
On the Use of Temperature Measurements as a Process Analytical Technology (PAT) for the Monitoring of a Pharmaceutical Freeze-Drying Process





Abstract
:1. Introduction
- to monitor product temperature, as it must remain below the threshold value to preserve final product quality. Usually, both product temperature at the bottom of the vial (TB) and that at the interface of sublimation (Ti) should be known.
- to identify the occurrence of the ending point of the primary drying stage, corresponding to the time instant when no more ice is present or the thickness of the frozen layer (Lfrozen) approaches zero, if a flat and planar interface is assumed.
- to estimate the value of the parameters of a mathematical model of the process. The heat transfer coefficient (Kv) and the resistance to mass transfer (Rp), which allow us to describe, respectively, the heat transfer to the product and the mass transfer from the product to the drying chamber, are generally selected. They can be used to predict the effect of the operating conditions and thus to optimize the process, minimizing the duration of the drying stage [14,15].
2. Procedure for Process Monitoring Using Temperature Measurement
2.1. Determination of the End of Primary Drying
2.2. Identification of Process Parameters
- (a)
- The value of the coefficient Kv can be calculated in a preliminary experiment using the measurement of product temperature. In fact, from the integral energy balance for the frozen product, where Kv can be obtained as the ratio of the total energy supplied to the sample and then used to sublimate the initial mass m of ice:
- (b)
- The cake resistance Rp and its variation with Ldried can be calculated using the Kv value calculated in the previous step. The proposed algorithm is the following:
- The heat flux Jq to the product is calculated with Equation (2),using the estimated value of Kv and the measured values of Tshelf and of the temperature of the product at the bottom of the vial (TB), whose difference represents the driving force.
- The sublimation flux Jw is then calculated using Equation (3),the energy balance at the interface of sublimation, considering that in steady-state the heat flow is equivalent to the mass flow times the heat of ice sublimation ∆Hs.
- The cake resistance can be calculated from the sublimation flux, which, similarly to the heat flux, can be described as the product of a mass transfer coefficient (1/Rp) times a driving force (the difference between water vapor partial pressure at different locations: the interface and the chamber average):During primary drying, it can be assumed that pw,c is equal to chamber pressure (the fraction of air or inert gas is generally negligible), while the water partial pressure at the interface, pw,i, can be calculated: the interface temperature is not known, but TB is measured, and Ti can be estimated by iteratively repeating steps (iii) to (v), neglecting the temperature gradient in the vial as a first attempt, or taking a value (1÷2) °C lower than TB on the basis of the experience.
- The sublimation flux in the time interval considered also allows us to estimate the evolution of Lfrozen (and thus, as difference to total thickness, of Ldried) using a mass balance to the frozen layer:and thus, to relate Rp to Ldried for each time interval. Integrating Equation (5), it is also possible to monitor the progress of the drying process in-line.
- The temperature at the interface can be estimated precisely from the product temperature at the bottom of the container, which is the variable usually measured with thermocouples inserted in vials, considering the heat balance for the frozen product and previous relationships:This, once Lfrozen has been calculated from (iv), allows us to calculate pw,i with greater accuracy, and then to calculate Rp using Equation (4).
- The values of Rp depend on the type of product (and freezing history), but also on the thickness of the dried product, as shown before. To model this dependence, an equation like the following one is usually adopted:
3. Experimental Set-Up
3.1. Experimental Apparatus
3.2. Case Study
4. Results and Discussions
4.1. Evaluation of PAT Based on Thermocouples
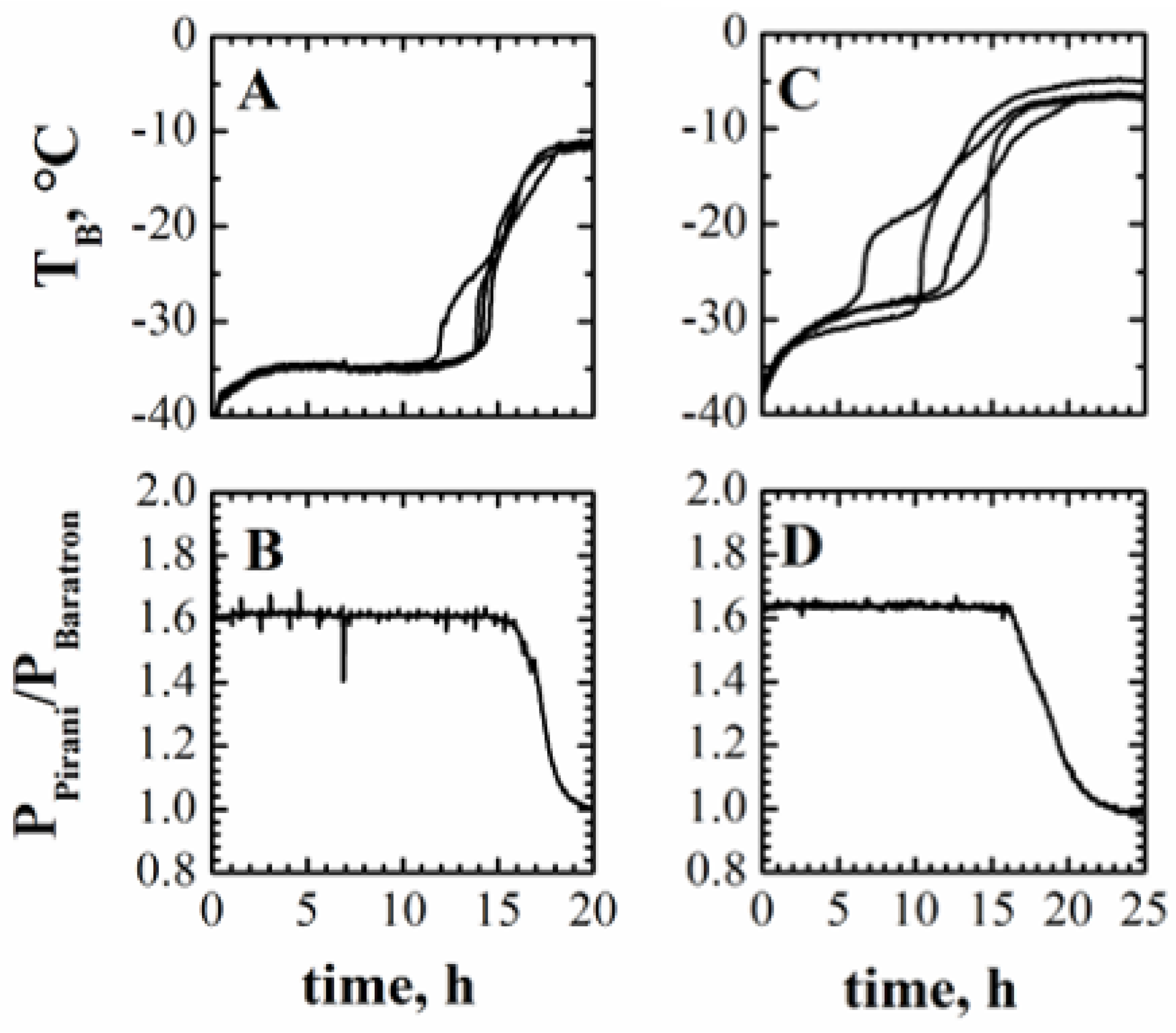
4.1.1. End of Primary Drying
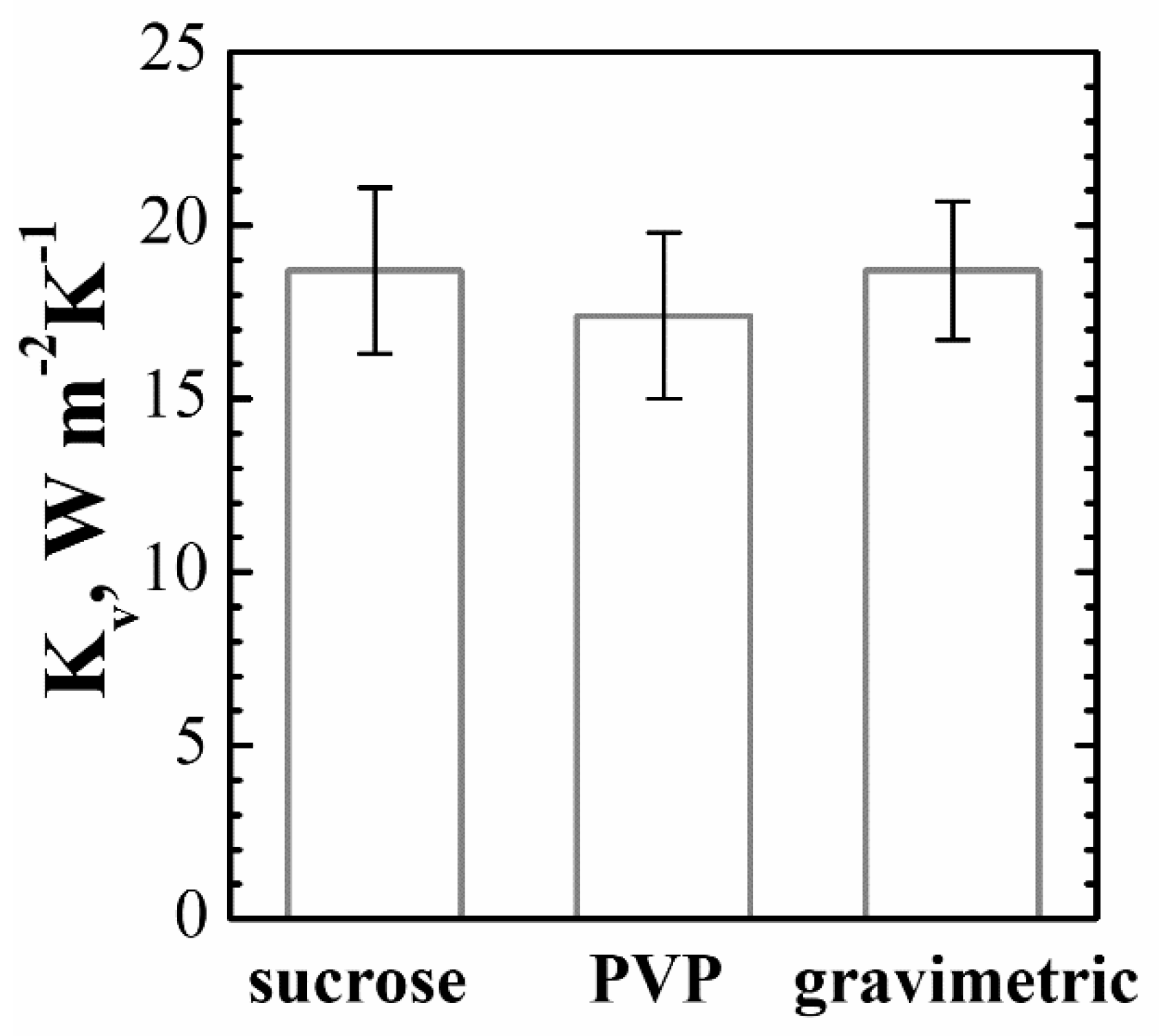
4.1.2. Kv Estimation and Uncertainty
- The drying time t1 is defined by the user, and its uncertainty is negligible.
- A set of about 100 vials was identified as representative of the full batch. The vials were weighed before and after the test, thus obtaining a set of Δm measurements and, with Equation (1b), a set of Kv coefficients. The mean value is 18.7 W·m−2·K−1 and the dispersion can be treated using a Type-A method, thus obtaining a standard deviation of about 2 W·m−2·K−1 and a standard uncertainty, that is, the standard deviation of the mean value, of about uA(vial) = 0.2 W·m−2·K−1.
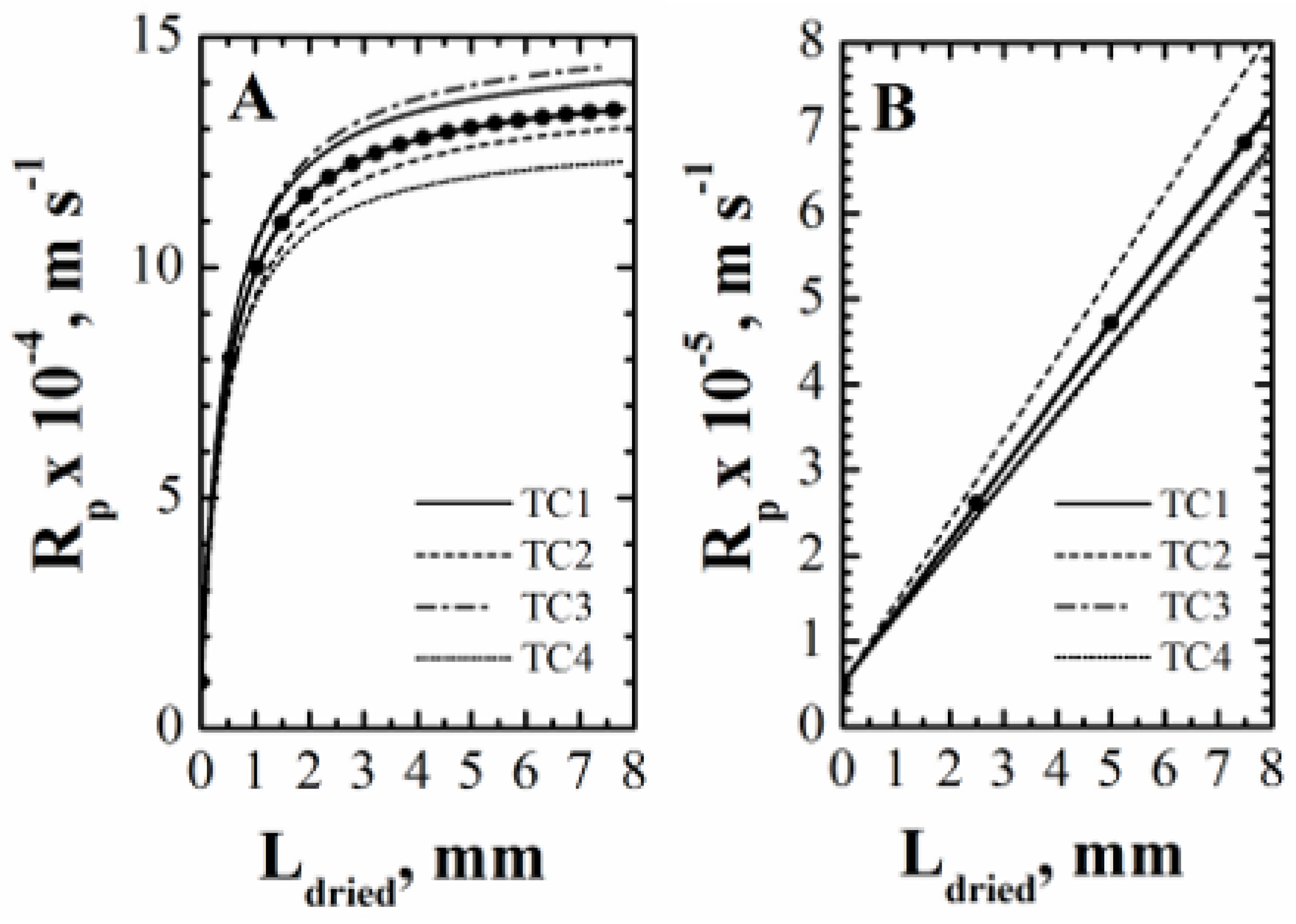
4.1.3. Rp Estimation and Monitoring of Primary Drying Progress
- estimate the values of Rp,0, , and from the curve of Rp vs. Ldried obtained from the first temperature measurement;
- use the previously obtained values of Rp,0 and to calculate the value of in such a way that the data of Rp vs. Ldried obtained from the other temperature measurements can be best-fitted;
- calculate the mean value of and its standard deviation.
4.2. Strengths and Weaknesses of Thermocuples for Pharmaceutical Applications in Comparison with a Contactless Device (IR Camera)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Symbols
| fitting parameter for cake resistance relationship in Equation (7) | |
| Av | cross-section area of the vial |
| fitting parameter for cake resistance relationship in Equation (7) | |
| ∆Hs | heat of ice sublimation |
| Jq | heat flux to the product |
| Jw | sublimation flux |
| kfrozen | thermal conductivity of the frozen layer |
| Kv | heat transfer coefficient |
| L | thickness of the product |
| Ldried | thickness of the dried product |
| Lfrozen | thickness of the frozen layer |
| m | mass of ice in the vial |
| ∆m | variation of ice by sublimation in the test |
| pw,i | water vapor partial pressure at the interface of sublimation |
| pw,c | water vapor partial pressure in the drying chamber |
| Rp | resistance to mass transfer |
| Rp,0 | fitting parameter for cake resistance relationship (7) |
| sensitivity coefficient of the output quantity y with respect to the input quantity x evaluated at the measurement values | |
| TB | product temperature at the bottom of the vial |
| Ti | product temperature at the interface of sublimation |
| Tshelf | shelf (or fluid) temperature |
| t | time |
| tdrying | time required to complete the ice sublimation |
| U | expanded uncertainty |
| uA(x) | standard uncertainty of the quantity x evaluated with type-A method |
| uB(x) | standard uncertainty of the quantity x evaluated with type-B method |
| uc(x) | combined uncertainty of the quantity x |
| vi | interface retreating velocity |
| ρdried | density of the dried product |
| ρfrozen | density of the frozen product |
References
- Jennings, T.A. Lyophilization: Introduction and Basic Principles; Interpharm/CRC Press: Boca Raton, FL, USA, 1999. [Google Scholar]
- Oetjen, G.W.; Haseley, P. Freeze-Drying, 2nd ed.; Wiely-VHC: Weinheim, Germany, 2004. [Google Scholar]
- Franks, F.; Auffret, T. Freeze-Drying of Pharmaceuticals and Biopharmaceuticals; The Royal Society of Chemistry: London, UK, 2007. [Google Scholar] [CrossRef]
- Fissore, D. Freeze Drying of Pharmaceuticals. In Encyclopedia of Pharmaceutical Science and Technology; CRC Press: Boca Raton, FL, USA, 2013; pp. 1723–1737. [Google Scholar] [CrossRef]
- Bellows, R.J.; King, C. Freeze-drying of aqueous solutions: Maximum allowable operating temperature. Cryobiology 1972, 9, 559–561. [Google Scholar] [CrossRef] [PubMed]
- Pikal, M.J. Freeze-drying of proteins: Process, formulation, and stability. In Formulation and Delivery of Proteins and Peptides; Cleland, J.L., Langer, R., Eds.; American Chemical Society: Washington DC, USA, 1994; pp. 120–133. [Google Scholar]
- Franks, F. Freeze-drying of bioproducts: Putting principles into practice. Eur. J. Pharm. Biopharm. 1998, 45, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mutukuri, T.T.; Wilson, N.E.; Zhou, Q. Pharmaceutical protein solids: Drying technology, solid-state characterization and stability. Adv. Drug Deliv. Rev. 2021, 172, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Pikal, M.J. Stabilization of lyophilized pharmaceuticals by process optimization: Challenges and opportunities. Am. Pharm. Rev. 2012, 15, 82. Available online: http://www.americanpharmaceuticalreview.com/Featured-Articles/122325-Stabilization-of-Lyophilized-Pharmaceuticals-by-Process-Optimization-Challenges-and-Opportunities/ (accessed on 10 February 2023).
- Tsourouflis, S.; Flink, J.M.; Karel, M. Loss of structure in freeze-dried carbohydrates solutions: Effect of temperature, moisture content and composition. J. Sci. Food Agric. 1976, 27, 509–519. [Google Scholar] [CrossRef]
- Depaz, R.A.; Pansare, S.; Patel, S.M. Freeze-drying above the glass transition temperature in amorphous protein formulations while maintaining product quality and improving process efficiency. J. Pharm. Sci. 2016, 105, 40–49. [Google Scholar] [CrossRef]
- Patel, S.M.; Nail, S.L.; Pikal, M.J.; Geidobler, R.; Winter, G.; Hawe, A.; Davagnino, J.; Gupta, S.R. Lyophilized drug product cake appearance: What is acceptable? J. Pharm. Sci. 2017, 106, 1706–1721. [Google Scholar] [CrossRef]
- Johnson, R.; Lewis, L. Freeze-drying protein formulations above their collapse temperatures: Possible issues and concerns. Am. Pharm. Rev. 2011, 14, 50–54. Available online: http://www.americanpharmaceuticalreview.com/images/article/7_Johnson.pdf (accessed on 10 February 2023).
- Barresi, A.A.; Fissore, D. In-line product quality control of pharmaceuticals. In Freeze-Drying Processes; Wiley: Hoboken, NJ, USA, 2011; pp. 91–154. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A.A. Using mathematical modeling and prior knowledge for QbD. In Freeze-Drying Processes; Wiley: Hoboken, NJ, USA, 2015; pp. 565–593. [Google Scholar] [CrossRef]
- Food and Drug Administration. Guidance for Industry PAT—A Framework for Innovative Pharmaceutical Manufacturing and Quality Assurance. 2004. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm070305.pdf (accessed on 10 February 2023).
- Barresi, A.; Pisano, R.; Fissore, D.; Rasetto, V.; Velardi, S.A.; Vallan, A.; Parvis, M.; Galan, M. Monitoring of the primary drying of a lyophilization process in vials. Chem. Eng. Process. Process. Intensif. 2009, 48, 408–423. [Google Scholar] [CrossRef] [Green Version]
- Nail, S.; Tchessalov, S.; Shalaev, E.; Ganguly, A.; Renzi, E.; Dimarco, F.; Wegiel, L.; Ferris, S.; Kessler, W.; Pikal, M.; et al. Recommended best practices for process monitoring instrumentation in pharmaceutical freeze drying—2017. AAPS PharmSciTech 2017, 18, 2379–2393. [Google Scholar] [CrossRef] [Green Version]
- Fissore, D.; Pisano, R.; Barresi, A.A. Process analytical technology for monitoring pharmaceuticals freeze-drying—A comprehensive review. Dry. Technol. 2018, 36, 1839–1865. [Google Scholar] [CrossRef]
- Patel, S.M.; Doen, T.; Pikal, M.J. Determination of end point of primary drying in freeze-drying process control. AAPS PharmSciTech 2010, 11, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisano, R. Automatic control of a freeze-drying process: Detection of the end point of primary drying. Dry. Technol. 2022, 40, 140–157. [Google Scholar] [CrossRef]
- Milton, N.; Pikal, M.J.; Roy, M.L.; Nail, S.L. Evaluation of manometric temperature measurement as a method of monitoring product temperature during lyophilization. PDA J. Pharm. Sci. Technol. 1997, 51, 7–16. [Google Scholar]
- Chouvenc, P.; Vessot, S.; Andrieu, J.; Vacus, P. Optimization of the freeze-drying cycle: A new model for pressure rise analysis. Dry. Technol. 2004, 22, 1577–1601. [Google Scholar] [CrossRef]
- Velardi, S.A.; Rasetto, V.; Barresi, A.A. Dynamic Parameters Estimation method: Advanced Manometric Temperature Measurement approach for freeze-drying monitoring of pharmaceutical solutions. Ind. Eng. Chem. Res. 2008, 47, 8445–8457. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A. On the methods based on the Pressure Rise Test for monitoring a freeze-drying process. Dry. Technol. 2011, 29, 73–90. [Google Scholar] [CrossRef] [Green Version]
- Pisano, R.; Ferri, G.; Fissore, D.; Barresi, A.A. Freeze-drying monitoring via Pressure Rise Test: The role of the pressure sensor dynamics. In Proceedings of the Instrumentation and Measurement Technology Conference—IMTC 2017, Torino, Italy, 22–25 May 2017; pp. 1282–1287. [Google Scholar] [CrossRef]
- Schneid, S.C.; Gieseler, H.; Kessler, W.J.; Pikal, M.J. Non-invasive product temperature determination during primary drying using Tunable Diode Laser Absorption Spectroscopy. J. Pharm. Sci. 2009, 98, 3406–3418. [Google Scholar] [CrossRef]
- Rasetto, V.; Marchisio, D.L.; Fissore, D.; Barresi, A.A. On the use of a dual-scale model to improve understanding of a pharmaceutical freeze-drying process. J. Pharm. Sci. 2010, 99, 4337–4350. [Google Scholar] [CrossRef]
- Barresi, A.A.; Pisano, R.; Fissore, D. Advanced control in freeze-drying. In Intelligent Control in Drying; Martynenko, A., Bück, A., Eds.; CRC Press: Boca Raton, FL, USA, 2018; Chapter 19; pp. 367–401. [Google Scholar] [CrossRef]
- Vollrath, I.; Pauli, V.; Friess, W.; Freitag, A.; Hawe, A.; Winter, G. Evaluation of heat flux measurement as a new process analytical technology monitoring tool in freeze drying. J. Pharm. Sci. 2017, 106, 1249–1257. [Google Scholar] [CrossRef]
- Schneid, S.; Gieseler, H. Evaluation of a new wireless Temperature Remote Interrogation System (TEMPRIS) to measure product temperature during freeze drying. AAPS PharmSciTech 2008, 9, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Kasper, J.C.; Wiggenhorn, M.; Resch, M.; Friess, W. Implementation and evaluation of an optical fiber system as novel process monitoring tool during lyophilization. Eur. J. Pharm. Biopharm. 2013, 83, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Bosca, S.; Barresi, A.A.; Fissore, D. Use of a soft sensor for the fast estimation of dried cake resistance during a freeze-drying cycle. Int. J. Pharm. 2013, 451, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassini, S.; Parvis, M.; Barresi, A.A. Inert thermocouple with nanometric thickness for lyophilization monitoring. IEEE Trans. Instrum. Meas. 2013, 62, 1276–1283. [Google Scholar] [CrossRef]
- Parvis, M.; Grassini, S.; Fulginiti, D.; Pisano, R.; Barresi, A.A. Sputtered thermocouple array for vial temperature mapping. In Proceedings of the 2014 IEEE International Instrumentation and Measurement Technology Conference (I2MTC) Proceedings, Montevideo, Uruguay, 12–15 May 2014; pp. 1465–1470. [Google Scholar] [CrossRef]
- Oddone, I.; Fulginiti, D.; Barresi, A.A.; Grassini, S.; Pisano, R. Non-invasive temperature monitoring in freeze drying: Control of freezing as a case study. Dry. Technol. 2015, 33, 1621–1630. [Google Scholar] [CrossRef]
- Jiang, X.; Kazarin, P.; Sinanis, M.D.; Darwish, A.; Raghunathan, N.; Alexeenko, A.; Peroulis, D. A non-invasive multipoint product temperature measurement for pharmaceutical lyophilization. Sci. Rep. 2022, 12, 12010. [Google Scholar] [CrossRef]
- Corbellini, S.; Parvis, M.; Vallan, A. In-process temperature mapping system for industrial freeze dryers. IEEE Trans. Instrum. Meas. 2010, 59, 1134–1140. [Google Scholar] [CrossRef]
- Bosca, S.; Corbellini, S.; Barresi, A.A.; Fissore, D. Freeze-drying monitoring using a new Process Analytical Technology: Toward a “zero defect” process. Dry. Technol. 2013, 31, 1744–1755. [Google Scholar] [CrossRef]
- Emteborg, H.; Zeleny, R.; Charoud-Got, J.; Martos, G.; Lüddeke, J.; Schellin, H.; Teipel, K. Infrared thermography for monitoring of freeze-drying processes: Instrumental developments and preliminary results. J. Pharm. Sci. 2014, 103, 2088–2097. [Google Scholar] [CrossRef]
- Emteborg, H.; Charoud-Got, J.; Seghers, J. Infrared thermography for monitoring of freeze drying processes—Part 2: Monitoring of temperature on the surface and vertically in cuvettes during freeze drying of a pharmaceutical formulation. Pharmaceutics 2022, 14, 1007. [Google Scholar] [CrossRef]
- Van Bockstal, P.-J.; Corver, J.; De Meyer, L.; Vervaet, C.; De Beer, T. Thermal imaging as a noncontact inline Process Analytical Tool for product temperature monitoring during continuous freeze-drying of unit doses. Anal. Chem. 2018, 90, 13591–13599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lietta, E.; Colucci, D.; Distefano, G.; Fissore, D. On the use of infrared thermography for monitoring a vial freeze-drying process. J. Pharm. Sci. 2019, 108, 391–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colucci, D.; Morra, L.; Zhang, X.; Fissore, D.; Lamberti, F. An automatic computer vision pipeline for the in-line monitoring of freeze-drying processes. Comput. Ind. 2020, 115, 103184. [Google Scholar] [CrossRef]
- Harguindeguy, M.; Fissore, D. Temperature/end point monitoring and modelling of a batch freeze-drying process using an infrared camera. Eur. J. Pharm. Biopharm. 2021, 158, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Fissore, D.; Pisano, R.; Barresi, A.A. On the use of temperature measurement to monitor a freeze-drying process for pharmaceuticals. In Proceedings of the 2017 IEEE International Instrumentation and Measurement Technology Conference (I2MTC), Turin, Italy, 22–25 May 2017; pp. 1276–1281. [Google Scholar] [CrossRef]
- Velardi, S.A.; Barresi, A.A. Development of simplified models for the freeze-drying process and investigation of the optimal operating conditions. Chem. Eng. Res. Des. 2008, 86, 9–22. [Google Scholar] [CrossRef]
- Giordano, A.; Barresi, A.A.; Fissore, D. On the use of mathematical models to build the design space for the primary drying phase of a pharmaceutical lyophilization process. J. Pharm. Sci. 2011, 100, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Fissore, D.; Pisano, R.; Barresi, A.A. Advanced approach to build the design space for the primary drying of a pharmaceutical freeze-drying process. J. Pharm. Sci. 2011, 100, 4922–4933. [Google Scholar] [CrossRef]
- Pisano, R.; Fissore, D.; Velardi, S.A.; Barresi, A. In-line optimization and control of an industrial freeze-drying process for pharmaceuticals. J. Pharm. Sci. 2010, 99, 4691–4709. [Google Scholar] [CrossRef]
- Fissore, D.; Pisano, R.; Barresi, A.A. Model-based framework for the analysis of failure consequences in a freeze-drying process. Ind. Eng. Chem. Res. 2012. [Google Scholar] [CrossRef]
- Oddone, I.; Van Bockstal, P.-J.; De Beer, T.; Pisano, R. Impact of vacuum-induced surface freezing on inter- and intra-vial heterogeneity. Eur. J. Pharm. Biopharm. 2016, 103, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Capozzi, L.C.; Pisano, R. Looking inside the ‘black box’: Freezing engineering to ensure the quality of freeze-dried biopharmaceuticals. Eur. J. Pharm. Biopharm. 2018, 129, 58–65. [Google Scholar] [CrossRef] [PubMed]
- BIPM, IEC, IFCC, ILAC, ISO, IUPAC, IUPAP, OIML. Evaluation of Measurement Data – Guide to the Expression of Uncertainty in Measurement, JCGM 100:2008. Available online: https://www.bipm.org/documents/20126/2071204/JCGM_100_2008_E.pdf/cb0ef43f-baa5-11cf-3f85-4dcd86f77bd6 (accessed on 10 February 2023).
- ISO/IEC Guide 98-3:2008; Uncertainty of Measurement—Part 3: Guide to the Expression of Uncertainty in Measurement (GUM:1995). Available online: https://www.iso.org/standard/50461.html (accessed on 10 February 2023).
- ASTM E2846-20; Standard Guide for Thermocouple Verification. American National Standard ASTM International: West Conshohocken, PA, USA, 2021. [CrossRef]
- International Standard ISO 18434-1:2008; Condition Monitoring and Diagnostics of Machines—Thermography—Part 1: General Procedures. Available online: https://www.iso.org/obp/ui/#iso:std:iso:18434:-1:ed-1:v1:en (accessed on 10 February 2023).
- Colucci, D.; Maniaci, R.; Fissore, D. Monitoring of the freezing stage in a freeze-drying process using IR thermography. Int. J. Pharm. 2019, 566, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Harguindeguy, M.; Stratta, L.; Fissore, D.; Pisano, R. Investigation of the freezing phenomenon in vials using an infrared camera. Pharmaceutics 2021, 13, 1664. [Google Scholar] [CrossRef] [PubMed]
- Demichela, M.; Barresi, A.A.; Baldissone, G. The effect of human error on the temperature monitoring and control of freeze drying processes by means of thermocouples. Front. Chem. 2018, 6, 419. [Google Scholar] [CrossRef]
- Bosca, S.; Barresi, A.A.; Fissore, D. Design of a robust soft-sensor to monitor in-line a freeze-drying process. Dry. Technol. 2015, 33, 1039–1050. [Google Scholar] [CrossRef]
- Harguindeguy, M.; Fissore, D. Micro freeze-dryer and infrared-based PAT: Novel tools for primary drying design space determination of freeze-drying processes. Pharm. Res. 2021, 38, 707–719. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tdrying (h) | Sucrose—Vial | ||||
|---|---|---|---|---|---|
| #1 | #2 | #3 | #4 | ||
| 17 | 20.07 | 19.65 | 20.17 | 19.45 | ±0.36 (±1.8%) |
| 18 | 18.95 | 18.56 | 19.04 | 18.35 | ±0.35 (±1.9%) |
| 19 | 18.04 | 17.66 | 18.13 | 17.82 | ±0.34 (±1.9%) |
() | ±1.0 (±5.4%) | ±1.0 (±5.4%) | ±1.0 (±5.3%) | ±1.0 (±5.4%) | |
| tdrying (h) | PVP—Vial | ||||
|---|---|---|---|---|---|
| #1 | #2 | #3 | #4 | ||
| 17 | 19.34 | 18.69 | 17.93 | 19.13 | ±0.71 (±3.8%) |
| 18.5 | 17.91 | 17.26 | 16.57 | 17.71 | ±0.67 (±3.9%) |
| 20 | 16.64 | 15.98 | 15.36 | 16.44 | ±0.64 (±4.0%) |
() | ±1.4 (±7.5%) | ±1.4 (±7.8%) | ±1.3 (±7.8%) | ±1.3 (±7.6%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallan, A.; Fissore, D.; Pisano, R.; Barresi, A.A. On the Use of Temperature Measurements as a Process Analytical Technology (PAT) for the Monitoring of a Pharmaceutical Freeze-Drying Process. Pharmaceutics 2023, 15, 861. https://doi.org/10.3390/pharmaceutics15030861
Vallan A, Fissore D, Pisano R, Barresi AA. On the Use of Temperature Measurements as a Process Analytical Technology (PAT) for the Monitoring of a Pharmaceutical Freeze-Drying Process. Pharmaceutics. 2023; 15(3):861. https://doi.org/10.3390/pharmaceutics15030861
Chicago/Turabian StyleVallan, Alberto, Davide Fissore, Roberto Pisano, and Antonello A. Barresi. 2023. "On the Use of Temperature Measurements as a Process Analytical Technology (PAT) for the Monitoring of a Pharmaceutical Freeze-Drying Process" Pharmaceutics 15, no. 3: 861. https://doi.org/10.3390/pharmaceutics15030861
APA StyleVallan, A., Fissore, D., Pisano, R., & Barresi, A. A. (2023). On the Use of Temperature Measurements as a Process Analytical Technology (PAT) for the Monitoring of a Pharmaceutical Freeze-Drying Process. Pharmaceutics, 15(3), 861. https://doi.org/10.3390/pharmaceutics15030861