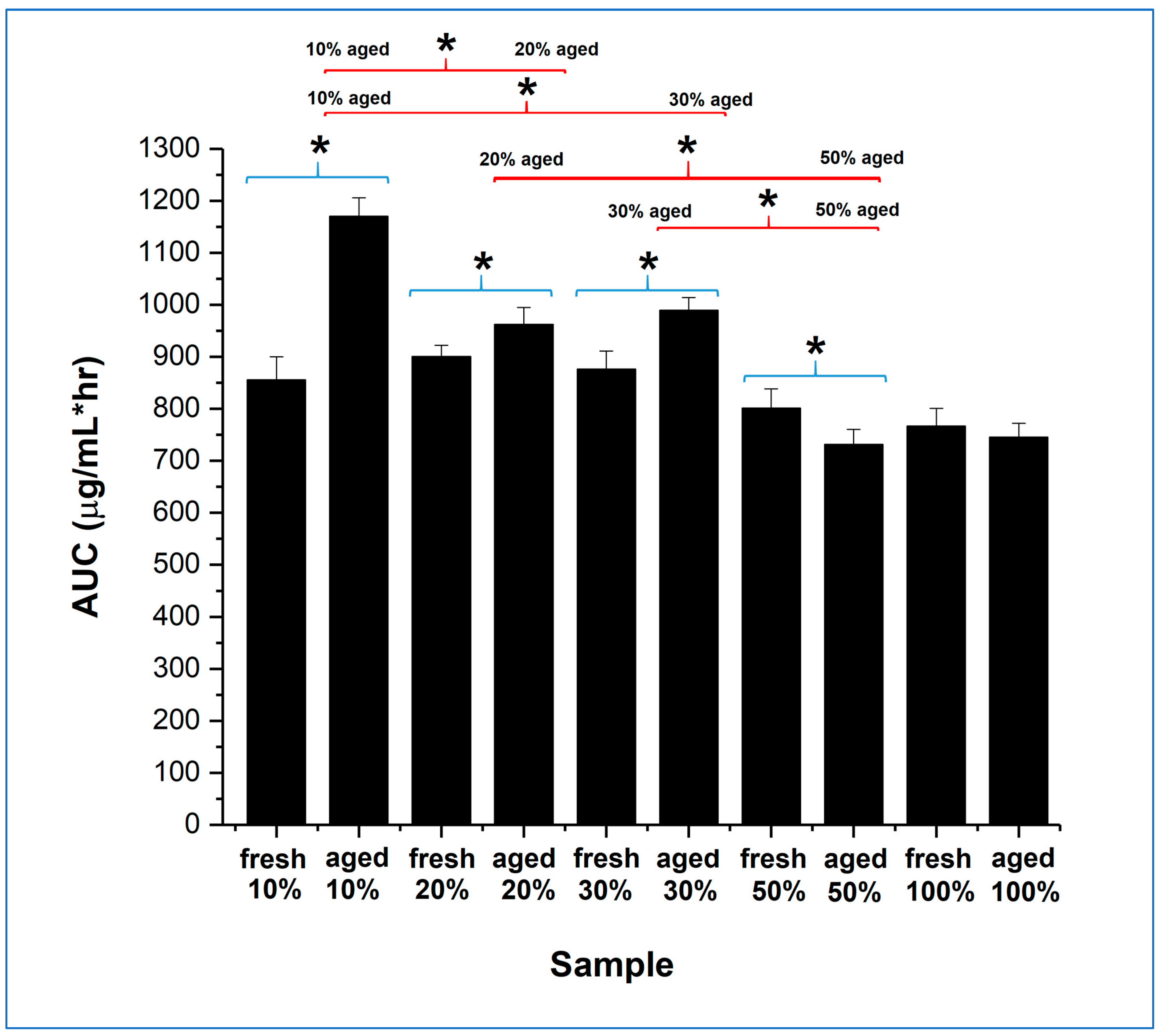
The overall dissolution performance can be further evaluated by comparing the area under the curve (AUC) of the dissolution–supersaturation profiles for the same quantity of drug, displayed in
Figure 8, for the full 24 h of study. The 10, 20, and 30%
w/
w itraconazole-loaded aged samples all showed a statistically significant increase in AUC compared with their fresh counterparts, although the effect was much greater for the 10%
w/
w drug-loaded sample. Importantly, the aged 10%
w/
w drug-loaded samples showed a statistically significant better AUC performance than all other aged samples. Overall, the dissolution advantage of the microfibre generation and humidity treatment seems to be greater for the lower drug loading samples, but this must be balanced against the downstream formulation constraints and product size issues, as will be discussed in the next section.
3.5. Tablet Development—Powder Characterisation
Tablet development focussed on the 10 and 30% w/w drug-loaded aged microfibres, as these respectively showed the greatest dissolution/supersaturation advantage and the likely benefits of size reduction of the final product due to greater drug incorporation. Wet granulation was not considered to be a desirable method of tablet production here due to the risk of damage to the microfibres because of the presence of water (or other granulation solvents) and heat; a direct compression method was therefore selected. However, this approach requires careful consideration of the mixing and flow properties of the individual components and the formulation as a whole; hence, the collapsed microfibres were studied without further processing.
Both the aged microfibre samples showed poor flow characteristics, as would be expected given their morphologies, with Carr indices of > 30%. Binary mixtures of the aged microfibre samples with Compressol SM® or StarTab® showed significant improvements in flow behaviour: even at 60% w/w microfibre content, the Carr indices were < 20%, indicating good to fair flow. Conversely, binary mixtures with Avicel PH102® showed no improvement in flow properties compared with the microfibre samples alone, except at the lowest microfibre incorporation (10% w/w). The segregation potential of the binary mixtures was assessed using a novel segregation cell based on a tapped density apparatus. Here, the arrangement of an outer plastic cylinder with a removable base and a set of stacking inner plastic cylinders, rather than the normal single measuring cylinder, allows for ease of sampling at specific heights in the powder bed, after tapping to simulate the vibration and particle movement expected in the tabletting processes.
The mixing and segregation profiles of the aged microfibres were assessed using the 10% w/w itraconazole-loaded system. No segregation was seen with binary mixtures based on StarTab® containing 10 to 60%w/w microfibres, with drug content values of all individual unit-sized samples (300 mg total weight) comfortably within the range of 95 to 105% of theoretical, significantly better than the pharmacopoeial content uniformity specification of 85 to 115% of theoretical. Binary mixtures based on Avicel PH102® or Compressol SM® showed inappropriate segregation behaviour at low microfibre loadings (10% w/w), with content uniformity data outside the pharmacopoeial limits, but this improved as the microfibre loadings increased, such that at 60% w/w microfibre loading, the content uniformity values were within the range of 95 to 105% of theoretical. The lack of segregation seen with StarTab® is attributable to its morphology, with the high specific surface area and porosity providing greater opportunity for mechanical interlocking of the microfibres and the carrier particles. Subsequent experiments showed that a pre-mixing step of microfibres, equivalent to 10% w/w loading in the final mix, with StarTab® prior to dilution with either Avicel PH102®, Compressol SM®, or a 1:1 mix of these two excipients, led to non-segregating powders, with all measured drug content values being in the range of 95 to 105% of theoretical. These results suggest that, as long as the processed microfibres are initially mixed with StarTab®, other components with alternative functionalities may be added to generate a fully functional tablet formulation.
3.6. Tablet Development—Physical Characterisation
Using the aged 10%
w/
w itraconazole-loaded microfibres, nine different tablet formulations, labelled F1 to F9 and shown in
Table 1, were produced via a direct compression process, i.e., pre-mixing of the microfibres with StarTab
® to prevent segregation, dry mixing with other ingredients, lubrication, and compression. In all formulations, the level of the disintegrant (crospovidone (Kollidon CL-F
®)) was kept constant at 5%
w/
w, and the level of the lubricant (glyceryl dibehenate (Compritol 888 ATO
®)) was maintained at 1.5%
w/
w. The formulations are divided into three groups (Groups 1, 2, and 3) based on the content of the drug-loaded microfibres (10, 30, and 50%
w/
w, respectively). Within each group, the amount of StarTab
® relative to Avicel PH102
® and Compressol SM
® was increased in the ratios of 1:1:1 (formulations F1, F4, and F7), 2:1:1 (formulations F2, F5, and F8), and 3:1:1 (formulations F3, F6, and F9). Finally, for comparative purposes, Group 4 included the highest possible loading of the microfibres (93.5%
w/
w) with just the disintegrant and lubricant in formulation F10 and the raw, unprocessed ingredients in the same ratios in formulation F11. All formulations were compressed at four compression forces: 10, 16, 20, and 26 kN.
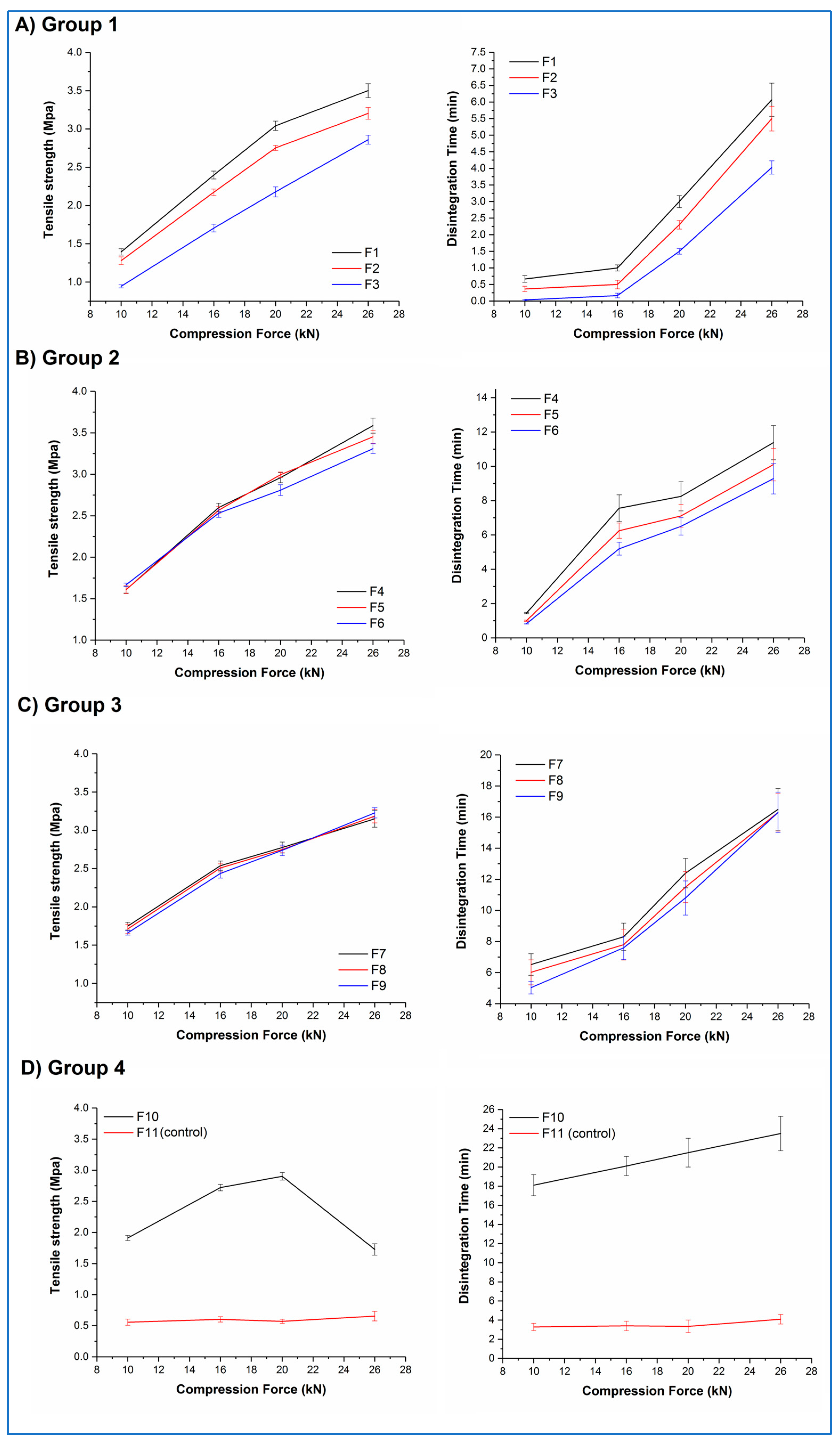
Figure 9A–D shows the tensile strength–compression force and disintegration time–compression force curves for tablets from all 11 formulations.
Both F10 and F11 mixtures stuck to the tablet punches during compression, illustrating the adhesive nature of their major constituents. F11 tablets showed low tensile strengths (circa 0.6 MPa) and correspondingly short disintegration times (circa 3.5 min) at all compression forces, along with lamination during the tensile strength testing. In contrast, F10 tablets showed an increase in tensile strength as a function of compression force up to 20 kN (ranging from 1.9 to 2.9 MPa), then a decrease at 26 kN, along with lamination during testing at this compression force. No significant differences (p > 0.05) in the disintegration time were observed for F10 tablets, with all tablets disintegrating in 20 to 23 min. All F11 tablet batches and the F10 tablet batch showing lamination failed the pharmacopoeial friability test; all other F10 tablet batches passed this test.
Within Groups 1, 2, and 3, a roughly linear increase in tablet tensile strength with increasing compression force was observed, with a concomitant increase in disintegration time. A closer inspection of the data highlights that both the percentage of aged microfibres and the ratio of the excipients affected the behaviour of the tablets. Within each group, tablets with the lowest relative content of StarTab® (F1, F4, F7) generally showed numerically greater values of tensile strength than the formulations with the middle (F2, F5, F8) and highest (F3, F6, F9) relative content of this excipient, although the differences were statistically significant (p < 0.05) only for Group 1 formulations at all compression forces and Group 2 formulations at the highest compression forces. A similar pattern was observed with the disintegration times. Comparing between groups, increasing the aged microfibre content from 10% w/w to 30% w/w resulted in an increase in tensile strength and disintegration time for formulations compressed at the same force and with the same excipient ratio. For example, F2 (Group 1) tablets compressed at 16 kN showed mean values of 2.1 MPa and 30 s, respectively, whereas the mean values for F5 (Group 2) tablets were significantly higher (p < 0.05) at 2.6 MPa and 6.2 min. However, as the microfibre content increases still further to 50% w/w, the differences become less significant. For example, F4 (Group 2) tablets compressed at 20 kN show statistically (p > 0.05) similar mean tensile strength and disintegration time values (2.9 MPa and 8.2 min, respectively) compared with the equivalent F7 (Group 3) tablets (2.8 MPa and 10.2 min, respectively). No sticking or lamination was observed for any of these formulations (F1 to F9), and all batches passed the pharmacopoeial friability test.
These results suggest that, although it is possible to directly compress the aged microfibres into tablets, a more considered formulation approach is required to produce robust tablets. A complex relationship exists between the formulation components in terms of the effect on the tensile strength and dissolution time of the resultant tablets, but the dominant formulation factor appears to be the content of the aged microfibres, with the excipient ratio playing a smaller role, most obviously observed at lower microfibre contents when the excipient content is correspondingly greater. The effect of increasing the compression force is predictable in that increasing compression force leads to increased tensile strength and disintegration time. However, these findings also demonstrate that it is possible to fine-tune the tensile strength and disintegration time of the tablets by varying the microfibre content, excipient ratios, and compression forces, potentially allowing the development of tablets for different purposes.
3.7. Tablet Development—Non-Sink Dissolution Testing
Formulations F3 and F9 were chosen for the non-sink dissolution study, representing the extremes of microfibre content, tensile strength, and disintegration time and having the same excipient ratio. Replicate formulations, labelled as F3* and F9*, containing the aged 30%
w/
w itraconazole-loaded microfibres were also studied. This sample selection allows investigation of the effects of both drug content and rate of supersaturation generation on the overall dissolution performance of the tablets.
Table 3 shows the characterisation data of all these batches.
Drug content uniformity and weight uniformity data were excellent, with all tablet batches clearly passing the relevant pharmacopoeial specifications. No significant differences (p > 0.05) were observed in any test between equivalent batches varying only in the drug content, i.e., F3/F3* and F9/F9*. This is a significant result, as it demonstrates that the microfibres show the same mechanical and formulation behaviour irrespective of drug content in the range of 10 to 30% w/w.
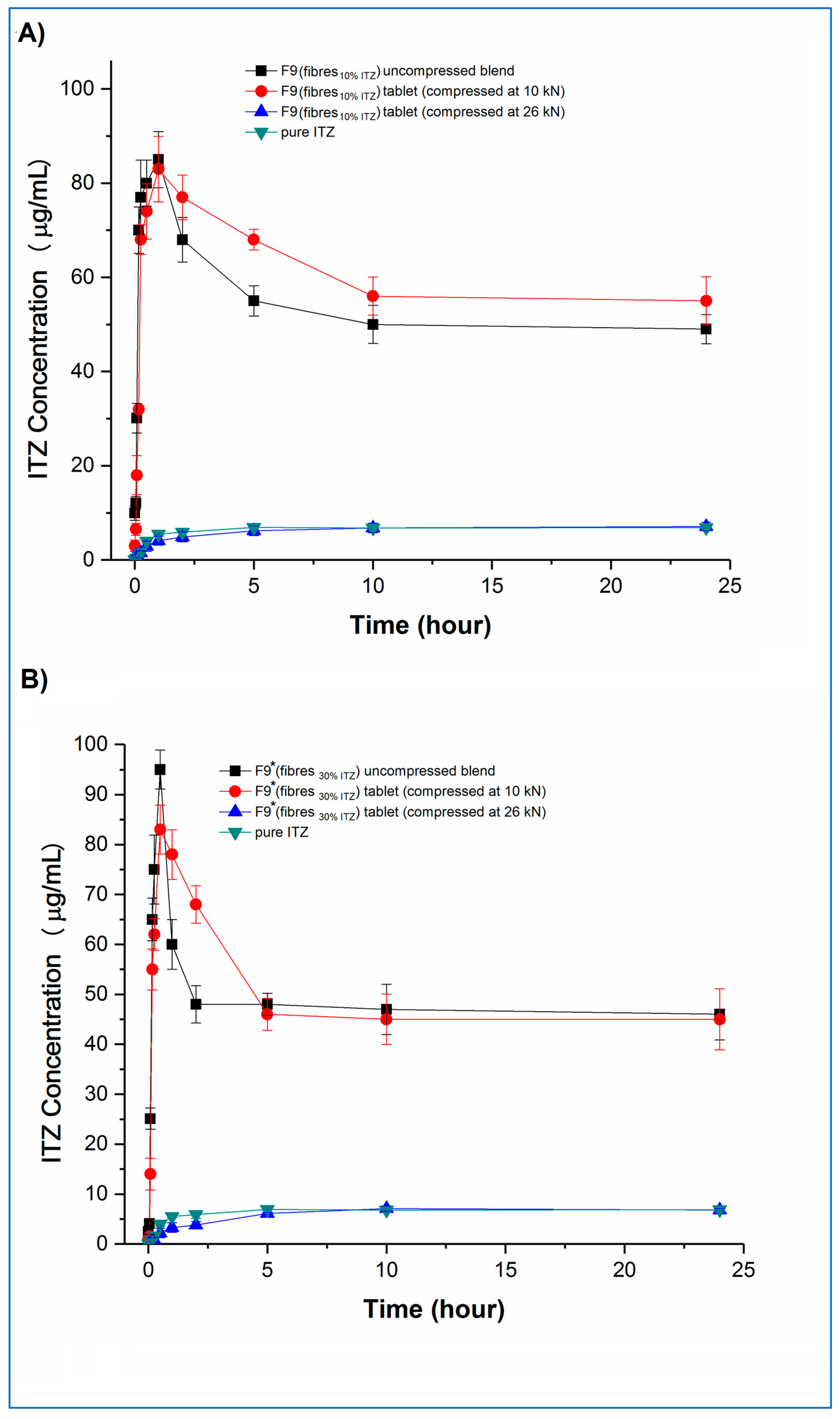
Concentration–time profiles, generated under the same non-sink conditions as used earlier for the microfibres, of the tablets and the corresponding uncompressed blends and pure crystalline itraconazole are shown in
Figure 10 (F3 and F3*) and
Figure 11 (F9 and F9*), respectively.
Examining the fast-disintegrating F3 formulations first, the uncompressed microfibres and the tablets compressed at 10, 16, and 20 kN showed broadly equivalent dissolution performance, achieving and maintaining supersaturation (Cmax) at levels of approximately 20 µg/mL, i.e., approximately 2.8-fold higher than the pure crystalline drug solubility, although there was a trend towards slightly lower rates of drug dissolution from the tablets as the compression force increased. Tablets compressed at the highest compression force (26 kN) did not show supersaturation, and the corresponding dissolution profiles were similar to those of the pure crystalline drug. F3* microfibres and tablets showed similar dissolution behaviour to the equivalent F3 microfibres and tablets in relation to the effects of compression force and supersaturation generation. However, for F3* tablets compressed at forces up to and including 20 kN, some drug precipitation occurred shortly after the initial high values of Cmax (in the range of 58 to 68 µg/mL) were attained, leading to the maintenance supersaturation levels being approximately 50 µg/mL, i.e., approximately 7.1-fold higher than the pure crystalline drug solubility and approximately 25% higher than the solubility of amorphous itraconazole established earlier. There was a trend toward lower peak Cmax levels and lower maintenance supersaturation levels with increasing compression force. The plateau supersaturation levels obtained for the F3* tablets, which contain 9 mg itraconazole, were approximately 2.5-fold higher than those for the F3* tablets, which contain 3 mg itraconazole, demonstrating that the dissolution advantage of the microfibres is preserved at the higher drug loading, even if there is not a completely linear relationship between drug loading and plateau drug concentrations.
Both F9 and F9* tablets compressed at 26 kN force showed dissolution profiles indistinguishable from the raw crystalline drug. F9 uncompressed microfibres and tablets compressed at 10 kN both showed an initial fast dissolution and high C
max (approximately 85 and 83 µg/mL, respectively), with subsequent precipitation and stabilisation of supersaturation levels at approximately 50 and 56 µg/mL, respectively. The higher drug-loaded F9* formulation showed similar behaviour. In this case, the C
max values were approximately 83 and 95 µg/mL, respectively, for the uncompressed microfibres and tablets compressed at 10 kN, with the supersaturation concentration plateauing for both products at approximately 48 µg/mL. Closer inspection of the data reveals that, for both F9 and F9*, the tablets compressed at 10 kN showed significantly (
p < 0.05) lower dissolution at the very early stages of the experiment, up to 15 min for F9 and 30 min for F9*, than the corresponding powder blend, which may be attributable to the 5 min disintegration time for these tablets reducing the initial rate of dissolution. This slower buildup of supersaturation is then responsible for the retention of the higher supersaturation concentrations in the tablets compared with the powder blends, following the model of Han and Lee [
37].
Taken together, these results indicate that the rate and extent of supersaturation can be controlled by varying the drug content of the formulation and the disintegration time of the tablets, which will then affect the total amount of drug available for absorption in the intestine. The overall effect of this may be quantified by calculating the 24 h AUC values of the dissolution–supersaturation profiles: values (mean ± sd) of 480 ± 14, 1223 ± 18, 1448 ± 44 and 1178 ± 36 µg/mL.h were observed for F3, F3*, F9, and F9* tablets compressed at 10 kN, respectively, with the equivalent value for crystalline itraconazole being 158 ± 4 µg/mL.h. The 3-fold increase in overall drug content between F3 and F3*, achieved via an increase in the drug loading of the microfibres (10 and 30% w/w), led to a 2.5-fold increase in both final sustained concentration and AUC. However, a similarly derived 3-fold itraconazole content increase between F9 and F9* did not result in a similar dissolution advantage, and indeed, a lower AUC was observed for the F9* tablets, indicating that, above a certain limit, increasing the drug content has no effect on the final dissolution profile. The 5-fold increase in overall drug content between F3 and F9, achieved via an increase in the microfibre content of the tablets (10 and 50% w/w), led to an almost 3-fold increase in both final sustained concentration and AUC. Conversely, the similarly derived 5-fold itraconazole content increase between F3* and F9* tablets resulted in a slight decrease in overall AUC, again suggesting that there is a maximal effect of increasing the content of the drug. This dose effect is attributable to the initial supersaturated concentrations exceeding the critical value whereby the energy barrier to recrystallisation is sufficiently low to allow precipitation to occur. From these data, F9 would appear to be the most beneficial formulation. However, the absorption site for itraconazole is the small intestine, which has a commonly accepted transit time of 3 to 5 h. Therefore, considering only the first five hours of the dissolution experiment to simulate the likely absorption window, the corresponding AUC (mean ± SD) values for F3, F3*, F9, F9* tablets, and crystalline drug are 103 ± 5, 272 ± 9, 361 ± 14, 307 ± 12, and 18 ± 2 µg/mL∙h, confirming that F9 is the best of the current formulations.
3.8. Tablet Development—Physical State of the Drug in the Compressed Tablets
The non-sink dissolution study highlighted that all tablet batches compressed at 26 kN showed dissolution profiles indistinguishable from the raw crystalline itraconazole, suggesting that compression at this force had resulted in the recrystallisation of the amorphous itraconazole in the microfibres. This was confirmed by
13C CP/MAS SSNMR analysis.
Figure 12 shows the responses of F3 and F9* tablets, with the lowest (1%
w/
w) and highest (15%
w/
w) itraconazole content, respectively, compressed at 10 and 26 kN, and the peaks attributable to crystalline and amorphous drug [
4] shown by arrows. All tablets compressed at 10, 16, and 20 kN showed the amorphous response, and all tablets compressed at 26 kN showed the crystalline response, indicating that the recrystallisation process is compression force dependent, with the critical force being between 20 and 26 kN. These results highlight the need to understand the effect of process variables on the physical structure and behaviour of the formulation and, by extension, the likely effect on the biological performance of the drug.
4. General Discussion and Future Perspectives
Building on our previous work [
3,
4], this study has shown that it is possible to prepare fully amorphous, one-phase itraconazole-loaded sucrose microfibres using melt centrifugal spinning, up to a drug content of 30%
w/
w. At 50%
w/
w drug loading, phase separation was seen, including the generation of pure itraconazole microfibres in the amorphous state. Fresh microfibres with up to 30%
w/
w itraconazole showed similar non-sink dissolution behaviour to that previously observed for the 10%
w/
w drug-loaded samples, i.e., a fast initial dissolution with high supersaturated concentrations being generated and subsequent partial drug precipitation with lower supersaturation concentrations being maintained for up to 24 h. Exposure for 30 days to 25 °C/75% RH resulted in the recrystallisation of the sucrose component of the microfibres, but the itraconazole remained in the amorphous state. The dissolution–supersaturation advantage seen in the fresh samples was maintained and even enhanced in the aged systems, ascribed to the slower initial dissolution of crystalline sucrose preventing a too-rapid rise in supersaturation and hence reducing the extent of precipitation thereafter.
An in-depth tablet formulation development study was carried out using aged itraconazole-loaded microfibres and commonly used direct compression tabletting excipients. To enhance mixing and prevent segregation, a pre-mixing step of the microfibres with StarTab
®, a partially pregelatinised starch, was necessary prior to subsequent mixing with the remainder of the excipients, lubrication, and compression. The content of the aged microfibres was observed to have the greatest influence on the physical behaviour of the tablets, with the excipient mix playing a lesser role. Non-sink dissolution studies showed that the dissolution–supersaturation advantage of the itraconazole-loaded (10 and 30%
w/
w) aged microfibres was maintained in the tablet formulations containing 10%
w/
w microfibres and even enhanced for a microfibre content of 50%
w/
w in the final tablets when tablets were compressed at low and medium forces. However, compression at the highest force resulted in the recrystallisation of the amorphous drug, and the dissolution profiles became indistinguishable from those of the crystalline raw drug. All tablet batches tested for dissolution passed all relevant pharmacopoeial tests, with excellent drug content uniformity and weight uniformity being demonstrated. Tablet formulation F9, containing 50%
w/
w microfibres composed of 10%
w/
w itraconazole, showed the greatest AUC advantage when measured over 5 h to simulate intestinal transit and itraconazole’s likely absorption window, or 24 h to simulate transit through the entire gastro-intestinal tract. This was attributed to a relatively slow rate of supersaturation generation and a subsequent slow precipitation rate, which led to a high maintenance supersaturation level. The lack of further increase in C
max or AUC seen with the microfibres and tablets with the highest drug loading (F9* compared with F9) demonstrates the overall limit of supersaturation that may be achieved with this approach and this drug. Additionally, it allows a rough estimation of the critical supersaturation concentration, above which drug precipitation occurs rapidly, as described by Han and Lee [
37]. This value can be estimated here as being in the range of 90 to 100 µg/mL.
F9 and F9* tablets, which contain 50% w/w microfibres, showed longer disintegration times (around 5 min) compared with their equivalent formulations containing 10% w/w microfibres (a few seconds), which was thought to contribute to the slower onset of supersaturation. Extending this approach and developing a tablet with an even longer disintegration time may then lead to greater overall dissolution performance. In this case, the rate of supersaturation generation and hence the potential for drug precipitation would be expected to be reduced further while still maintaining the overall dissolution advantage of the amorphous state of the drug and the increase in solubility due to the presence of sucrose in the microfibres. This may be achieved by reducing the content of disintegrant in the tablet formulation. Increasing the compression force, which would also be expected to increase disintegration time and slow the initial dissolution rate, carries the risk of causing the in situ recrystallisation of the amorphous itraconazole, thus losing all benefit of this formulation approach.
From a commercial perspective, the potential scalability and speed of the centrifugal melt spinning process are important considerations. On a lab scale, 10 g of powder mix can be converted into microfibres in approximately 5 min once the equipment is at the correct temperature. The spinning process has the potential to be made into a (semi)-continuous process, as long as there is a balance between feeding of the starting materials (the powder mix) and collection of the product (the microfibres); hence, it is potentially scalable into a commercially viable process. However, as there is a need to humidity-treat the microfibres prior to formulation into tablets, the overall process will need to remain a batch process.



 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}