

Platelet Activation by Antisense Oligonucleotides (ASOs) in the Göttingen Minipig, including an Evaluation of Glycoprotein VI (GPVI) and Platelet Factor 4 (PF4) Ontogeny

 ,
,  , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antisense Oligonucleotides
2.2. Study Design
2.3. Blood Collection and Sample Preparation
2.4. Platelet Activation Assay in Platelet-Rich Plasma and Whole Blood
2.5. Thrombospondin 1 (TSP-1) Immunoassay
2.6. 96-Well Plate Platelet Aggregometry in Platelet-Rich Plasma
2.7. GPVI Pull-Down with ASO-Coated Streptavidin Beads
2.8. GPVI and PF4 Quantification by ELISA
2.9. Statistical Analysis
3. Results
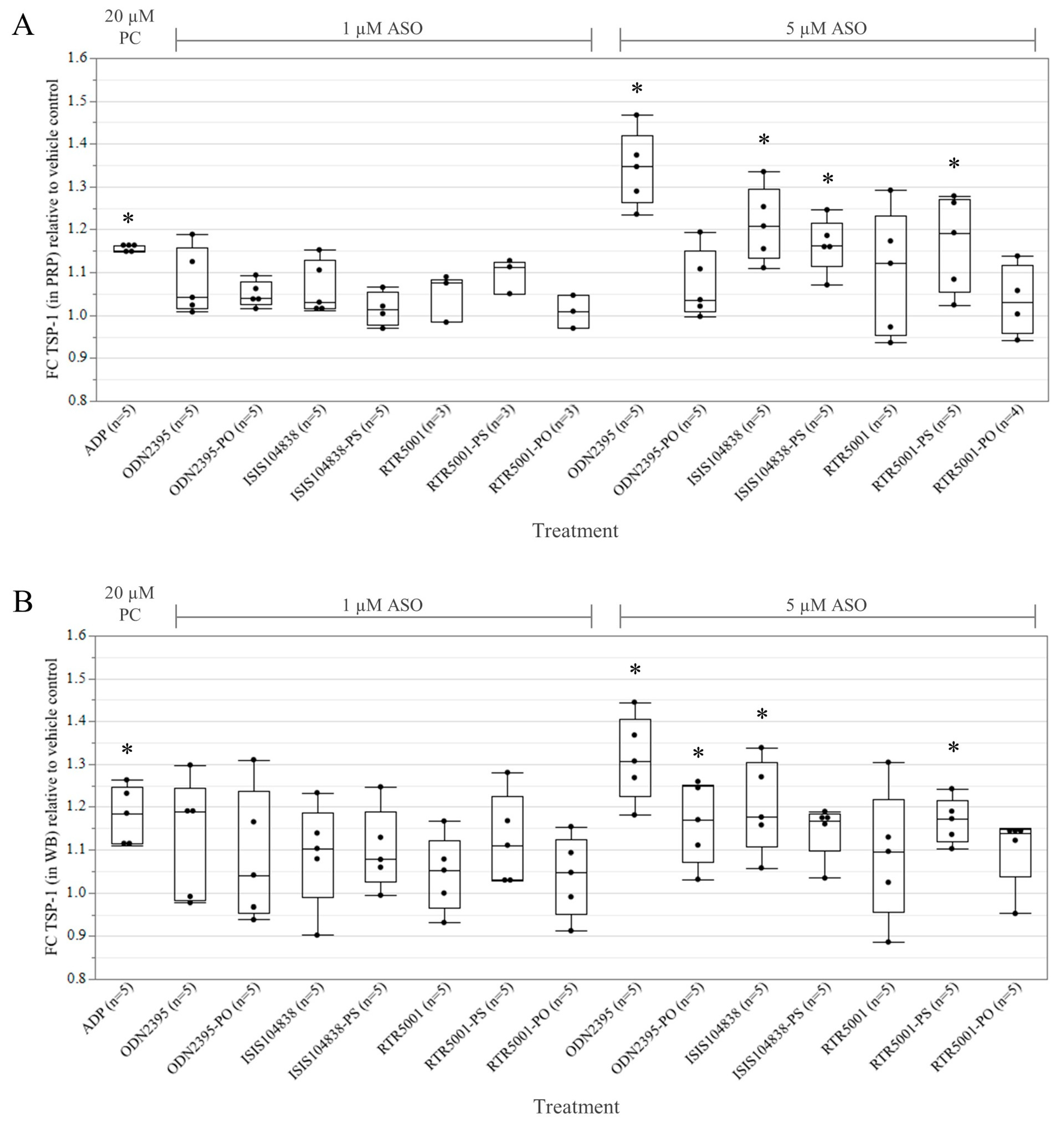
3.1. ASO-Induced Platelet Activation in Platelet-Rich Plasma and Whole Blood
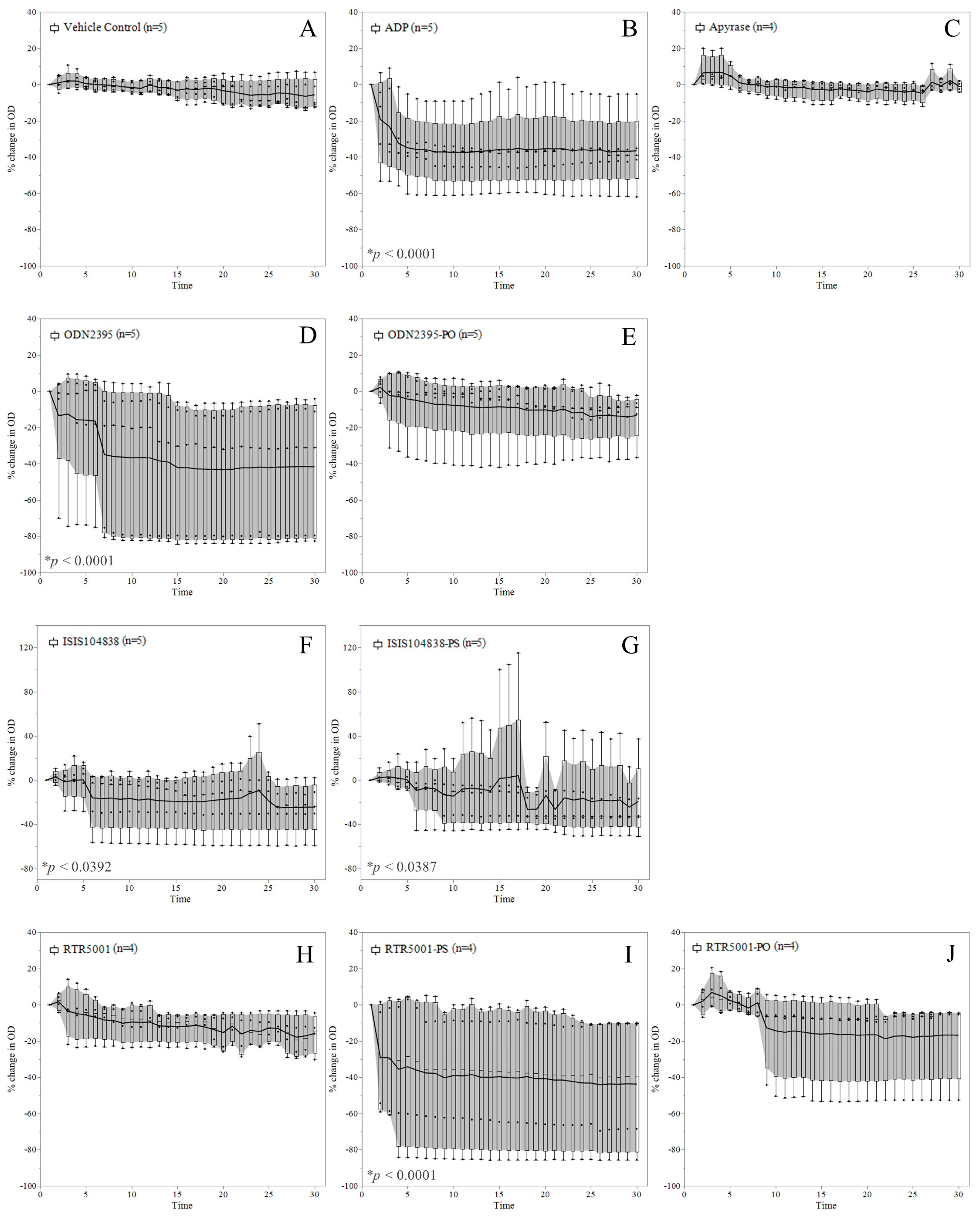
3.2. ASO-Induced Platelet Aggregation in Platelet-Rich Plasma
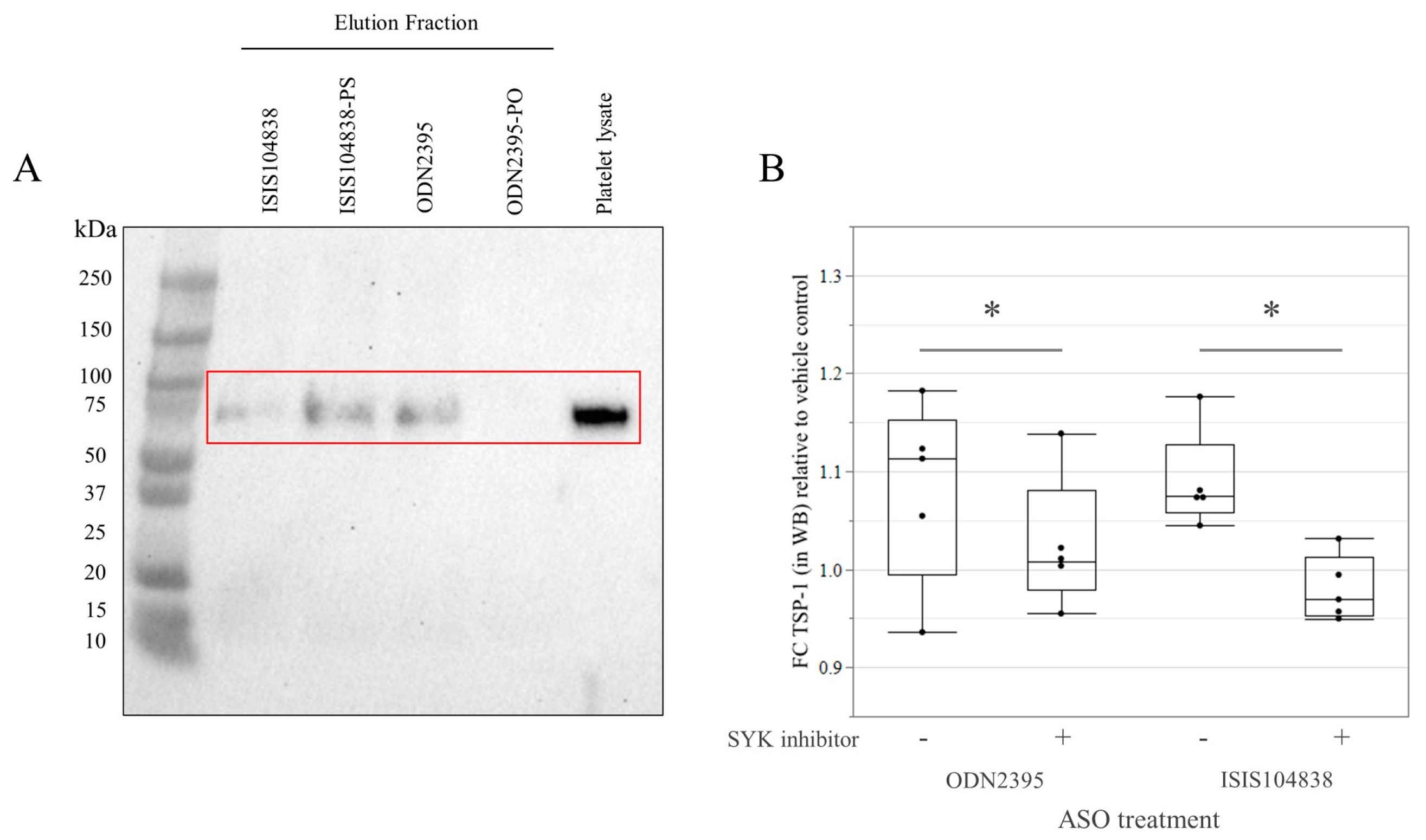
3.3. GPVI Protein Binding and Signaling in Göttingen Minipig Platelets
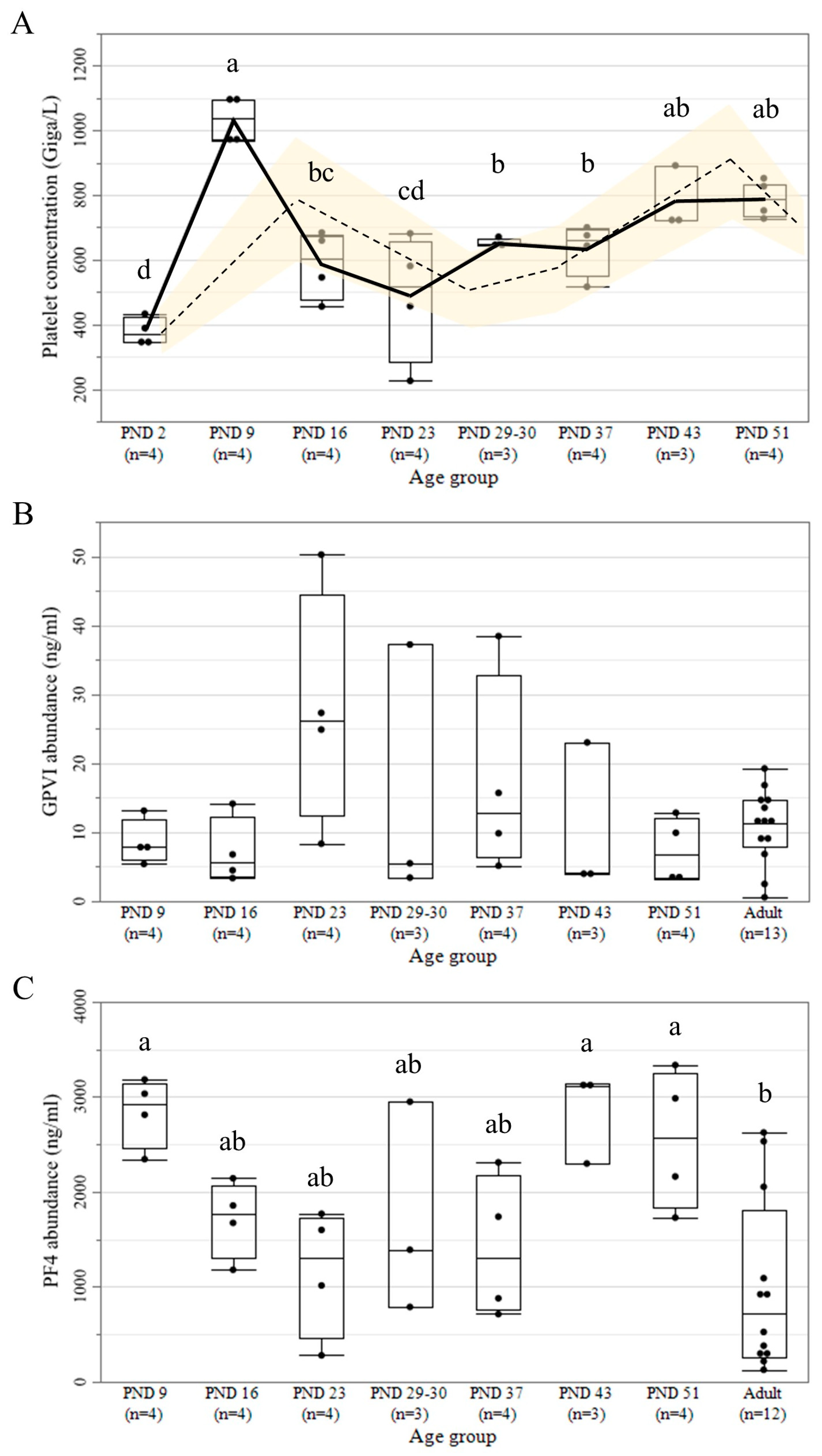
3.4. Platelet Count and Protein Abundance of GPVI and PF4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, S.F.; Meisler, M.H. Antisense Oligonucleotide Therapy for Neurodevelopmental Disorders. Dev. Neurosci. 2021, 43, 247–252. [Google Scholar] [CrossRef]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.D. The Powerful World of Antisense Oligonucleotides: From Bench to Bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.K. The Medicinal Chemistry of Antisense Oligonucleotides. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 39–69. ISBN 9781119070290. [Google Scholar]
- Krishnan, A.V.; Mishra, D. Antisense Oligonucleotides: A Unique Treatment Approach. Indian Pediatr. 2020, 57, 165–171. [Google Scholar] [CrossRef]
- Swayze, E.; Bhat, B. The Medicinal Chemistry of Oligonucleotides. In Antisense Drug Technology: Principles, Strategies, and Applications; Crooke, S., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 143–182. [Google Scholar]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Østergaard, M.E.; de Hoyos, C.L.; Wan, W.B.; Shen, W.; Low, A.; Berdeja, A.; Vasquez, G.; Murray, S.; Migawa, M.T.; Liang, X.H.; et al. Understanding the Effect of Controlling Phosphorothioate Chirality in the DNA Gap on the Potency and Safety of Gapmer Antisense Oligonucleotides. Nucleic Acids Res. 2020, 48, 1691–1700. [Google Scholar] [CrossRef]
- Laxton, C.; Brady, K.; Moschos, S.; Turnpenny, P.; Rawal, J.; Pryde, D.C.; Sidders, B.; Corbau, R.; Pickford, C.; Murray, E.J. Selection, Optimization, and Pharmacokinetic Properties of a Novel, Potent Antiviral Locked Nucleic Acid-Based Antisense Oligomer Targeting Hepatitis C Virus Internal Ribosome Entry Site. Antimicrob. Agents Chemother. 2011, 55, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Ho, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and Nontoxic Antisense Oligonucleotides. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Wood, M.J.A. Antisense Oligonucleotides: The next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol. 2018, 14, 9–22. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharm. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; de Hoyos, C.L.; Migawa, M.T.; Vickers, T.A.; Sun, H.; Low, A.; Bell, T.A.; Rahdar, M.; Mukhopadhyay, S.; Hart, C.E.; et al. Chemical Modification of PS-ASO Therapeutics Reduces Cellular Protein-Binding and Improves the Therapeutic Index. Nat. Biotechnol. 2019, 37, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, N. Hybridization-Independent Effects. In Oligonucleotide-Based Drugs and Therapeutics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018. [Google Scholar]
- Frazier, K.S. Antisense Oligonucleotide Therapies:The Promise and the Challenges from a Toxicologic Pathologist’s Perspective. Toxicol. Pathol. 2015, 43, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Senín, L.D.; Garro, C.B.; Gómez, E.S.; Santos-Rubio, M.D. Inotersen and Severe Thrombocytopenia: 2 Case Reports and Review. Int. J. Clin. Pharmacol. Ther. 2022, 60, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Henry, S.P.; Narayanan, P.; Shen, L.; Bhanot, S.; Younis, H.S.; Burel, S.A. Assessment of the Effects of 2′-Methoxyethyl Antisense Oligonucleotides on Platelet Count in Cynomolgus Nonhuman Primates. Nucleic Acid Ther. 2017, 27, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, P.; Shen, L.; Curtis, B.R.; Bourdon, M.A.; Nolan, J.P.; Gupta, S.; Hoffmaster, C.; Zhou, F.; Christian, B.; Schaubhut, J.L.; et al. Investigation into the Mechanism(s) That Leads to Platelet Decreases in Cynomolgus Monkeys during Administration of ISIS 104838, a 20-MoE-Modified Antisense Oligonucleotide. Toxicol. Sci. 2018, 164, 613–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.; Gatti, P.; Papoian, T. Safety of Antisense Oligonucleotide and SiRNA-Based Therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Witztum, J.L.; Kwoh, T.J.; Pham, N.C.; Salgado, N.; McEvoy, B.W.; Cheng, W.; Hughes, S.G.; Bhanot, S.; et al. The Effects of 2′-O-Methoxyethyl Containing Antisense Oligonucleotides on Platelets in Human Clinical Trials. Nucleic Acid Ther. 2017, 27, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Flierl, U.; Nero, T.L.; Lim, B.; Arthur, J.F.; Yao, Y.; Jung, S.M.; Gitz, E.; Pollitt, A.Y.; Zaldivia, M.T.K.; Jandrot-Perrus, M.; et al. Phosphorothioate Backbone Modifications of Nucleotide-Based Drugs Are Potent Platelet Activators. J. Exp. Med. 2015, 212, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Jaax, M.E.; Krauel, K.; Marschall, T.; Brandt, S.; Gansler, J.; Fürll, B.; Appel, B.; Fischer, S.; Block, S.; Helm, C.A.; et al. Complex Formation with Nucleic Acids and Aptamers Alters the Antigenic Properties of Platelet Factor 4. Blood 2013, 122, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.; Antonsson, M.; Elebring, M.; Jansson-Löfmark, R.; Weidolf, L. Drug Metabolism and Pharmacokinetic Strategies for Oligonucleotide- and MRNA-Based Drug Development. Drug Discov. Today 2018, 23, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Lightfoot, H.; Schneider, A.; Hall, J. Pharmacokinetics and Pharmacodynamics of Antisense Oligonucleotides. In Oligonucleotide-Based Drugs and Therapeutics; Ferrari, N., Seguin, R., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; pp. 107–136. ISBN 9781119070290. [Google Scholar]
- Crooke, S.T.; Vickers, T.A.; Liang, X.H. Phosphorothioate Modified Oligonucleotide-Protein Interactions. Nucleic Acids Res. 2021, 48, 5235–5253. [Google Scholar] [CrossRef] [PubMed]
- Slingsby, M.H.L.; Vijey, P.; Tsai, I.T.; Roweth, H.; Couldwell, G.; Wilkie, A.R.; Gaus, H.; Goolsby, J.M.; Okazaki, R.; Terkovich, B.E.; et al. Sequence-Specific 2’-O-Methoxyethyl Antisense Oligonucleotides Activate Human Platelets through Glycoprotein VI, Triggering Formation of Platelet-Leukocyte Aggregates. Haematologica 2022, 107, 519–531. [Google Scholar] [CrossRef]
- Sewing, S.; Roth, A.B.; Winter, M.; Dieckmann, A.; Bertinetti-Lapatki, C.; Tessier, Y.; McGinnis, C.; Huber, S.; Koller, E.; Ploix, C.; et al. Assessing Single-Stranded Oligonucleotide Drug-Induced Effects in Vitro Reveals Key Risk Factors for Thrombocytopenia. PLoS ONE 2017, 12, e0187574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braendli-Baiocco, A.; Festag, M.; Erichsen, K.D.; Persson, R.; Mihatsch, M.J.; Fisker, N.; Funk, J.; Mohr, S.; Constien, R.; Ploix, C.; et al. The Minipig Is a Suitable Non-Rodent Model in the Safety Assessment of Single Stranded Oligonucleotides. Toxicol. Sci. 2017, 157, 112–128. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, A.; Tardiveau, C.; Ayuso, M.; Buyssens, L.; Bars, C.; van Ginneken, C.; Fant, P.; Leconte, I.; Braendli-Baiocco, A.; Parrott, N.; et al. Safety Testing of an Antisense Oligonucleotide Intended for Pediatric Indications in the Juvenile Göttingen Minipig, Including an Evaluation of the Ontogeny of Key Nucleases. Pharmaceutics 2021, 13, 1442. [Google Scholar] [CrossRef]
- Baenziger, N.L.; Brodie, G.N.; Majerus, P.W. Isolation and Properties of a Thrombin-Sensitive Protein of Human Platelets. J. Biol. Chem. 1972, 247, 2723–2731. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Jackson, S.P. Platelet Thrombus Formation in Flowing Blood. In Platelets; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar] [CrossRef]
- Starlinger, P.; Moll, H.P.; Assinger, A.; Nemeth, C.; Hoetzenecker, K.; Gruenberger, B.; Gruenberger, T.; Kuehrer, I.; Schoppmann, S.F.; Gnant, M.; et al. Thrombospondin-1: A Unique Marker to Identify in Vitro Platelet Activation When Monitoring in Vivo Processes. J. Thromb. Haemost. 2010, 8, 1809–1819. [Google Scholar] [CrossRef]
- Yun, S.H.; Sim, E.H.; Goh, R.Y.; Park, J.I.; Han, J.Y. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed Res. Int. 2016, 2016, 9060143. [Google Scholar] [CrossRef] [Green Version]
- Wiedmeier, S.E.; Henry, E.; Sola-Visner, M.C.; Christensen, R.D. Platelet Reference Ranges for Neonates, Defined Using Data from over 47000 Patients in a Multihospital Healthcare System. J. Perinatol. 2009, 29, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Faustini, M.; Bronzo, V.; Maffeo, G.; Russo, V.; Munari, E.; Vigo, D. Reference Intervals and Age-Related Changes for Platelet Count, Mean Platelet Volume and Plateletcrit in Healthy Pre-Weaning Piglets in Italy. J. Vet. Med. Ser. A Physiol. Pathol. Clin. Med. 2003, 50, 466–469. [Google Scholar] [CrossRef] [PubMed]
- van Poelgeest, E.P.; Hodges, M.R.; Moerland, M.; Tessier, Y.; Levin, A.A.; Persson, R.; Lindholm, M.W.; Dumong Erichsen, K.; Ørum, H.; Cohen, A.F.; et al. Antisense-Mediated Reduction of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9): A First-in-Human Randomized, Placebo-Controlled Trial. Br. J. Clin. Pharm. 2015, 80, 1350–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, M.W.; Elmén, J.; Fisker, N.; Hansen, H.F.; Persson, R.; Møller, M.R.; Rosenbohm, C.; Ørum, H.; Straarup, E.M.; Koch, T. PCSK9 LNA Antisense Oligonucleotides Induce Sustained Reduction of LDL Cholesterol in Nonhuman Primates. Mol. Ther. 2012, 20, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell, K.L.; Geary, R.S.; Baker, B.F.; Glover, J.M.; Mant, T.G.K.; Yu, R.Z.; Tami, J.A.; Dorr, F.A. Phase I Trial of ISIS 104838, a 2′-Methoxyethyl Modified Antisense Oligonucleotide Targeting Tumor Necrosis Factor-α. J. Pharmacol. Exp. Ther. 2002, 303, 1334–1343. [Google Scholar] [CrossRef]
- Burel, S.A.; Machemer, T.; Baker, B.F.; Kwoh, T.J.; Paz, S.; Younis, H.; Henry, S.P. Early-Stage Identification and Avoidance of Antisense Oligonucleotides Causing Species-Specific Inflammatory Responses in Human Volunteer Peripheral Blood Mononuclear Cells. Nucleic Acid Ther. 2022, 32, 457–472. [Google Scholar] [CrossRef]
- Chan, M.V.; Warner, T.D. Standardised Optical Multichannel (Optimul) Platelet Aggregometry Using High-Speed Shaking and Fixed Time Point Readings. Platelets 2012, 23, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Vinholt, P.J.; Nybo, M.; Nielsen, C.B.; Hvas, A.M. Light Transmission Aggregometry Using Pre-Coated Microtiter Plates and a Victor X5 Plate Reader. PLoS ONE 2017, 12, e0185675. [Google Scholar] [CrossRef] [Green Version]
- Bollen, P.J.A.; Madsen, L.W.; Meyer, O.; Ritskes-Hoitinga, J. Growth Differences of Male and Female Göttingen Minipigs during Ad Libitum Feeding: A Pilot Study. Lab. Anim. 2005, 39, 80–93. [Google Scholar] [CrossRef]
- Lambert, M.; Reznikov, A.; Nguyen, Y.; Rauova, L.; Poncz, M. Platelet Factor 4 Levels Inversely Correlate with Platelet Transfusion Needs In Pediatric Patients Treated for Standard Risk Acute Lymphoblastic Leukemia. Blood 2010, 116, 725. [Google Scholar] [CrossRef]
- Cabeza, N.; Li, Z.; Schulz, C.; Kremmer, E.; Massberg, S.; Bültmann, A.; Gawaz, M. Surface Expression of Collagen Receptor Fc Receptor-γ/Glycoprotein VI Is Enhanced on Platelets in Type 2 Diabetes and Mediates Release of CD40 Ligand and Activation of Endothelial Cells. Diabetes 2004, 53, 2117–2121. [Google Scholar] [CrossRef] [Green Version]
- Feyen, B.; Penard, L.; van Heerden, M.; Fant, P.; Marsden, E.; de Jonghe, S.; Desmidt, M.; Mousa, S.M.; Bailey, G.P. “All Pigs Are Equal” Does the Background Data from Juvenile Göttingen Minipigs Support This? Reprod. Toxicol. 2016, 64, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Grossi, A.B.; Zeltner, A.; Christoffersen, C.; Søndergaard, A.C. Reference Data of Clinical Chemistry and Hematology in Juvenile Göttingen Minipigs. Toxicol. Lett. 2016, 258, S235. [Google Scholar] [CrossRef]
- Pawlowsky, K.; Ernst, L.; Steitz, J.; Stopinski, T.; Kögel, B.; Henger, A.; Kluge, R.; Tolba, R. The Aachen Minipig: Phenotype, Genotype, Hematological and Biochemical Characterization, and Comparison to the Göttingen Minipig. Eur. Surg. Res. 2017, 58, 193–203. [Google Scholar] [CrossRef] [PubMed]
- van Donge, T.; Evers, K.; Koch, G.; van den Anker, J.; Pfister, M. Clinical Pharmacology and Pharmacometrics to Better Understand Physiological Changes during Pregnancy and Neonatal Life. Handb. Exp. Pharm. 2020, 261, 325–337. [Google Scholar] [CrossRef]
- Yu, R.Z.; Grundy, J.S.; Geary, R.S. Clinical Pharmacokinetics of Second Generation Antisense Oligonucleotides. Expert Opin. Drug Metab. Toxicol. 2013, 9, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, A.; Adams, M.; Cao, X.; Yamaguchi, A.; Henderson, J.; Busch-Østergren, P.; Udager, A.; Pitchiaya, S.; Tourdot, B.; Kasputis, T.; et al. Antisense Oligonucleotides and Nucleic Acids Generate Hypersensitive Platelets. Thromb. Res. 2021, 200, 64–71. [Google Scholar] [CrossRef]
- Karaki, S.; Paris, C.; Rocchi, P. Antisense Oligonucleotides, A Novel Developing Targeting Therapy. In Antisense Therapy; BoD—Books on Demand GmbH: Norderstedt, Germany, 2019. [Google Scholar]
- Liang, X.H.; Shen, W.; Sun, H.; Kinberger, G.A.; Prakash, T.P.; Nichols, J.G.; Crooke, S.T. Hsp90 Protein Interacts with Phosphorothioate Oligonucleotides Containing Hydrophobic 2′-Modifications and Enhances Antisense Activity. Nucleic Acids Res. 2016, 44, 3892–3907. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, S.I.; Miyake, K. CHAPTER 13: Nucleic Acid Innate Immune Receptors. In RSC Drug Discovery Series; The Royal Society of Chemistry: London, UK, 2019; Volume 2019-January. [Google Scholar]
- Agrawal, S.; Kandimalla, E.R. CHAPTER 14: Synthetic Agonists of Toll-like Receptors and Therapeutic Applications. In RSC Drug Discovery Series; The Royal Society of Chemistry: London, UK, 2019; Volume 2019-January. [Google Scholar]
- Mangsbo, S.M.; Sanchez, J.; Anger, K.; Lambris, J.D.; Ekdahl, K.N.; Loskog, A.S.; Nilsson, B.; Tötterman, T.H. Complement Activation by CpG in a Human Whole Blood Loop System: Mechanisms and Immunomodulatory Effects. J. Immunol. 2009, 183, 6724–6732. [Google Scholar] [CrossRef] [Green Version]
- Henry, S.P.; Kim, T.W.; Kramer-Stickland, K.; Zanardi, T.A.; Fey, R.A.; Levin, A.A. Toxicologic Properties of 2′-o-Methoxyethyl Chimeric Antisense Inhibitors in Animals and Man. In Antisense Drug Technology: Principles, Strategies, and Applications, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Zhang, X.; Kimura, Y.; Fang, C.; Zhou, L.; Sfyroera, G.; Lambris, J.D.; Wetsel, R.A.; Miwa, T.; Song, W.C. Regulation of Toll-like Receptor-Mediated Inflammatory Response by Complement in Vivo. Blood 2007, 110, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Andersson, P.; den Besten, C. Preclinical and Clinical Drug-Metabolism, Pharmacokinetics and Safety of Therapeutic Oligonucleotides. In RSC Drug Discovery Series; Agrawal, S., Gait, M.J., Eds.; Royal Society of Chemistry: London, UK, 2019; Volume 2019-Januaay, pp. 474–517. ISBN 978-1-78801-571-4. [Google Scholar]
- Chan, M.V.; Armstrong, P.C.; Warner, T.D. 96-Well Plate-Based Aggregometry. Platelets 2018, 29, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filkova, A.A.; Martyanov, A.A.; Garzon Dasgupta, A.K.; Panteleev, M.A.; Sveshnikova, A.N. Quantitative Dynamics of Reversible Platelet Aggregation: Mathematical Modelling and Experiments. Sci. Rep. 2019, 9, 6217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, C.J.; Larson, M.G.; Feng, D.L.; Sutherland, P.A.; Lindpaintner, K.; Myers, R.H.; D’Agostino, R.A.; Levy, D.; Tofler, G.H. Genetic and Environmental Contributions to Platelet Aggregation: The Framingham Heart Study. Circulation 2001, 103, 3051–3056. [Google Scholar] [CrossRef] [Green Version]
- Yee, D.L.; Sun, C.W.; Bergeron, A.L.; Dong, J.F.; Bray, P.F. Aggregometry Detects Platelet Hyperreactivity in Healthy Individuals. Blood 2005, 106, 2723–2729. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.I.; Garner, S.F.; Angenent, W.; Bernard, A.; Berzuini, C.; Burns, P.; Farndale, R.W.; Hogwood, J.; Rankin, A.; Stephens, J.C.; et al. Mapping the Platelet Profile for Functional Genomic Studies and Demonstration of the Effect Size of the GP6 Locus. J. Thromb. Haemost. 2007, 5, 1756–1765. [Google Scholar] [CrossRef] [Green Version]
- Panzer, S.; Höcker, L.; Koren, D. Agonists-Induced Platelet Activation Varies Considerably in Healthy Male Individuals: Studies by Flow Cytometry. Ann. Hematol. 2006, 85, 121–125. [Google Scholar] [CrossRef]
- Kunicki, T.J.; Nugent, D.J. The Genetics of Normal Platelet Reactivity. Blood 2010, 116, 2627–2634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, P.K.; Henry, S.; Li, N. Drug-Induced Thrombocytopenia: Mechanisms and Relevance in Preclinical Safety Assessment. Curr. Opin. Toxicol. 2019, 17, 23–30. [Google Scholar] [CrossRef]
- Jung, S.M.; Moroi, M.; Soejima, K.; Nakagaki, T.; Miura, Y.; Berndt, M.C.; Gardiner, E.E.; Howes, J.M.; Pugh, N.; Bihan, D.; et al. Constitutive Dimerization of Glycoprotein VI (GPVI) in Resting Platelets Is Essential for Binding to Collagen and Activation in Flowing Blood. J. Biol. Chem. 2012, 287, 30000–30013. [Google Scholar] [CrossRef] [Green Version]
- Curtis, B.R.; Swyers, J.; Divgi, A.; McFarland, J.G.; Aster, R.H. Thrombocytopenia after Second Exposure to Abciximab Is Caused by Antibodies That Recognize Abciximab-Coated Platelets. Blood 2002, 99, 2054–2059. [Google Scholar] [CrossRef]
- Santostefano, M.J.; Kirchner, J.; Vissinga, C.; Fort, M.; Lear, S.; Pan, W.J.; Prince, P.J.; Hensley, K.M.; Tran, D.; Rock, D.; et al. Off-Target Platelet Activation in Macaques Unique to a Therapeutic Monoclonal Antibody. Toxicol. Pathol. 2012, 40, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Warkentin, T.E. Heparin-Induced Thrombocytopenia: Pathogenesis and Management. Br. J. Haematol. 2003, 121, 535–555. [Google Scholar] [CrossRef] [PubMed]
- Furihata, K.; Clemetson, K.J.; Deguchi, H.; Kunicki, T.J. Variation in Human Platelet Glycoprotein VI Content Modulates Glycoprotein VI-Specific Prothrombinase Activity. Arter. Thromb. Vasc. Biol. 2001, 21, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Barrachina, M.N.; Sueiro, A.M.; Izquierdo, I.; Hermida-Nogueira, L.; Guitián, E.; Casanueva, F.F.; Farndale, R.W.; Moroi, M.; Jung, S.M.; Pardo, M.; et al. GPVI Surface Expression and Signalling Pathway Activation Are Increased in Platelets from Obese Patients: Elucidating Potential Anti-Atherothrombotic Targets in Obesity. Atherosclerosis 2019, 281, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadova-Bach, E.; Gil-Pulido, J.; Sarukhanyan, E.; Burkard, P.; Shityakov, S.; Schonhart, C.; Stegner, D.; Remer, K.; Nurden, P.; Nurden, A.T.; et al. Platelet Glycoprotein VI Promotes Metastasis through Interaction with Cancer Cell–Derived Galectin-3. Blood 2020, 135, 1146–1160. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, P.K.; Curtis, B.R.; Shen, L.; Schneider, E.; Tami, J.A.; Paz, S.; Burel, S.A.; Tai, L.J.; Machemer, T.; Kwoh, T.J.; et al. Underlying Immune Disorder May Predispose Some Transthyretin Amyloidosis Subjects to Inotersen-Mediated Thrombocytopenia. Nucleic Acid Ther. 2020, 30, 94–103. [Google Scholar] [CrossRef]
- Al-Tamimi, M.; Mu, F.T.; Moroi, M.; Gardiner, E.E.; Berndt, M.C.; Andrews, R.K. Measuring Soluble Platelet Glycoprotein VI in Human Plasma by ELISA. Platelets 2009, 20, 143–149. [Google Scholar] [CrossRef]
- Al-Tamimi, M.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K. Focusing on Plasma Glycoprotein VI. Thromb. Haemost. 2012, 107, 648–655. [Google Scholar]
- Rajasekhar, D.; Kestin, A.S.; Bednarek, F.J.; Ellis, P.A.; Barnard, M.R.; Michelson, A.D. Neonatal Platelets Are Less Reactive than Adult Platelets to Physiological Agonists in Whole Blood. Thromb. Haemost. 1994, 72, 957–963. [Google Scholar] [CrossRef]
- Rajasekhar, D.; Barnard, M.R.; Bednarek, F.J.; Michelson, A.D. Platelet Hyporeactivity in Very Low Birth Weight Neonates. Thromb. Haemost. 1997, 77, 1002–1007. [Google Scholar] [CrossRef]
- Israels, S.J.; Daniels, M.; McMillan, E.M. Deficient Collagen-Induced Activation in the Newborn Platelet. Pediatr. Res. 1990, 27, 337–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Marin, F.; Liu, Z.J.; Gutti, R.; Sola-Visner, M. Neonatal Thrombocytopenia and Megakaryocytopoiesis. Semin. Hematol. 2010, 47, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corby, D.G.; O’Barr, T.P. Decreased Alpha-Adrenergic Receptors in Newborn Platelets: Cause of Abnormal Response to Epinephrine. Dev. Pharm. 1981, 2, 215–225. [Google Scholar] [CrossRef]
- Caparrós-Pérez, E.; Teruel-Montoya, R.; López-Andreo, M.J.; Llanos, M.C.; Rivera, J.; Palma-Barqueros, V.; Blanco, J.E.; Vicente, V.; Martínez, C.; Ferrer-Marín, F. Comprehensive Comparison of Neonate and Adult Human Platelet Transcriptomes. PLoS ONE 2017, 12, e0183042. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Rauova, L.; Bailey, M.; Sola-Visner, M.C.; Kowalska, M.A.; Poncz, M. Platelet Factor 4 Is a Negative Autocrine in Vivo Regulator of Megakaryopoiesis: Clinical and Therapeutic Implications. Blood 2007, 110, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Wang, Y.; Bdeir, K.H.; Nguyen, Y.; Kowalska, M.A.; Poncz, M. Platelet Factor 4 Regulates Megakaryopoiesis through Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) on Megakaryocytes. Blood 2009, 114, 2290–2298. [Google Scholar] [CrossRef]
- Gewirtz, A.; Calabretta, B.; Rucinski, B.; Niewiarowski, S.; Xu, W.Y. Inhibition of Human Megakaryocytopoiesis in Vitro by Platelet Factor 4 (PF4) and a Synthetic COOH-Terminal PF4 Peptide. J. Clin. Investig. 1989, 83, 1477–1486. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ASO | Modifications | Sequence (5’-3’) | Length | PS Load | Platelet Effect |
|---|---|---|---|---|---|
| ODN2395 | PS | t*c*g*t*c*g*t*t*t*t*c*g*g*c*g*c*g*c*g*c*c*g* | 22 | 21 | platelet activation |
| ODN2395-PO | unmodified | tcgtcgttttcggcgcgcgccg | 22 | 0 | no effect |
| ISIS104838 | 2’MOE/PS/2’MOE | G*C*T*G*A*t*t*a*g*a*g*a*g*a*g*G*T*C*C*C* | 20 | 19 | platelet activation |
| ISIS104838-PS | PS | g*c*t*g*a*t*t*a*g*a*g*a*g*a*g*g*t*c*c*c* | 20 | 19 | unknown |
| RTR5001 | LNA/PS/LNA | T*G*C*t*a*c*a*a*a*a*c*C*C*A* | 14 | 13 | no effect |
| RTR5001-PS | PS | t*g*c*t*a*c*a*a*a*a*c*c*c*a* | 14 | 13 | unknown |
| RTR5001-PO | unmodified | tgctacaaaaccca | 14 | 0 | unknown |
| ASO | Modifications | Length | PS Load | Platelet Effect | |||
|---|---|---|---|---|---|---|---|
| Human, NHP, Göttingen minipig | Göttingen minipig | ||||||
| In Vivo | In Vitro H | In Vitro | |||||
| Activation Assay, Aggregometry | Activation Assay | Aggregometry | |||||
| ODN2395 | PS | 22 | 21 | unknown | activation, aggregation | activation | aggregation |
| ODN2395-PO | unmodified | 22 | 0 | unknown | no effect | PRP: no effectWB: activation | no effect |
| ISIS104838 | 2′MOE/PS/2′MOE | 20 | 19 | thrombocytopenia H,N | activation | activation | aggregation |
| ISIS104838-PS | PS | 20 | 19 | unknown | unknown | PRP: no effectWB: activation | aggregation |
| RTR5001 | LNA/PS/LNA | 14 | 13 | no effect H,N,M | unknown | no effect | no effect |
| RTR5001-PS | PS | 14 | 13 | unknown | unknown | activation | aggregation |
| RTR5001-PO | unmodified | 14 | 0 | unknown | unknown | no effect | no effect |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenzuela, A.; Ayuso, M.; Buyssens, L.; Bars, C.; Van Ginneken, C.; Tessier, Y.; Van Cruchten, S. Platelet Activation by Antisense Oligonucleotides (ASOs) in the Göttingen Minipig, including an Evaluation of Glycoprotein VI (GPVI) and Platelet Factor 4 (PF4) Ontogeny. Pharmaceutics 2023, 15, 1112. https://doi.org/10.3390/pharmaceutics15041112
Valenzuela A, Ayuso M, Buyssens L, Bars C, Van Ginneken C, Tessier Y, Van Cruchten S. Platelet Activation by Antisense Oligonucleotides (ASOs) in the Göttingen Minipig, including an Evaluation of Glycoprotein VI (GPVI) and Platelet Factor 4 (PF4) Ontogeny. Pharmaceutics. 2023; 15(4):1112. https://doi.org/10.3390/pharmaceutics15041112
Chicago/Turabian StyleValenzuela, Allan, Miriam Ayuso, Laura Buyssens, Chloé Bars, Chris Van Ginneken, Yann Tessier, and Steven Van Cruchten. 2023. "Platelet Activation by Antisense Oligonucleotides (ASOs) in the Göttingen Minipig, including an Evaluation of Glycoprotein VI (GPVI) and Platelet Factor 4 (PF4) Ontogeny" Pharmaceutics 15, no. 4: 1112. https://doi.org/10.3390/pharmaceutics15041112
APA StyleValenzuela, A., Ayuso, M., Buyssens, L., Bars, C., Van Ginneken, C., Tessier, Y., & Van Cruchten, S. (2023). Platelet Activation by Antisense Oligonucleotides (ASOs) in the Göttingen Minipig, including an Evaluation of Glycoprotein VI (GPVI) and Platelet Factor 4 (PF4) Ontogeny. Pharmaceutics, 15(4), 1112. https://doi.org/10.3390/pharmaceutics15041112