

Modulation of the Cytotoxic Properties of Pd(II) Complexes Based on Functionalized Carboxamides Featuring Labile Phosphoryl Coordination Sites

 ,
,  ,
,
Abstract
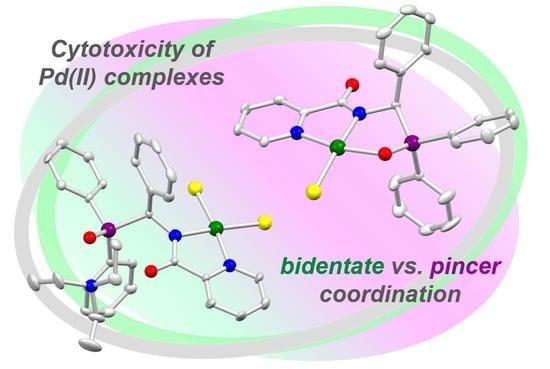
:
1. Introduction
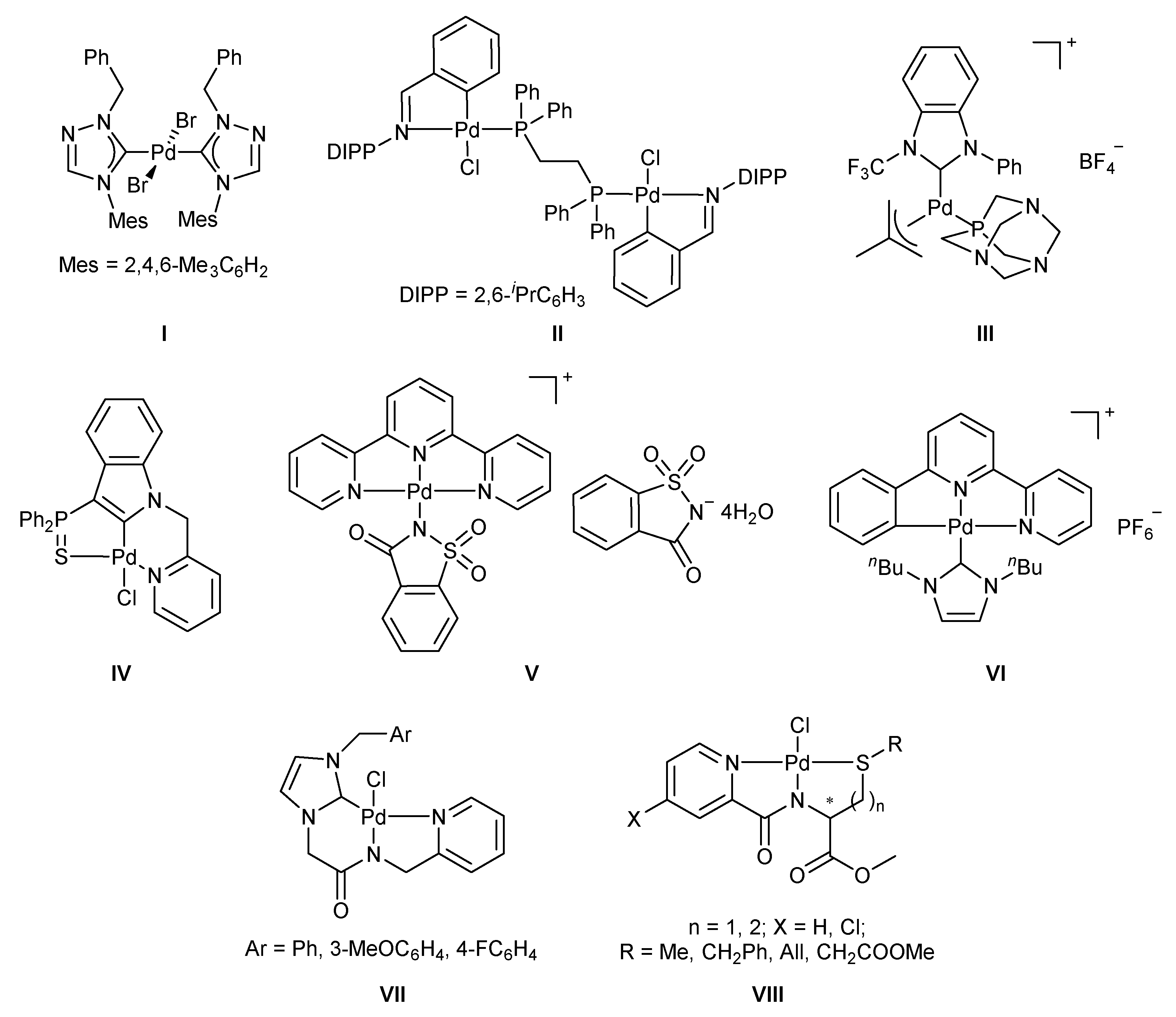
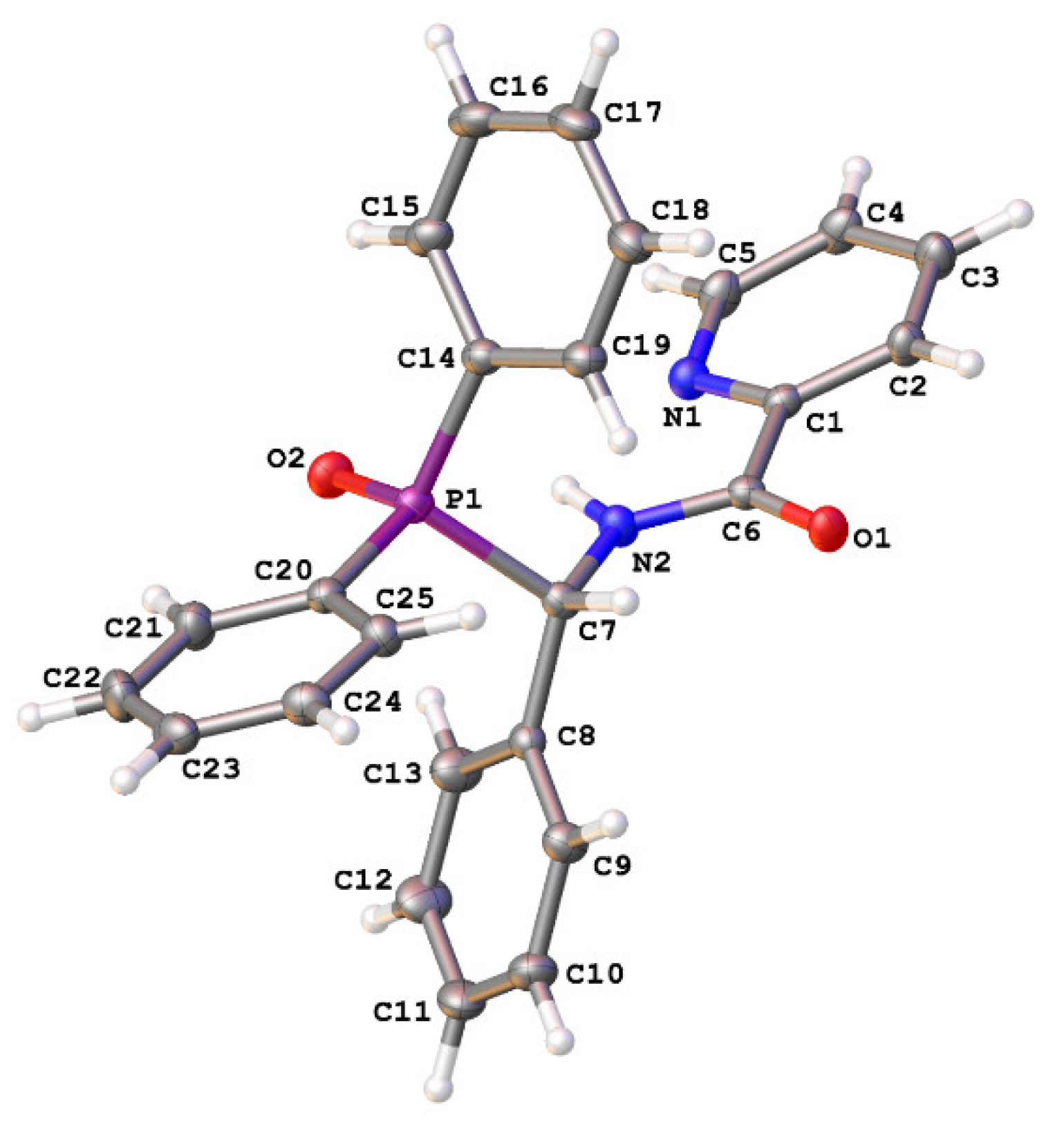
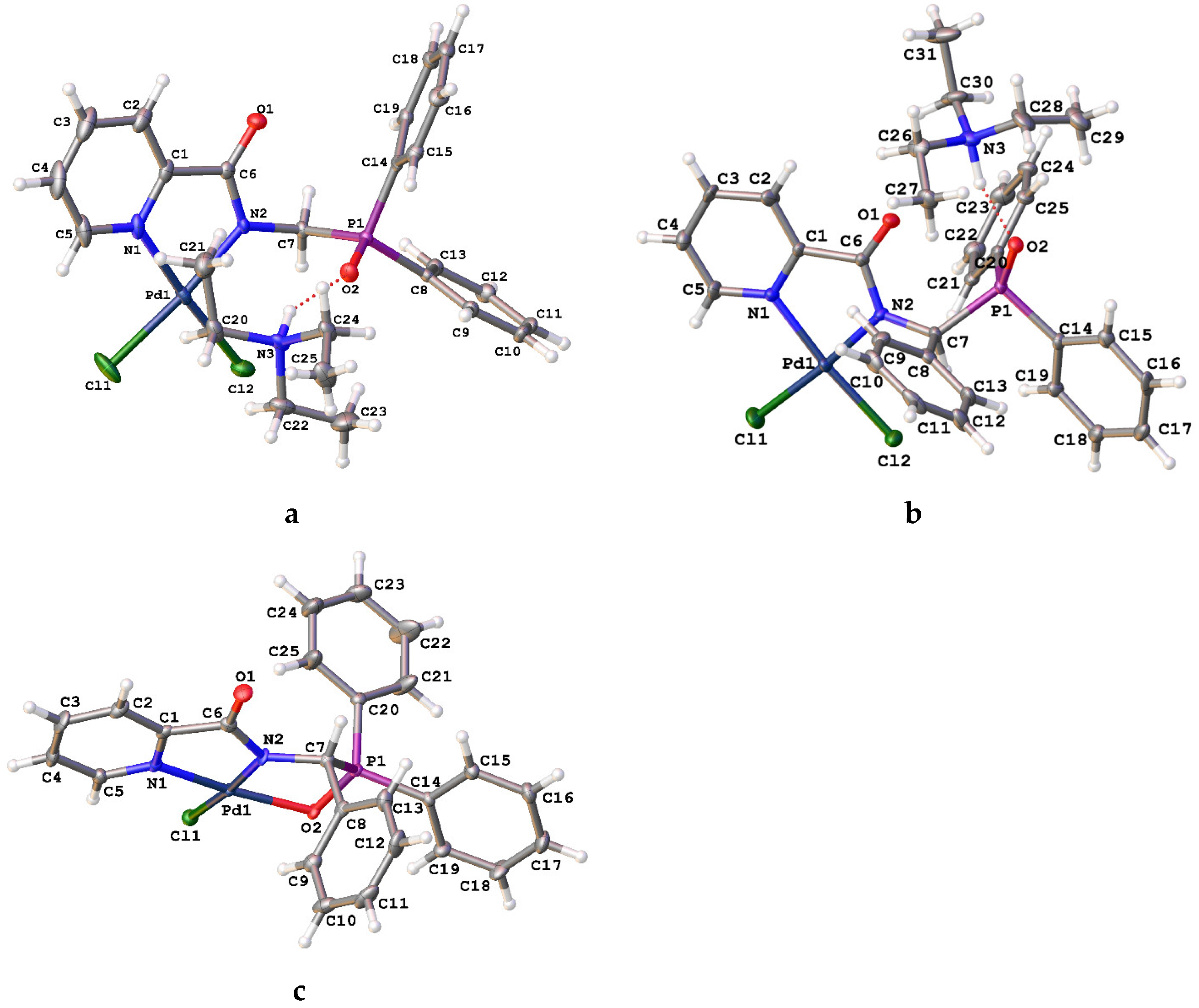
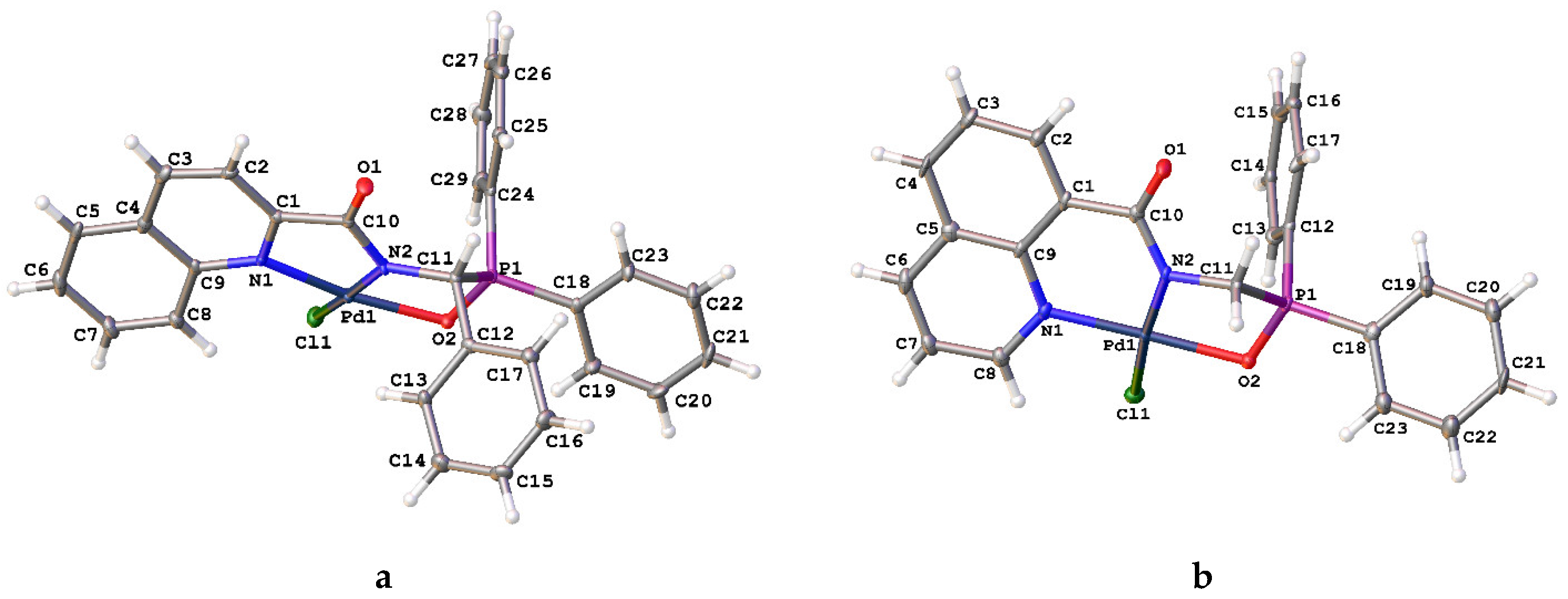
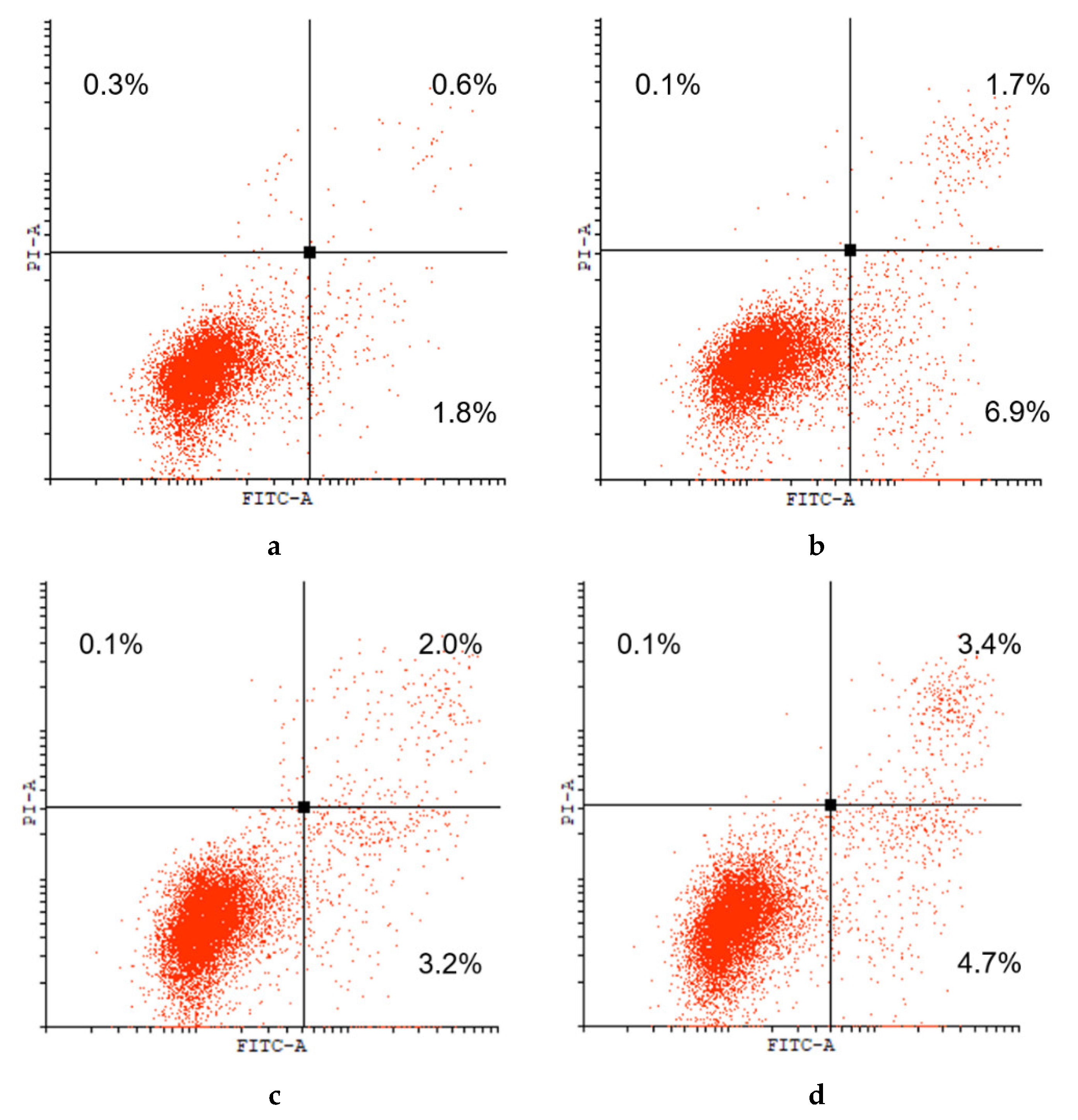
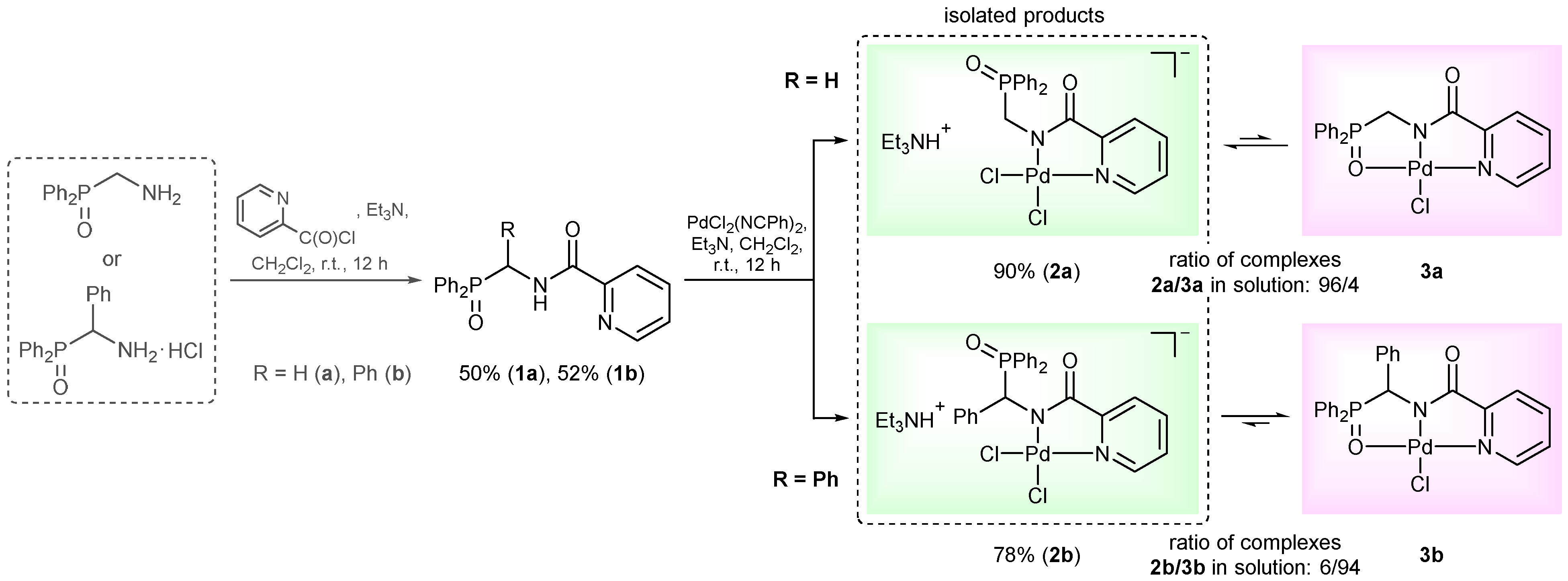
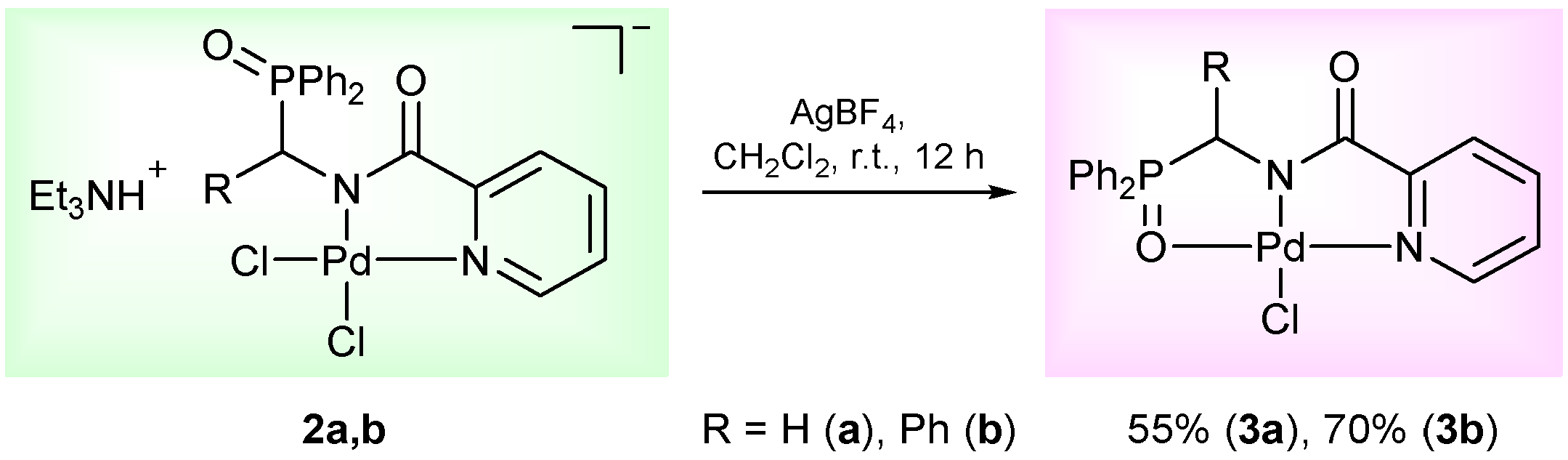
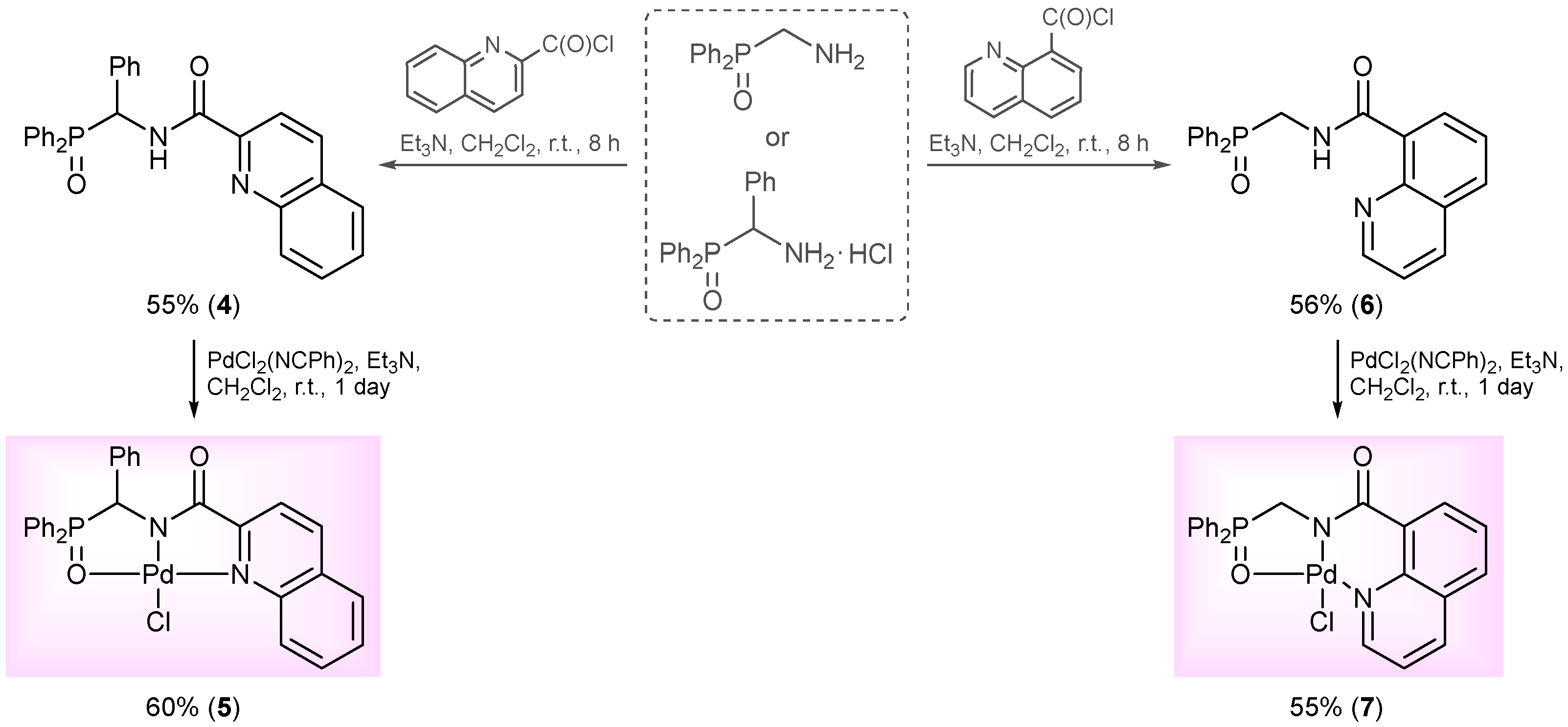
2. Results and Discussion
3. Conclusions
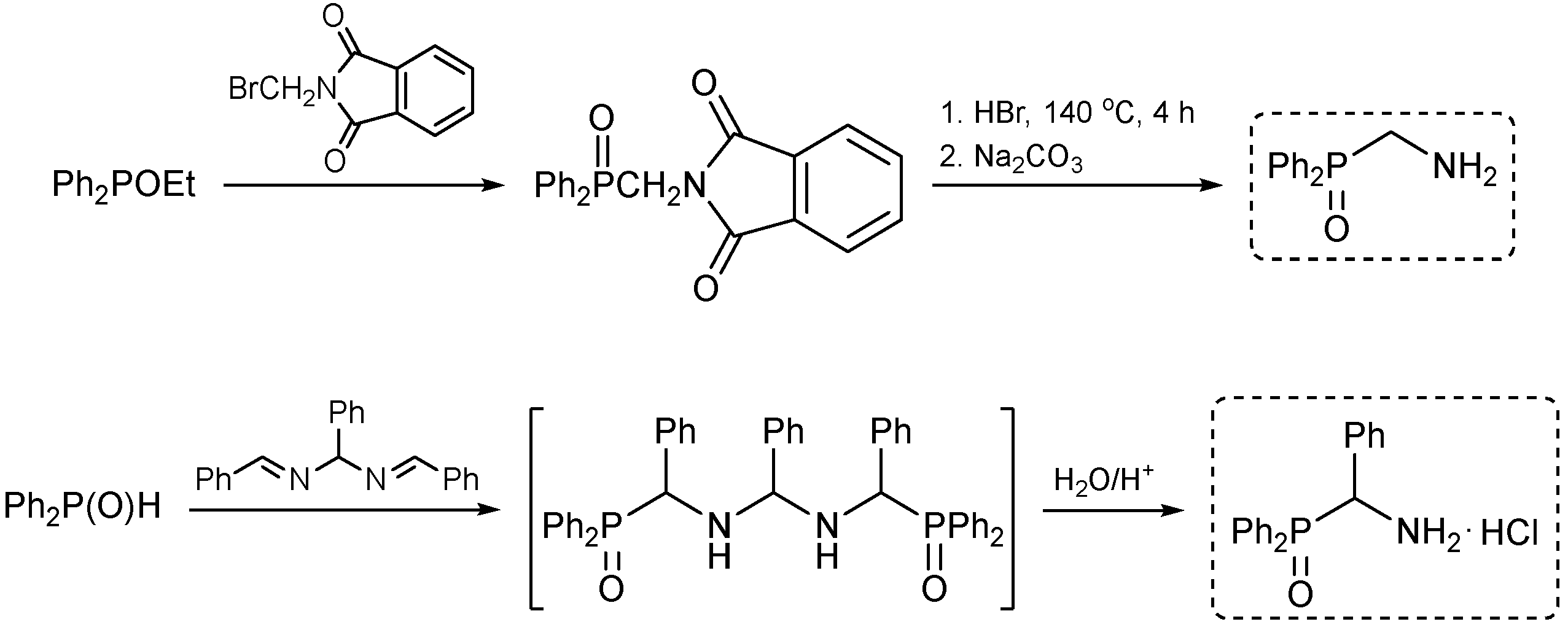
4. Experimental Section
4.1. General Remarks
4.2. Syntheses
4.2.1. N-[(Diphenylphosphoryl)methyl]picolinamide, 1a

4.2.2. N-[(Diphenylphosphoryl)(phenyl)methyl]picolinamide, 1b

4.2.3. Complex [κ2-N,N-(L)Pd(II)Cl] 2a

4.2.4. Complex [κ2-N,N-(L)Pd(II)Cl] 2b

4.2.5. Complex [κ3-O,N,N-(L)Pd(II)Cl] 3a

4.2.6. Complex [κ3-O,N,N-(L)Pd(II)Cl] 3b

4.2.7. N-[(Diphenylphosphoryl)(phenyl)methyl]quinoline-2-carboxamide, 4

4.2.8. Complex [κ3-O,N,N-(L)Pd(II)Cl] 5

4.2.9. N-[(Diphenylphosphoryl)methyl]quinoline-8-carboxamide, 6

4.2.10. Complex [κ3-O,N,N-(L)Pd(II)Cl] 7

4.3. X-ray Crystallography
4.4. Cytotoxicity Studies
4.5. Apoptosis Induction Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xu, C.; Gao, X.; Yao, Q. Platinum-based drugs for cancer therapy and anti-tumor strategies. Theranostics 2022, 12, 2115–2132. [Google Scholar] [CrossRef] [PubMed]
- Ravera, M.; Gabano, E.; McGlinchey, M.J.; Osella, D. Pt(IV) antitumor prodrugs: Dogmas, paradigms, and realities. Dalton Trans. 2022, 51, 2121–2134. [Google Scholar] [CrossRef]
- Czarnomysy, R.; Radomska, D.; Szewczyk, O.K.; Roszczenko, P.; Bielawski, K. Platinum and palladium complexes as promising sources for antitumor treatments. Int. J. Mol. Sci. 2021, 22, 8271. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Z.; Deng, Z.; Zhu, G. Recent advances in the synthesis, stability, and activation of platinum(IV) anticancer prodrugs. Coord. Chem. Rev. 2021, 442, 213991. [Google Scholar] [CrossRef]
- Jin, S.; Guo, Y.; Guo, Z.; Wang, X. Monofunctional platinum(II) anticancer agents. Pharmaceuticals 2021, 14, 133. [Google Scholar] [CrossRef]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Crespo, M. Cyclometallated platinum(IV) compounds as promising antitumour agents. J. Organomet. Chem. 2019, 879, 15–26. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkova, D.; Ivanova, S. Application of approved cisplatin derivatives in combination therapy against different cancer diseases. Molecules 2022, 27, 2466. [Google Scholar] [CrossRef]
- Yu, C.; Wang, Z.; Sun, Z.; Zhang, L.; Zhang, W.; Xu, Y.; Zhang, J.-J. Platinum-based combination therapy: Molecular rationale, current clinical uses, and future perspectives. J. Med. Chem. 2020, 63, 13397–13412. [Google Scholar] [CrossRef]
- Xiao, X.; Oswald, J.T.; Wang, T.; Zhang, W.; Li, W. Use of anticancer platinum compounds in combination therapies and challenges in drug delivery. Curr. Med. Chem. 2020, 27, 3055–3078. [Google Scholar] [CrossRef]
- Peng, K.; Liang, B.-B.; Liu, W.; Mao, Z.-W. What blocks more anticancer platinum complexes from experiment to clinic: Major problems and potential strategies from drug design perspectives. Coord. Chem. Rev. 2021, 449, 214210. [Google Scholar] [CrossRef]
- Lucaciu, R.L.; Hangan, A.C.; Sevastre, B.; Oprean, L.S. Metallo-drugs in cancer therapy: Past, present and future. Molecules 2022, 27, 6485. [Google Scholar] [CrossRef]
- Ferraro, M.G.; Piccolo, M.; Misso, G.; Santamaria, R.; Irace, C. Bioactivity and development of small non-platinum metal-based chemotherapeutics. Pharmaceutics 2022, 14, 954. [Google Scholar] [CrossRef]
- Paprocka, R.; Wiese-Szadkowska, M.; Janciauskiene, S.; Kosmalski, T.; Kulik, M.; Helmin-Basa, A. Latest developments in metal complexes as anticancer agents. Coord. Chem. Rev. 2022, 452, 214307. [Google Scholar] [CrossRef]
- Murray, B.S.; Dyson, P.J. Recent progress in the development of organometallics for the treatment of cancer. Curr. Opin. Chem. Biol. 2020, 56, 28–34. [Google Scholar] [CrossRef]
- Simpson, P.V.; Desai, N.M.; Casari, I.; Massi, M.; Falasca, M. Metal-based antitumor compounds: Beyond cisplatin. Future Med. Chem. 2019, 11, 119–135. [Google Scholar] [CrossRef]
- Scattolin, T.; Voloshkin, V.A.; Visentin, F.; Nolan, S.P. A critical review of palladium organometallic anticancer agents. Cell Rep. Phys. Sci. 2021, 2, 100446. [Google Scholar] [CrossRef]
- Bangde, P.; Prajapati, D.; Dandekar, P.; Fairlamb, I.J.S.; Kapdi, A.R. Palladacycles as potential anticancer agents. In Palladacycles. Catalysis and Beyond; Kapdi, A., Maiti, D., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 9; pp. 343–370. [Google Scholar] [CrossRef]
- Vojtek, M.; Marques, M.P.M.; Ferreira, I.M.P.L.V.O.; Mota-Filipe, H.; Diniz, C. Anticancer activity of palladium-based complexes against triple-negative breast cancer. Drug Discov. Today 2019, 24, 1044–1058. [Google Scholar] [CrossRef]
- Ryabov, A.D. Cyclopalladated compounds as enzyme prototypes and anticancer drugs. In Palladacycles: Synthesis, Characterization and Applications; Dupont, J., Pfeffer, M., Eds.; Wiley: Weinheim, Germany, 2008; Chapter 13; pp. 307–339. [Google Scholar] [CrossRef]
- Bugarčić, Ž.D.; Bogojeski, J.; van Eldik, R. Kinetics, mechanism and equilibrium studies on the substitution reactions of Pd(II) in reference to Pt(II) complexes with bio-molecules. Coord. Chem. Rev. 2015, 292, 91–106. [Google Scholar] [CrossRef]
- Van-Ha, N.; Hien, P.T.T.; Dat, D.T.; Thao, D.T. Highly cytotoxic palladium(II) complexes with 1,2,4-triazole-derived carbene ligands. Mendeleev Commun. 2022, 32, 594–596. [Google Scholar] [CrossRef]
- Aliwaini, S.; Peres, J.; Kröger, W.L.; Blanckenberg, A.; de la Mare, J.; Edkins, A.L.; Mapolie, S.; Prince, S. The palladacycle, AJ-5, exhibits anti-tumour and anti-cancer stem cell activity in breast cancer cells. Cancer Lett. 2015, 357, 206–218. [Google Scholar] [CrossRef]
- Scattolin, T.; Bortolamiol, E.; Visentin, F.; Palazzolo, S.; Caligiuri, I.; Perin, T.; Canzonieri, V.; Demitri, N.; Rizzolio, F.; Togni, A. Palladium(II)-η3-allyl complexes bearing N-trifluoromethyl N-heterocyclic carbenes: A new generation of anticancer agents that restrain the growth of high-grade serous ovarian cancer tumoroids. Chem. Eur. J. 2020, 26, 11868–11876. [Google Scholar] [CrossRef] [PubMed]
- Aleksanyan, D.V.; Spiridonov, A.A.; Churusova, S.G.; Rybalkina, E.Y.; Danshina, A.A.; Peregudov, A.S.; Klemenkova, Z.S.; Kozlov, V.A. Thiophosphorylated indoles as a promising platform for the creation of cytotoxic Pd(II) pincer complexes. Inorg. Chim. Acta 2023, 548, 121369. [Google Scholar] [CrossRef]
- Cetin, Y.; Adiguzel, Z.; Polat, H.U.; Akkoc, T.; Tas, A.; Cevatemre, B.; Celik, G.; Carikci, B.; Yilmaz, V.T.; Ulukaya, E.; et al. A palladium(II)–saccharinate complex of terpyridine exerts higher anticancer potency and less toxicity than cisplatin in a mouse allograft model. Anti-Cancer Drugs 2017, 28, 898–910. [Google Scholar] [CrossRef]
- Fong, T.T.-H.; Lok, C.-N.; Chung, C.Y.-S.; Fung, Y.-M.E.; Chow, P.-K.; Wan, P.-K.; Che, C.-M. Cyclometalated palladium(II) N-heterocyclic carbene complexes: Anticancer agents for potent in vitro cytotoxicity and in vivo tumor growth suppression. Angew. Chem., Int. Ed. 2016, 55, 11935–11939. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Lee, J.-Y.; Chang, Y.-Y.; Hu, C.-H.; Wang, N.M.; Lee, H.M. Palladium complexes with tridentate N-heterocyclic carbene ligands: Selective “normal” and “abnormal” bindings and their anticancer activities. Organometallics 2015, 34, 4359–4368. [Google Scholar] [CrossRef]
- Churusova, S.G.; Aleksanyan, D.V.; Rybalkina, E.Y.; Susova, O.Y.; Peregudov, A.S.; Brunova, V.V.; Gutsul, E.I.; Klemenkova, Z.S.; Nelyubina, Y.V.; Glushko, V.N.; et al. Palladium(II) pincer complexes of functionalized amides with S-modified cysteine and homocysteine residues: Cytotoxic activity and different aspects of their biological effect on living cells. Inorg. Chem. 2021, 60, 9880–9898. [Google Scholar] [CrossRef]
- Churusova, S.G.; Aleksanyan, D.V.; Rybalkina, E.Y.; Susova, O.Y.; Brunova, V.V.; Aysin, R.R.; Nelyubina, Y.V.; Peregudov, A.S.; Gutsul, E.I.; Klemenkova, Z.S.; et al. Highly cytotoxic palladium(II) pincer complexes based on picolinylamides functionalized with amino acids bearing ancillary S-donor groups. Inorg. Chem. 2017, 56, 9834–9850. [Google Scholar] [CrossRef]
- Kapdi, A.R.; Maiti, D. (Eds.) Palladacycles: Catalysis and Beyond; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Morales-Morales, D. (Ed.) Pincer Compounds: Chemistry and Applications; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Churusova, S.G.; Aleksanyan, D.V.; Rybalkina, E.Y.; Gutsul, E.I.; Peregudov, A.S.; Klemenkova, Z.S.; Nelyubina, Y.V.; Buyanovskaya, A.G.; Kozlov, V.A. Pincer–dipeptide and pseudodipeptide conjugates: Synthesis and bioactivity studies. J. Inorg. Biochem. 2022, 235, 111908. [Google Scholar] [CrossRef]
- Churusova, S.G.; Aleksanyan, D.V.; Rybalkina, E.Y.; Nelyubina, Y.V.; Peregudov, A.S.; Klemenkova, Z.S.; Kozlov, V.A. Non-classical N-metallated Pd(II) pincer complexes featuring amino acid pendant arms: Synthesis and biological activity. Polyhedron 2018, 143, 70–82. [Google Scholar] [CrossRef]
- Aleksanyan, D.V.; Churusova, S.G.; Rybalkina, E.Y.; Nelyubina, Y.V.; Kozlov, V.A. Pd(II) and Pt(II) pincer complexes of a benzothiazole-appended methylthioacetamide ligand: Synthesis and in vitro cytotoxicity. INEOS OPEN 2021, 4, 237–242. [Google Scholar] [CrossRef]
- Churusova, S.G.; Aleksanyan, D.V.; Vasil’ev, A.A.; Rybalkina, E.Y.; Susova, O.Y.; Klemenkova, Z.S.; Aysin, R.R.; Nelyubina, Y.V.; Kozlov, V.A. Design of pincer complexes based on (methylsulfanyl)acetic/propionic acid amides with ancillary S- and N-donors as potential catalysts and cytotoxic agents. Appl. Organomet. Chem. 2018, 32, e4360. [Google Scholar] [CrossRef]
- Aleksanyan, D.V.; Churusova, S.G.; Brunova, V.V.; Peregudov, A.S.; Shakhov, A.M.; Rybalkina, E.Y.; Klemenkova, Z.S.; Kononova, E.G.; Denisov, G.L.; Kozlov, V.A. Mechanochemistry for the synthesis of non-classical N-metalated palladium(II) pincer complexes. Dalton Trans. 2021, 50, 16726–16738. [Google Scholar] [CrossRef]
- Popoff, I.C.; Huber, L.K.; Block, B.P.; Morton, P.D.; Riordan, R.P. α-Aminophosphinic acids and α-aminophosphine oxides, I. Alkyl-α-aminoalkylphosphinic acids, α-aminoalkyl(aryl)phosphinic acids, and α-aminoalkyl(diaryl)phosphine oxides. J. Org. Chem. 1963, 28, 2898–2900. [Google Scholar] [CrossRef]
- Regits, M.; Eckes, H. Carbene, 22. Phosphene: Abfangreaktionen von (diphenylmethylen)phenyl-phosphan-oxid durch [2 + 2]-cycloaddition mit aldehyden. Chem. Ber. 1980, 113, 3303–3312. [Google Scholar] [CrossRef]
- Decken, A.; Gossage, R.A.; Yadav, P.N. Oxazoline chemistry. Part VIII. Synthesis and characterization of a new class of pincer ligands derived from the 2-(o-anilinyl)-2-oxazoline skeleton—Applications to the synthesis of group X transition metal catalysts. Can. J. Chem. 2005, 83, 1185–1189. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules; Wiley: New York, NY, USA, 1975. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete sructure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1b | 2a | 2b | 3b | |
|---|---|---|---|---|
| Pd1–Cl1 | – | 2.3031(7) | 2.305(2) | 2.3137(8) |
| Pd1–X 1 | – | 2.2977(7) | 2.309(2) | 2.060(2) |
| Pd1–N1 | – | 2.023(2) | 2.037(6) | 2.000(3) |
| Pd1–N2 | – | 2.0084(18) | 2.025(6) | 1.973(3) |
| P1–O2 | 1.4843(11) | 1.4934(17) | 1.502(5) | 1.536(2) |
| N1–C1 | 1.3438(18) | 1.349(3) | 1.341(9) | 1.367(4) |
| C1–C6 | 1.4997(19) | 1.494(3) | 1.509(10) | 1.519(5) |
| C6–O1 | 1.2295(17) | 1.245(3) | 1.242(8) | 1.241(4) |
| C6–N2 | 1.3373(18) | 1.332(3) | 1.336(9) | 1.329(5) |
| Cl1–Pd1–N2 | – | 174.56(6) | 174.66(18) | 173.19(8) |
| X–Pd1–N1 1 | – | 175.37(6) | 174.45(17) | 167.56(10) |
| N2–Pd1–N1 | – | 80.43(8) | 80.6(2) | 80.99(11) |
| N1–Pd1–Cl1 | – | 94.48(6) | 94.45(17) | 98.66(8) |
| Cl1–Pd1–X 1 | – | 89.83(3) | 90.52(8) | 93.78(6) |
| X–Pd1–N2 1 | – | 95.33(6) | 94.53(17) | 86.73(10) |
| Entry | Comp. | IC50 ± SD 1, μM | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HCT116 | MCF7 | PC3 | U251 | Scov3 | HEK293 | HBL100 | HBL100/Dox | ||
| 1 | 1a | >100.0 2 | 46% 3 | >100.0 2 | >100.0 2 | >100.0 2 | >100.0 2 | >100.0 2 | 35% 3 |
| 2 | 2a | 30.0 ± 5.0 | 33.0 ± 7.0 | 22.0 ± 10.0 | >80.0 4 | >80.0 4 | 24.0 ± 6.0 | >80.0 4 | >80.0 4 |
| 3 | 2b | 3.0 ± 0.5 | 11.0 ± 2.5 | 9.0 ± 1.5 | 43.0 ± 1.5 | 36.0 ± 2.5 | 6.8 ± 0.2 | 30.0 ± 3.5 | 26.0 ± 7.2 |
| 4 | 3a | 36.0 ± 2.0 | 45.0 ± 5.0 | 26.0 ± 6.0 | >80.0 4 | 30% 5 | 34.0 ± 3.0 | >80.0 4 | >80.0 4 |
| 5 | 3b | 4.0 ± 2.0 | 13.0 ± 1.5 | 16.0 ± 2.0 | 43.0 ± 5.0 | 40.0 ± 0.6 | 12.5 ± 3.5 | 17.8 ± 0.8 | 28.0 ± 2.6 |
| 6 | 7 | 38.0 ± 6.0 | 58.0 ± 12.0 | 52.0 ± 10.0 | n/d | n/d | 23.0 ± 3.0 | n/d | n/d |
| 7 | Cisplatin | 18.0 ± 2.0 | 25.0 ± 4.0 | 16.0 ± 3.0 | 16.5 ± 1.5 | 21.0 ± 3.0 | 12.5 ± 1.5 | 14.6 ± 3.6 | 23.6 ± 3.6 |
| Entry | Comp. | IC50 ± SD 1, μM | |||
|---|---|---|---|---|---|
| K562 | K562/iS9 | AMO1 | H9 | ||
| 1 | 1a | >80.0 2 | >80.0 2 | 41% 3 | >80.0 2 |
| 2 | 2a | 36.0 ± 2.0 | 44.0 ± 2.0 | 40.0 ± 2.0 | 32.0 ± 1.0 |
| 3 | 2b | 11.0 ± 3.4 | 8.5 ± 0.5 | 16.0 ± 1.0 | 12.5 ± 1.5 |
| 4 | 3a | 36.0 ± 4.0 | 40.0 ± 4.5 | 32.0 ± 2.0 | 23.0 ± 1.0 |
| 5 | 3b | 6.4 ± 0.4 | 7.2 ± 1.0 | 2.7 ± 0.5 | 3.2 ± 0.2 |
| 6 | Cisplatin | 15.5 ± 0.5 | 16.0 ± 2.0 | 3.2 ± 0.6 | 3.0 ± 1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aleksanyan, D.V.; Konovalov, A.V.; Churusova, S.G.; Rybalkina, E.Y.; Peregudov, A.S.; Aksenova, S.A.; Gutsul, E.I.; Klemenkova, Z.S.; Kozlov, V.A. Modulation of the Cytotoxic Properties of Pd(II) Complexes Based on Functionalized Carboxamides Featuring Labile Phosphoryl Coordination Sites. Pharmaceutics 2023, 15, 1088. https://doi.org/10.3390/pharmaceutics15041088
Aleksanyan DV, Konovalov AV, Churusova SG, Rybalkina EY, Peregudov AS, Aksenova SA, Gutsul EI, Klemenkova ZS, Kozlov VA. Modulation of the Cytotoxic Properties of Pd(II) Complexes Based on Functionalized Carboxamides Featuring Labile Phosphoryl Coordination Sites. Pharmaceutics. 2023; 15(4):1088. https://doi.org/10.3390/pharmaceutics15041088
Chicago/Turabian StyleAleksanyan, Diana V., Aleksandr V. Konovalov, Svetlana G. Churusova, Ekaterina Yu. Rybalkina, Alexander S. Peregudov, Svetlana A. Aksenova, Evgenii I. Gutsul, Zinaida S. Klemenkova, and Vladimir A. Kozlov. 2023. "Modulation of the Cytotoxic Properties of Pd(II) Complexes Based on Functionalized Carboxamides Featuring Labile Phosphoryl Coordination Sites" Pharmaceutics 15, no. 4: 1088. https://doi.org/10.3390/pharmaceutics15041088
APA StyleAleksanyan, D. V., Konovalov, A. V., Churusova, S. G., Rybalkina, E. Y., Peregudov, A. S., Aksenova, S. A., Gutsul, E. I., Klemenkova, Z. S., & Kozlov, V. A. (2023). Modulation of the Cytotoxic Properties of Pd(II) Complexes Based on Functionalized Carboxamides Featuring Labile Phosphoryl Coordination Sites. Pharmaceutics, 15(4), 1088. https://doi.org/10.3390/pharmaceutics15041088