
Hospital Production of Sterile 2% Propofol Nanoemulsion: Proof of Concept
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Propofol Nanoemulsion Production Processes
2.1.1. Extemporaneous Addition Method
2.1.2. “De Novo” Method
2.1.3. Sterilization Methods
2.1.4. Propofol Nanoemulsion Compositions
2.1.5. Physico-Chemical Characterizations
- Visual observation. NEs were visually inspected to detect any oiling out and phase separation.
- Granulometric analysis and zeta potential. The hydrodynamic size was determined using a DLS device from Malvern Instruments (Zetasizer® Nano ZS) in the NE diluted at 1:1500 (v/v). The mean hydrodynamic size was determined from 3 independent measurements performed at 25 °C. Additionally, to assess the lack of droplets above 1µm and 5µm, granulometric analysis was performed using a laser diffraction particle size analyzer (Mastersizer®; Malvern Instruments Ltd., Malvern, UK). Each sample was diluted in water to an appropriate concentration before measurement at 25 °C. Zeta potential measurement was performed on a 1:1500 (v/v) diluted NE sample using a Zetasizer Nano ZS coupled with a Folded Capillary Cell (DTS1060) from Malvern Instruments.
- pH and osmolality. The pH was measured by potentiometry (SevenCompact™ S210, Mettler Toledo, OH, USA). Osmolality was determined by cryoscopy (Löser™ type 15 osmometer, Löser Messtechnik, Berlin, Germany). Measures were repeated 3 times.
- HPLC assay. Propofol quantification was developed according to the following HPLC method. HPLC-UV UltiMate™ 3000 (Thermo Scientific™) with a DAD-3000 detector and a Phenomenex® Kinetex® (F5: 2.6 µm 100 Å 100 × 4.6 mm) column. The mobile phase was acetonitrile–methanol–water (13:54:33, v/v/v) delivered at a flow rate of 0.8 mL/min; the ultraviolet-light absorbance detector was set at 273 nm. The temperature of the column was set at 40 °C, and the run time was 13 min. The temperature of the autosampler was fixed at 10 °C to prevent the evaporation of the samples (methanol solvent). Prior to HPLC analysis, the NE was diluted 1/1000 in methanol, resulting in the complete destruction of the NE. The method was validated according to ICH guidelines Q2(R1) [12] by determining its linearity, accuracy and precision. Linearity was established using 5 calibration solutions of 12, 16, 20, 24 and 30 µg/mL, corresponding to 60–155 % of the working solution. Linear regression was performed using data analysis software. Linearity was determined using the correlation coefficient (R2). Precision was determined using the relative standard deviation or coefficient of variation (CV%), as defined by ICH. Precision (CV%) and accuracy (%) were determined using three quality controls (14, 22, 28 µg/mL) prepared with another stock solution. Intra-day repeatability (precision and accuracy) was determined using the three quality control solutions (in methanol) repeated six times per day. Inter-day repeatability (precision and accuracy) was determined by repeating the experiments on three days. Peak match values were determined using Thermo ScientificTM DionexTM ChromeleonTM 7 Chromatography Data System version 7.2.7.
- Free propofol determination. Propofol NEs were dialyzed on Rapid Equilibrium Dialysis (RED) Device Inserts (89809, Thermo Scientific™, Rockford, IL, USA), with a cut-off MW of 8000 daltons.
2.1.6. Stability Studies
- NE physical stability. Granulometric analysis was conducted on NE for 1 month. Additionally, to detect droplets larger than 1µm, NEs were analyzed by laser diffraction using a Mastersizer® 3000 particle size analyzer (Malvern Instruments, Malvern, UK). Values quoted are the average of 3 measurements +/− standard deviation (SD).
- Propofol chemical stability. Propofol quantification and research on the degradation products were performed using the HPLC method described previously. The stability-indicating nature of the HPLC analytical method was validated following the ICH and SFPC-GERPAC guidelines [13].
2.1.7. Sterility Test
2.1.8. Endotoxin Test
3. Results
3.1. NE Process Selection
3.1.1. Extemporaneous Addition
3.1.2. “De Novo” Formulation
3.2. Propofol HPLC-UV
3.2.1. Validation of the Method
3.2.2. Stability-Indicating Method
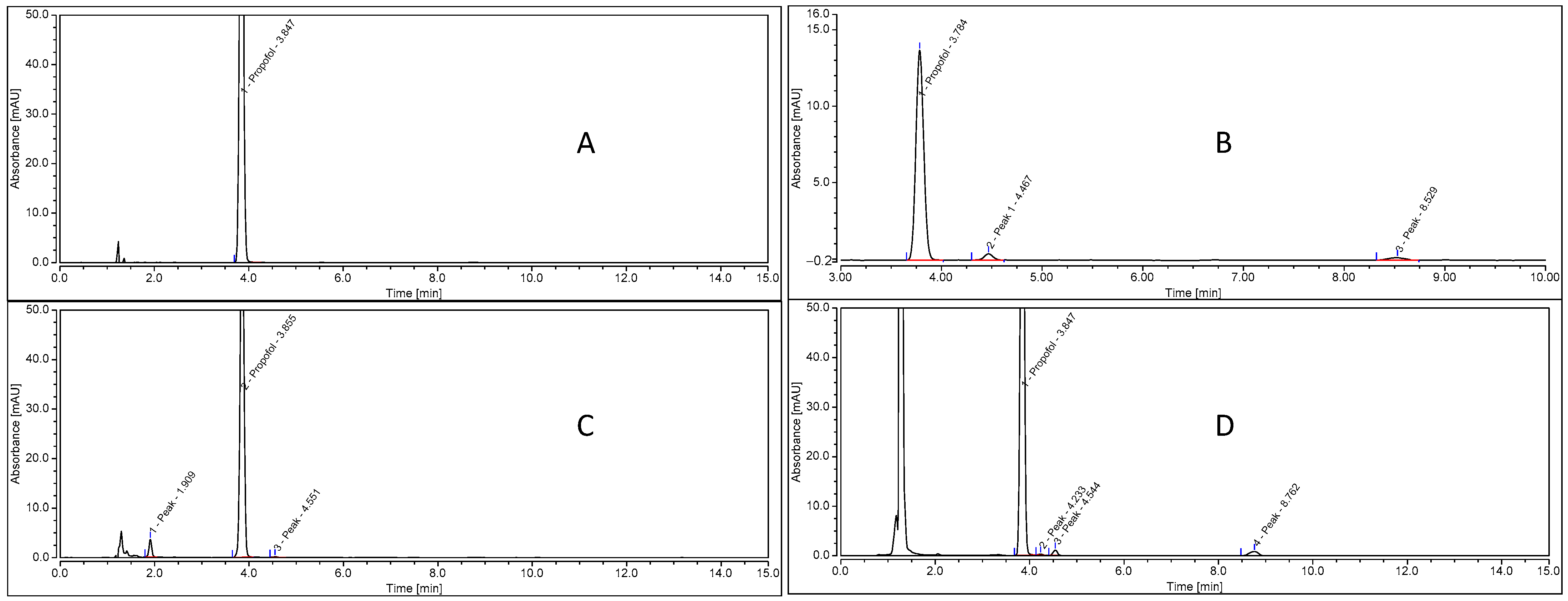
3.3. Impact of Heat Sterilization on Propofol Stability
3.4. Impact of EDTA in the Formulation on Propofol Stability after Heat Sterilization
3.5. Impact of Sterilization Processes on Physical and Chemical Stabilities
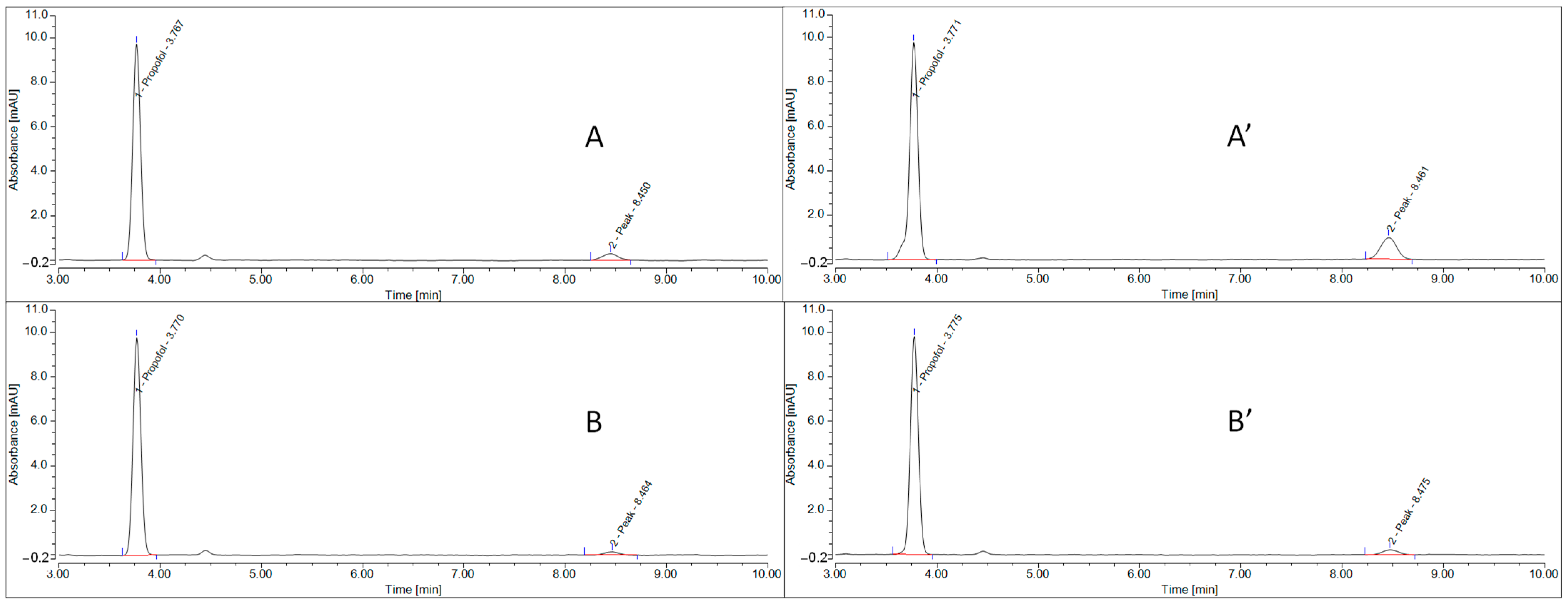
3.6. Short-Term Propofol Chemical Stability
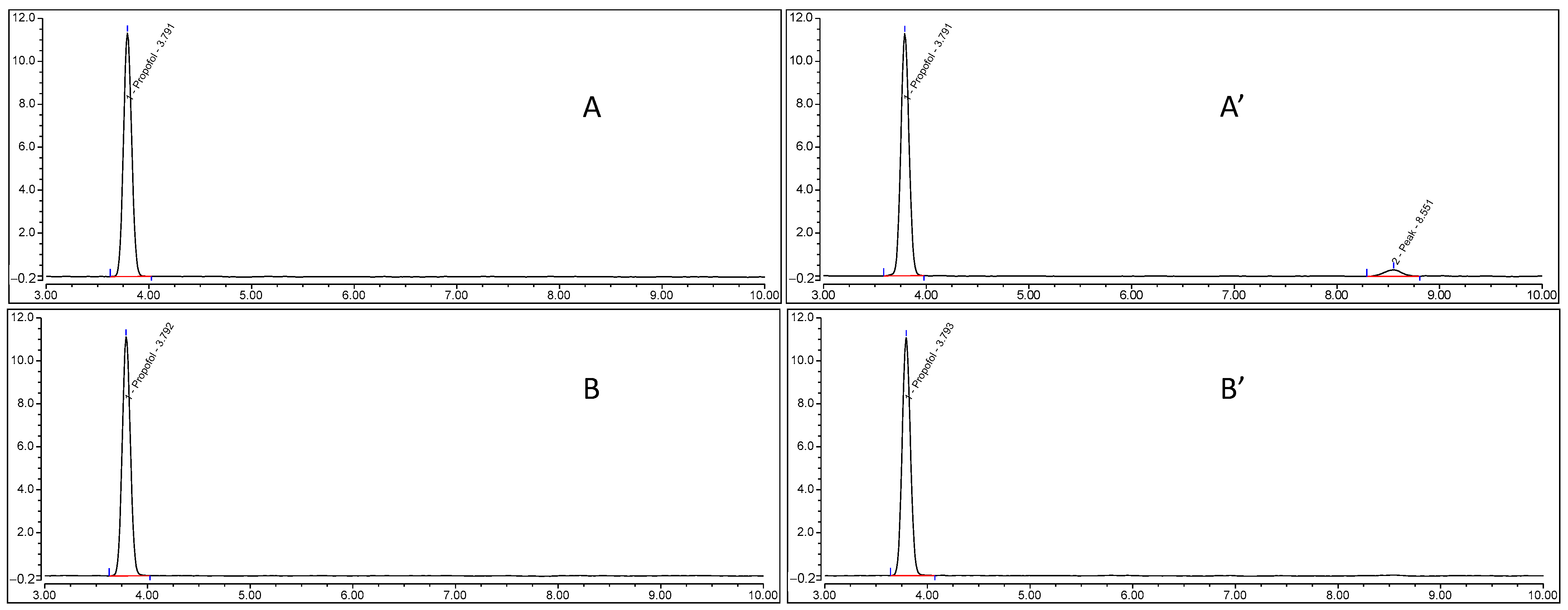
3.7. Free Propofol Determination
4. Discussion
4.1. Propofol Formulation Choice
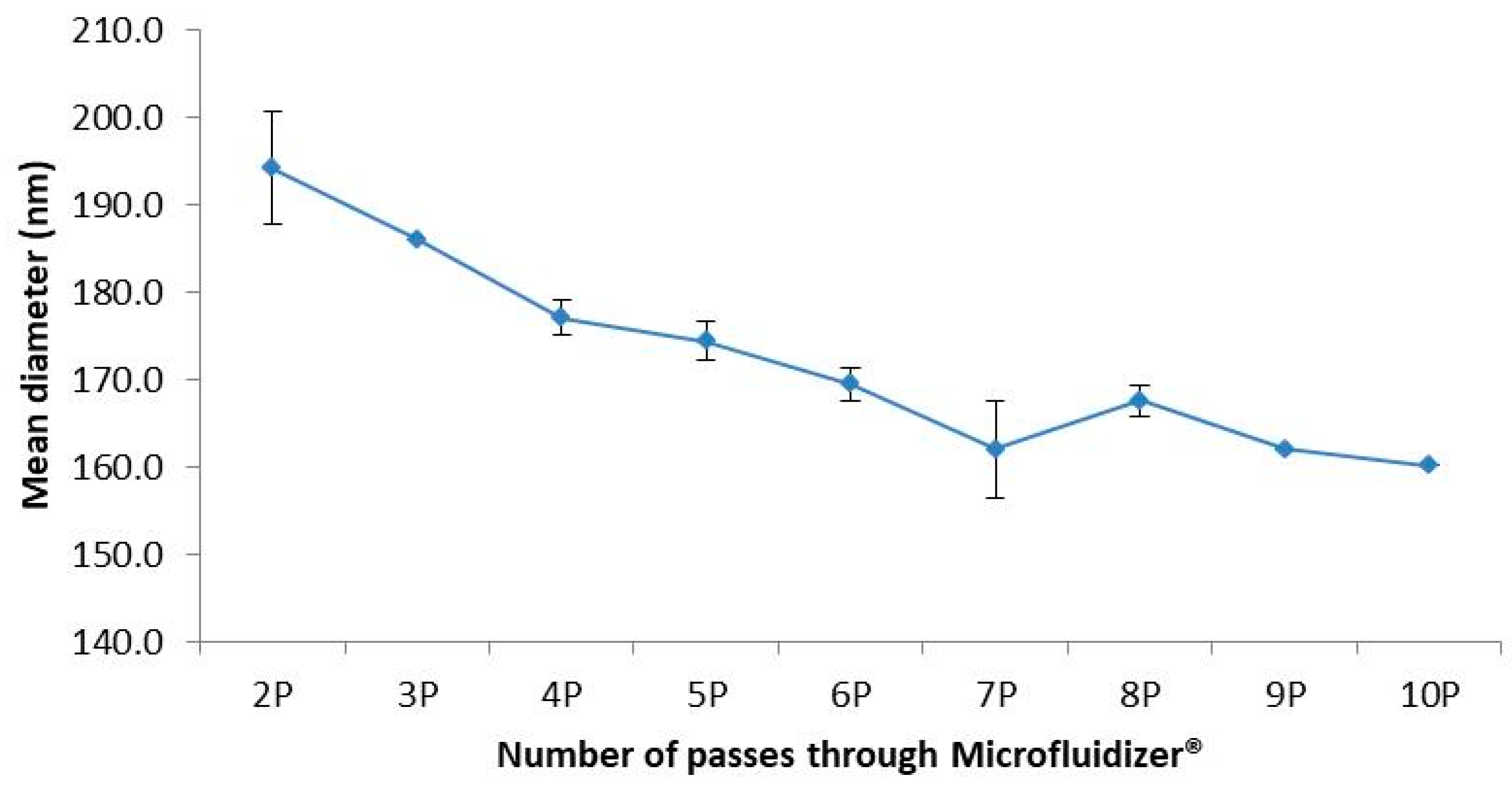
4.2. Process of Emulsification and Size Reduction
4.3. Safety Consideration with Regard to Free Propofol
4.4. Controls and Pharmaceutical Release
4.5. Feasibility of Propofol Production within Hospital Facilities
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sohn, J.-T. Propofol and sedation in patients with coronavirus disease. Am. J. Emerg. Med. 2021, 42, 250. [Google Scholar] [CrossRef]
- Norouzi, P.; Rastegari, A.; Mottaghitalab, F.; Farokhi, M.; Zarrintaj, P.; Saeb, M.R. Nanoemulsions for intravenous drug delivery. Nanoeng. Biomat. Adv. Drug. Del. 2020, 24, 582–601. [Google Scholar] [CrossRef]
- Mcclement, D. Nanoemulsions versus microemulsions: Terminology, differences, similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Hörmann, K.; Zimmer, A. Drug delivery and targeting with parenteral lipid nanoemulsion—A review. J. Control Release 2016, 223, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Washington, C.; Koosha, F.; Davis, S.S. Physical properties of parenteral fat emulsions containing 20% triglycerides, Intralipid, Ivelip. J. Clin. Pharm. Ther. 1993, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Prévot, G.; Mornet, S.; Jacobin-Valat, M.-J.; Mousli, Y.; Hemadou, A.; Duttine, M.; Trotier, A.; Sanchez, S.; Duonor-Cérutti, M.; et al. A nano-emulsion platform functionalized with a fully human scfv-fc antibody for atheroma targeting: Towards a theranostic approach to atherosclerosis. Int. J. Mol. Sci. 2021, 22, 5188. [Google Scholar] [CrossRef] [PubMed]
- Husson, E.; Crauste-Manciet, S.; Hadj-Salah, E.; Seguier, J.-C.; Brossard, D. Compatibility of parenteral drugs with commercialized total parenteral admixtures: Injection of drug inside the admixtures. Nutr. Clin. Metabol. 2003, 17, 8–14. [Google Scholar] [CrossRef]
- Husson, E.; Crauste-Manciet, S.; Hadj-Salah, E.; Seguier, J.-C.; Brossard, D. Compatibility of parenteral drugs with commercialized total parenteral admixtures during simulated Y-site infusion. Nutr. Clin. Metabol. 2003, 17, 72–79. [Google Scholar] [CrossRef]
- Hippalgaonkar, K.; Majumdar, S.; Kansara, V. Injectable Lipid Emulsion-advancements, opportunities and Challenges. AAPS PharmSciTech 2010, 11, 1526–1540. [Google Scholar] [CrossRef] [Green Version]
- Klement, W.; Arndt, J.O. Pain on injection of propofol: Effects of concentration and diluent. Br. J. Anaesth. 1991, 67, 281–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, R.H.; Harnisch, S. Physicochemical characterization of propofol-loaded emulsions and interaction with plasma proteins. Eur. Hosp. Pharm. 2000, 6, 24–31. [Google Scholar]
- ICH Harmonised Tripartite Guideline, 2022, Validation of Analytical Procedures: Text and Methodology. Q2 (R2). Available online: https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures (accessed on 8 January 2023).
- Sautou, V.; Brossard, D.; Crauste-Manciet, V.; Fleury-Souverain, S.; Lagarce, F.; Odou, P.; Roy, S.; Sadeghipour, F. Methodological Guidelines for Stability Studies of Hospital Pharmaceutical Preparations; SFPC GERPAC: Hyères, France, 2013; ISBN 978-2-9526010-6-1. Available online: https://gerpac.eu/IMG/pdf/guide_stabilite_anglais.pdf (accessed on 8 January 2023).
- ICH Harmonised Tripartite Guideline, 2004. Stability Testing of New Drug Substances and Products Q1A (R2). Available online: https://www.ema.europa.eu/en/ich-q1a-r2-stability-testing-new-drug-substances-drug-products (accessed on 8 January 2023).
- ICH Harmonised Tripartite Guideline, 2004. Evaluation of Stability Data, Q1E. Available online: https://www.ema.europa.eu/en/ich-q1e-evaluation-stability-data (accessed on 8 January 2023).
- European Pharmacopoeia. Sterility (2.6.1), 10th ed.; European Pharmacopoeia: Strasbourg, France, 2022. [Google Scholar]
- European Pharmacopeia. Bacterial Endotoxin (2.6.14), 10th ed.; European Pharmacopoeia: Strasbourg, France, 2022. [Google Scholar]
- USP Propofol Injectable Emulsion Monograph. Revision bulletin 2015 USP 39–NF 34. In The United States Pharmacopeia, 39 Rev., and the National Formulary, 34th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2015.
- USP 43-NF 38 -<85> Bacterial Endotoxins. In The United States Pharmacopeia, 43 Rev., and the National Formulary, 38th ed.; United States Pharmacopeial Convention: Rockville, MD, USA, 2020.
- Cèbe, A.; Dessane, B.; Venet, A.; Bernadou, J.M.; Xuereb, F.; Crauste-Manciet, S. Hospital Production of Propofol Injectable Nanoemulsion: Proof-of-Concept, European GERPAC Conference, October 2021, Hyères, France. Available online: https://www.gerpac.eu/hospital-production-of-propofol-injectable-nanoemulsion-proof-of-concept (accessed on 8 January 2023).
- Trapani, A.; Laquintana, V.; Lopodota, A.; Franco, M.; Latrofa, A.; Talani, G.; Sanna, E.; Trapani, G.; Liso, G. Evaluation of new propofol aqueous solutions for intravenous anesthesia. Int. J. Pharm. 2004, 278, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.T.; Naguib, M. Propofol: The challenges of formulation. Anesthesiology 2005, 103, 860–876. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.A.; Choi, B.M.; Cho, S.H.; Choe, S.M.; Ghim, J.L.; Lee, H.M.; Roh, Y.J.; Noh, G.J. Effectiveness safety and pharmacokinetic and pharmacodynamic characteristics of microemulsion propofol in patient undergoing elective surgery under total intravenous anesthesia. Br. J. Anaesth. 2010, 104, 563–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rites, J.C.; Cagno, G.; Vaz Perez, M.; Angrade, L.; Mathias, S.T. Comparative evaluation of propfol in nanoemulsion with solutol and soy lecithin for general anaesthesia. Rev. Bras. Anestesiol. 2016, 66, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.S.; Norton, J.R.; Guivarc’h, P.-H.; Litman, R.S. Pharmacodynamics and Pharmacokinetics of propofol in a medium chain triglyceride emulsion. Anesthesiology 2002, 97, 1401–1408. [Google Scholar] [CrossRef]
- Kazi, M.; Alqahtani, A.; Alharbi, M.; Ahmad, A.; Hussain, M.D.; Alothaid, H.; Aldughaim, M.S. The devolopment and optimization of lipid-based selfnanoemulsifying drug delivery systems for the intravenous delivery of propofol. Molecules 2023, 28, 1492. [Google Scholar] [CrossRef]
- Fukada, T.; Ozaki, M. Microbial growth in propofol formulations with disodium edetate and the influence of venous access system dead space. Anaesthesia 2007, 62, 575–580. [Google Scholar] [CrossRef]
- Sheskey, P.; Hancock, B.; Moss, G.; Goldfarb, D. Handbook of Pharmaceutical Excipients, 9th ed.; APhA/Pharmaceutical Press: London, UK, 2020. [Google Scholar]
- Han, J.; Washington, C. Partition of antimicrobial additives in an intraveinous emulsion and their effect on emulsion physical stability. Int. J. Pharm. 2005, 288, 263–271. [Google Scholar] [CrossRef]
- Herman, C.J.; Groves, M.J. The influence of free fatty acids formation on the pH of phospholipid-stabilized triglyceride emulsions. Pharm. Res. 1993, 10, 774–776. [Google Scholar] [CrossRef]
- Walker, S.; Tailor, S.N.A.; Lee, M.; Louie, L.; Louie, M.; Simor, A.E. Amphotericin B in Lipid Emulsion: Stability, Compatibility, and In Vitro Antifungal Activity. Antimicrob. Agent Chemother. 1998, 42, 762–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- do Egito, E.T.; Fessi, H.; Appel, M.; Barratt, G.; Legrand, P.; Bolard, J.; Devissaguet, J.P. A morphological study of an amphotericin B emulsion-based delivery system. Int. J. Pharm. 1996, 145, 17–27. [Google Scholar] [CrossRef]
- Nilsson, N.; Nezvalova-Henriksen, K.; Tho, I. Emulsion stability of different intravenous propofol formulations in simulated co-administration with remifentanil hydrochlorhide. Pharm. Technol. Hops. Pharm. 2019, 4, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Hota, S.S.; Pattnaik, S.; Mallick, S. Formulation and evaluation of multidose Propofol nanoemulsion using statistically designed experiments. Acta Chim. Slov. 2020, 67, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Lidgate, D.; Trattner, T.; Shultz, R.M.; Maskiewicz, R. Sterile filtration of parenteral emulsion. Pharm. Res. 1992, 9, 860–863. [Google Scholar] [CrossRef]
- EU Guidelines to Good Manufacturing Practice: Medicinal Products for Human and Veterinary Use. Annex 1 Manufacture of Sterile Medicinal Products. Available online: https://health.ec.europa.eu/system/files/2016-11/2008_11_25_gmp-an1_en_0.pdf (accessed on 8 January 2023).
- Ferezou, J.; Lai, N.-T.; Leray, C.; Hajri, T.; Frey, A.; Cabaret, Y.; Coutieu, J.; Lutton, C.; Bach, A.C. Lipid composition and structure of commercial parenteral emulsions. Biochim. Biophys. Acta 1994, 1213, 149–158. [Google Scholar] [CrossRef]
- Ferezou, J.; Gulik, A.; Domingo, N.; Milliat, F.; Dedieu, J.C.; Dunel-Erb, S.; Chevalier, C.; Bach, A.C. Intralipid 10%: Physicochemical characterization. Nutrition 2001, 17, 930–933. [Google Scholar] [CrossRef]
- Wu, Y.; Manna, S.; Petrochenko, P.; Koo, B.; Chen, L.; Xu, X.; Choi, S.; Kozak, D.; Zheng, J. Coexistence of oil droplets and lipid vesicles in propofol drug products. Int. J. Pharm. 2020, 577, 118998. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diprivan® | “>De Novo” Formulation | “Extemporaneous Addition” | |
|---|---|---|---|
| Propofol | 2 g | 2 g | 2 g |
| Intralipid® 20% | - | - | qs 100 mL |
| Soybean oil | 10 g | 10 g | |
| Egg lecithin | 1.2 g | 1.2 g | |
| Glycerol | 2.25 g | 2.25 g | |
| Edetate disodium | 0.0055 g | ±0.0055 g | |
| Sodium hydroxide | qs to adjust pH | qs to adjust pH | |
| Water for injection | qs 100 mL | qs 100 mL |
| Time (Minutes) | 240 | 330 | 420 | 480 |
|---|---|---|---|---|
| Concentration mg/mL | 0.0487 ± 0.0011 | 0.0540 ± 0.0019 | 0.0556 ± 0.0005 | 0.0543 ± 0.0037 |
| Raw Material | MDS (nm) | PDI | Droplets > 1 µm (%) | Droplets > 5 µm (%) | Zeta Potential (mV) | pH |
|---|---|---|---|---|---|---|
| Intralipid® | 290.6 ± 0.8 | 0.143 ± 0.002 | 0 | 0 | −44.5 ± 2.1 | 7.51 ± 0.02 |
| Propofol 1 | 290.7 ± 1.1 | 0.160 ± 0.005 | 2 | 0 | −43.1 ± 0.4 | 7.27 ± 0.11 |
| Propofol 2 | 294.0 ± 1.2 | 0.131 ± 0.019 | - | 7 | −42.2 ± 0.8 | 7.39 ± 0.05 |
| Before Heat Sterilization | After Heat Sterilization | |
|---|---|---|
| pH | 5.20 (±0.04) | 3.98 (±0.02) |
| mds | 161.6 (±1.2) | 816.9 (±67.1) |
| pdi | 0.0787 (±0.013) | 0.0987 (±0.0175) |
| pZ | −41.7 (±2.3) | −44.8 ((±0.5) |
| Visual observation | Homogeneous | Phase separation |
| pH | 7.59 (±0.01) | 4.74 (±0.05) |
| mds | 153.4 (±2.4) | 210.1 (±12.4) |
| pdi | 0.060 (±0.010) | 0.078 (±0.008) |
| pZ | −34.3 (±4.2) | −42.6 (±2.82) |
| Visual observation | Homogeneous | Homogeneous |
| pH | 8.49 (±0.04) | 5.59 (±0.05) |
| mds | 152.2 (±0.5) | 165.9 (±4.2) |
| pdi | 0.061 (±0.003) | 0.070 (±0.002) |
| pZ | −33.7 (±3.9) | −41.2 (±2.3) |
| Visual observation | Homogeneous | Homogeneous |
| pH | 8.97 (±0.01) | 5.95 (±0.05) |
| Mds (nm) | 150.9 (±0.8) | 155.3(±2.4) |
| pdi | 0.055 (±0.004) | 0.057 (±0.012) |
| pZ (mV) | −34.9(±2.2) | −39.7 (±2.4) |
| Visual observation | Homogeneous | Homogeneous |
| pH | 9.47 (±0.01) | 6.62 (±0.07) |
| Mds (nm) | 149.8(±1.8) | 152.4 (±1.2) |
| pdi | 0.055 (±0.009) | 0.064 (±0.007) |
| pZ (mV) | −33.3 (±4.4) | −39.0 (±1.0) |
| Visual observation | Homogeneous | Homogeneous |
| Day | Theoretical Concentration µg/mL | Observed Concentration µg/mL Mean ± SD | Accuracy % | Precision CV% | n |
|---|---|---|---|---|---|
| 1 | 14 | 14.40 ± 0.08 | 99.30 | 0.57 | 6 |
| 22 | 21.90 ± 0.11 | 100.89 | 0.48 | 6 | |
| 28 | 29.80 ± 0.17 | 99.7 | 0.58 | 6 | |
| 2 | 14 | 14.40 ± 0.07 | 99.41 | 0.47 | 6 |
| 22 | 22.20 ± 0.09 | 99.43 | 0.41 | 6 | |
| 28 | 28.80 ± 0.18 | 101.07 | 0.63 | 6 | |
| 3 | 14 | 13.90 ± 0.10 | 99.53 | 0.69 | 6 |
| 22 | 21.90 ± 0.12 | 98.06 | 0.55 | 6 | |
| 28 | 29.90 ± 0.15 | 100.28 | 0.49 | 6 |
| Over 3 Days | Theoretical Concentration % | Observed Concentration Mean % ±SD | Accuracy % | Precision CV% | n |
|---|---|---|---|---|---|
| QC1 | 100 | 99.41 ± 0.54 | 99.74 | 0.54 | 18 |
| QC2 | 100 | 99.46 ± 1.24 | 99.97 | 1.24 | 18 |
| QC3 | 100 | 99.41 ± 0.76 | 100.13 | 0.76 | 18 |
| Homogeneity test of variance | p = 0.3548 |
| Goodness of fit | R-Squared 0.9996, root-mean-square error 7.933 × 10−2 |
| CV% coefficient curves (3 days) | <0.2% |
| LOD | 0.00114 mg/mL |
| LOQ | 0.00381 mg/mL |
| Propofol Peak (%) | Degradation Peak at 8.5 min (%) | Degradation Peak Difference (%) before/after Heat Sterilization | |||
|---|---|---|---|---|---|
| Before heat sterilization | After heat sterilization | Before heat sterilization | After heat sterilization | ||
| pH 7.5 | 97.80 ± 0.37 | 93.73 ±0.66 | 2.20 ± 0.37 | 6.27 ± 0.66 | +4.07 |
| pH 8.5 | 97.97 ± 0.29 | 94.88 ± 0.66 | 2.03 ± 0.29 | 5.12 ± 0.44 | +3.09 |
| pH 9.0 | 97.90 ± 0.26 | 95.17± 0.59 | 2.10 ± 0.26 | 4.83 ± 0.59 | +2.73 |
| pH 9.5 | 97.93 ± 0.29 | 95.40 ± 0.29 | 2.07 ± 0.35 | 4.60 ± 0.29 | +2.53 |
| Before 0.22 µm Filtration | After 0.22 µm Filtration | |
|---|---|---|
| Mean diameter (nm) | 159.3 ± 0.3 | 164.2 ± 0.5 |
| PDI | 0.061 ± 0.015 | 0.064 ± 0.017 |
| Propofol concentration (µg/mL) | 20.90 ± 0.73 | 20.47 ± 0.82 |
| Propofol Peak (%) | Degradation Peak at 8.5 min (%) | Degradation Peak Difference (%) D0/D30 | |
|---|---|---|---|
| pH 7.5 | 94.86 ± 2.40 | 5.14 ± 2.40 | −2.82 |
| pH 8.5 | 95.72 ± 2.40 | 4.28 ± 1.53 | −0.58 |
| pH 9.0 | 95.83 ± 1.44 | 4.17 ± 1.44 | −0.66 |
| pH 9.5 | 95.86 ± 1.67 | 4.14 ± 1.67 | −0.91 |
| Free Propofol Concentration (mg/mL) | Free Propofol (%) | |
|---|---|---|
| 2% Propofol preparation | 0.0527 ± 0.0014 | 0.26 |
| Diprivan® 2% | 0.0534 ± 0.0004 | 0.27 |
| Diprivan® 1% | 0.0280 ± 0.0004 | 0.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cèbe, A.; Dessane, B.; Gohier, P.; Bernadou, J.-M.; Venet, A.; Xuereb, F.; Crauste-Manciet, S. Hospital Production of Sterile 2% Propofol Nanoemulsion: Proof of Concept. Pharmaceutics 2023, 15, 905. https://doi.org/10.3390/pharmaceutics15030905
Cèbe A, Dessane B, Gohier P, Bernadou J-M, Venet A, Xuereb F, Crauste-Manciet S. Hospital Production of Sterile 2% Propofol Nanoemulsion: Proof of Concept. Pharmaceutics. 2023; 15(3):905. https://doi.org/10.3390/pharmaceutics15030905
Chicago/Turabian StyleCèbe, Amélie, Bérangère Dessane, Pauline Gohier, Jean-Marc Bernadou, Arnaud Venet, Fabien Xuereb, and Sylvie Crauste-Manciet. 2023. "Hospital Production of Sterile 2% Propofol Nanoemulsion: Proof of Concept" Pharmaceutics 15, no. 3: 905. https://doi.org/10.3390/pharmaceutics15030905
APA StyleCèbe, A., Dessane, B., Gohier, P., Bernadou, J. -M., Venet, A., Xuereb, F., & Crauste-Manciet, S. (2023). Hospital Production of Sterile 2% Propofol Nanoemulsion: Proof of Concept. Pharmaceutics, 15(3), 905. https://doi.org/10.3390/pharmaceutics15030905