

New Insights into Alzheimer’s Disease: Novel Pathogenesis, Drug Target and Delivery
 ,
,
Abstract
:
1. Introduction
2. Developed Drugs Targeting Aβ and Tau for AD Therapy
2.1. Drugs Targeting Aβ
2.1.1. Drugs Used to Reduce Aβ Production

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Drug Target | Phase | Effect in Clinical Trials | Status | Refs. |
|---|---|---|---|---|---|
| Umibecestat (CNP520) | β-secretase | Phase 2/3 (NCT03131453; NCT02565511) | Cognitive function decreased slightly, brain atrophy increased, weight loss | Discontinued | [63,64] |
| CTS-21166 | β-secretase | Phase 1 (NCT00621010) | Reduced Aβ in plasma with long-lasting and well-tolerated effects | Discontinued | [65,66] |
| LY2811376 | β-secretase | Phase 1 (NCT00838084) | Reduced Aβ in CSF *; adverse effects: retinal toxicity | Discontinued | [67] |
| LY2886721 | β-secretase | Phase 1 Phase 2 (NCT01561430) | Reduced Aβ in plasma and CSF *; adverse effects: abnormal elevation of liver enzymes | Discontinued | [68] |
| AZD3839 | β-secretase | Phase 1 (NCT01348737) | Slightly reduced Aβ in plasma at doses that did not disrupt cardiac activity | Completed | [69] |
| Verubecestat (MK-8931) | β-secretase | Phase 3 (NCT01953601) | Well-tolerated; reduced Aβ40 levels in CSF * | Discontinued | [70] |
| Lanabecestat | β-secretase | Phase 3 (NCT02972658; NCT02783573) | Reduced Aβ40 and Aβ42 levels in plasma and CSF * | Discontinued | [71] |
| Elenbecestat (E2609) | β-secretase | Phase 3 (NCT02956486) | Well-tolerated; reduced Aβ levels in plasma and CSF*; reduced BACE1 enzyme activity in CSF *; did not alter BACE1 levels | Discontinued | [72,73] |
| Atabecestat (JNJ-54861911) | β-secretase | Phase 2 Phase 3 (NCT02569398) | Reduced Aβ levels in CSF * and plasma; adverse effects: cognitive deterioration, and elevated liver enzymes | Discontinued | [74,75,76,77,78] |
| LY3202626 | β-secretase | Phase 2 (NCT02791191; NCT03367403) | Resulted in high blood–brain barrier permeability; reduced Aβ1-42 in CSF *; no reduction in cognitive impairment and tau load | Discontinued | [79,80] |
| Semagacestat (LY450139) | γ-secretase | Phase 3 (NCT01035138; NCT00762411; NCT00594568) | Reduced the production of Aβ in patients; no reduction in cognitive impairment; adverse reactions: increased risk of skin cancer and infection | Discontinued | [41,81] |
| Avagacestat (BMS-708,163) | γ-secretase | Phase 2 (NCT00890890; NCT00810147) | Slightly reduced Aβ levels in CSF *; adverse reactions: gastrointestinal symptoms, skin diseases, and non-melanoma skin cancer | Discontinued | [82,83] |
| Tarenflurbil (R-flurbiprofen) | γ-secretase | Phase 3 (NCT00380276; NCT00380276; NCT00105547) | No reduction in cognitive impairment; adverse effects: dizziness, anemia and infection | Discontinued | [44,45] |
| PF-06648671 (Pfizer) | γ-secretase | Phase 1 (NCT02407353; NCT02440100) | Well-tolerated in healthy subjects; reduced plasma Aβ40 and Aβ42 and increased Aβ37 and Aβ38 | Discontinued | [84] |
| CHF5074 | γ-secretase | Phase 2 (NCT01303744) | Reduced inflammatory factor CD40 and TNF-α concentrations in CSF; improved executive function in ApoE4 gene carriers | Inactive | [85] |
| Bryostatin1 | α-secretase | Phase 2 (NCT02431468; NCT04538066) | Reduced Aβ40 and Aβ42 and cognitive impairment | Active, not recruiting | [50] |
| Isotretinoin | α-secretase | Phase 1 Phase 2 (NCT01560585) | Adverse events in 2/3 participants | Terminated | [47] |
| EHT0202 | α-secretase | Phase 2 (NCT00880412) | No significant effect | Completed | [86] |
| Acitretin | α-secretase | Phase 2 (NCT01078168) | Significantly increased CSF * APPs-α; safe and well-tolerated | Completed | [51] |
| Curcumin | α-secretase | Phase 2 (NCT00164749; NCT00099710; NCT01811381) | No effects on cognitive function and CFS * and plasma Aβ levels | Unknown | [47] |
2.1.2. Drugs Used to Prevent Aβ Aggregation
2.1.3. Drugs Used to Promote Aβ Clearance
2.2. Drugs Targeting Tau Protein
| Drug Name | Principle | Phase | Effect in Clinical Trials | Status | Refs. |
|---|---|---|---|---|---|
| TRx0237 (LMTM) | Inhibit Tau aggregation | Phase 3 (NCT01689233; NCT01689246; NCT02245568) | Did not significantly affect cognitive decline | Active, not recruiting | [130] |
| TPI-287 | microtubule stabilizer | Phase 1 (NCT01966666) | Severe hypersensitivity reactions | Completed | [131] |
| Tilavonemab (ABBV-8E12) | Passive immunity | Phase 2 (NCT03712787; NCT02880956) | Did not change the decline of cognitive, or lower brain atrophy or levels of plasma neurofilament light | Discontinued | [134] |
| BIIB076 (NI-105) | Passive immunity | Phase 1 (NCT03056729) | Reduced half the amount of mid-region-bearing tau in CSF * | Discontinued | [135] |
| Gosuranemab (BIIB092) | Passive immunity | Phase 2 (NCT03352557) | Lack of efficacy | Discontinued | https://www.clinicaltrials.gov/ (accessed on 28 March 2023) |
| Semorinemab (RO07105705) | Passive immunity | Phase 2 (NCT03289143; NCT03828747) | Caused 43.6% slowed decline in the ADAS-Cog11 coprimary, and did not change tangle accumulation | Active, not recruiting | https://www.clinicaltrials.gov/ (accessed on 28 March 2023) |
| Bepranemab (UCB0107) | Passive immunity | Phase 2 (NCT04867616) | No drug-related adverse events or changes in safety results were reported | Active, not recruiting | [136,137] |
| Zagotenemab (LY3303560) | Passive immunity | Phase 2 (NCT03518073) | Missed its primary endpoint | Discontinued | [138,139] |
| JNJ-63733657 | Passive immunity | Phase 2 (NCT04619420) | Dose-dependent reductions in free p217 tau in CSF * in volunteers. Adverse reactions: back pain and headache | Recruiting | https://www.clinicaltrials.gov/ (accessed on 28 March 2023) |
| AAD-vac1 | Active immunity | Phase 2 (NCT02579252) | Reduced brain atrophy and cognitive decline in mild to moderate AD * patients; reduced the levels of p-tau181 and p-tau217 | Completed | [132,133,140] |
| ACI-35 | Active immunity | Phase 1 (NCT04445831) | Developed antitau IgG and IgM antibodies preferentially against phosphorylated tau, with high IgG titers | Active, not recruiting | [141] |
2.3. Drugs Targeting Calcium Balance and Reactive Oxygen Species
2.4. New Hypotheses and Drug Targets for AD Treatments
2.4.1. Exogenous Formaldehyde Directly Induces AD-Like Pathology
2.4.2. Age-Related Endogenous Formaldehyde Induces Memory Decline
2.4.3. Formaldehyde Elicits Aβ Oligomerization and Fibrillation

2.4.4. Formaldehyde Promotes Tau Hyperphosphorylation and NFTs Formation
2.4.5. Endogenous Formaldehyde as a Target for AD Therapy
2.4.6. Formaldehyde-Degrading Enzyme-ALDH2 as a Target for AD Treatments
2.4.7. Formaldehyde-Degrading Enzyme-ALDH2 as a Target for AD Treatments
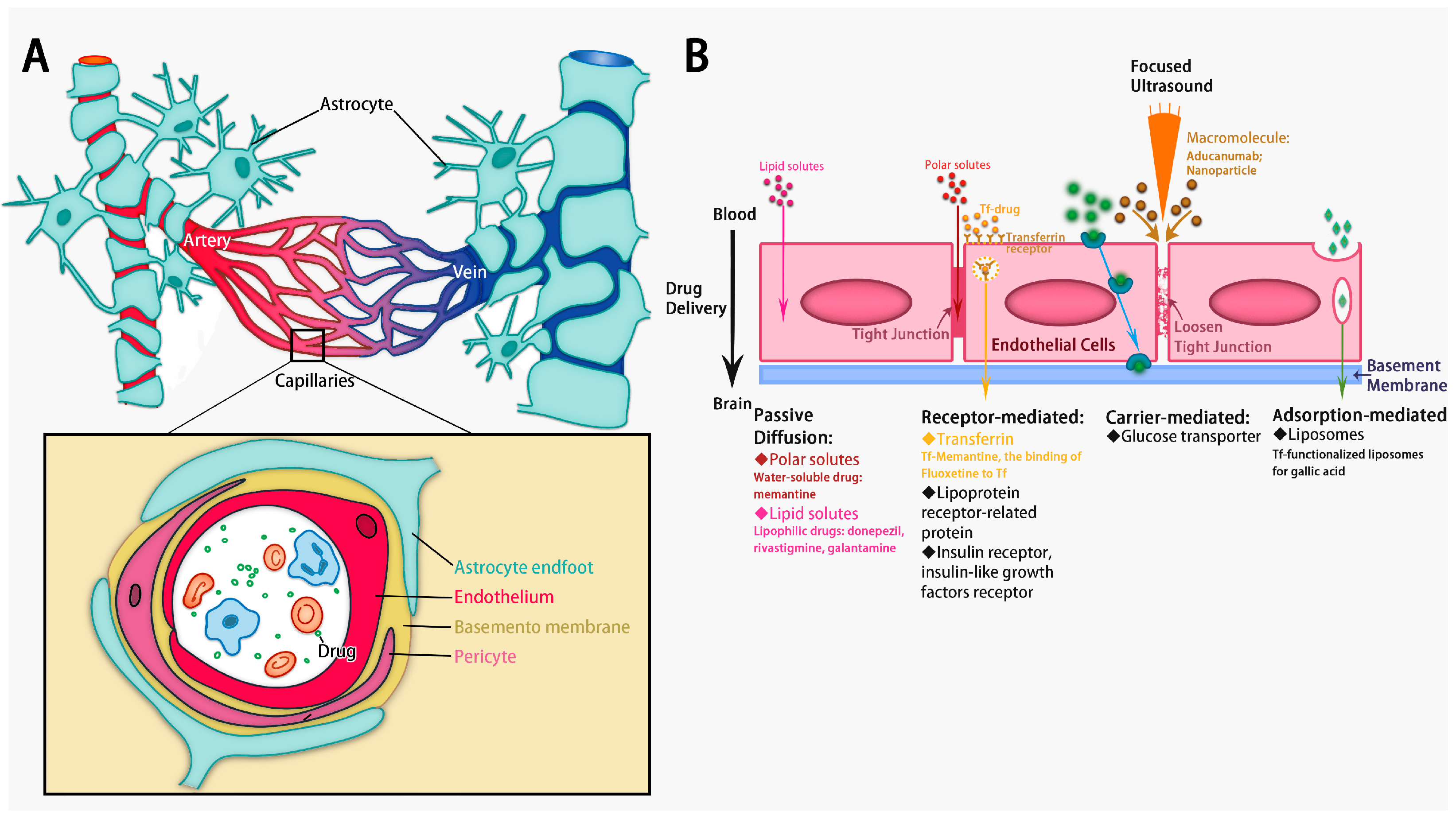
3. Enhancing BBB Penetration for Drug Delivery in AD
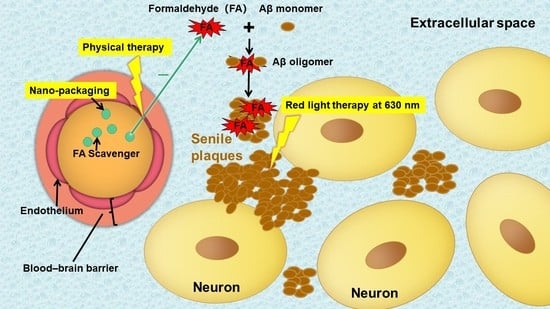
4. Aβ Plaques Deposition in ECS Blocks Drug Delivery in AD
5. Novel Drug Delivery for AD Treatments
5.1. Drug Delivery via Brain ECS
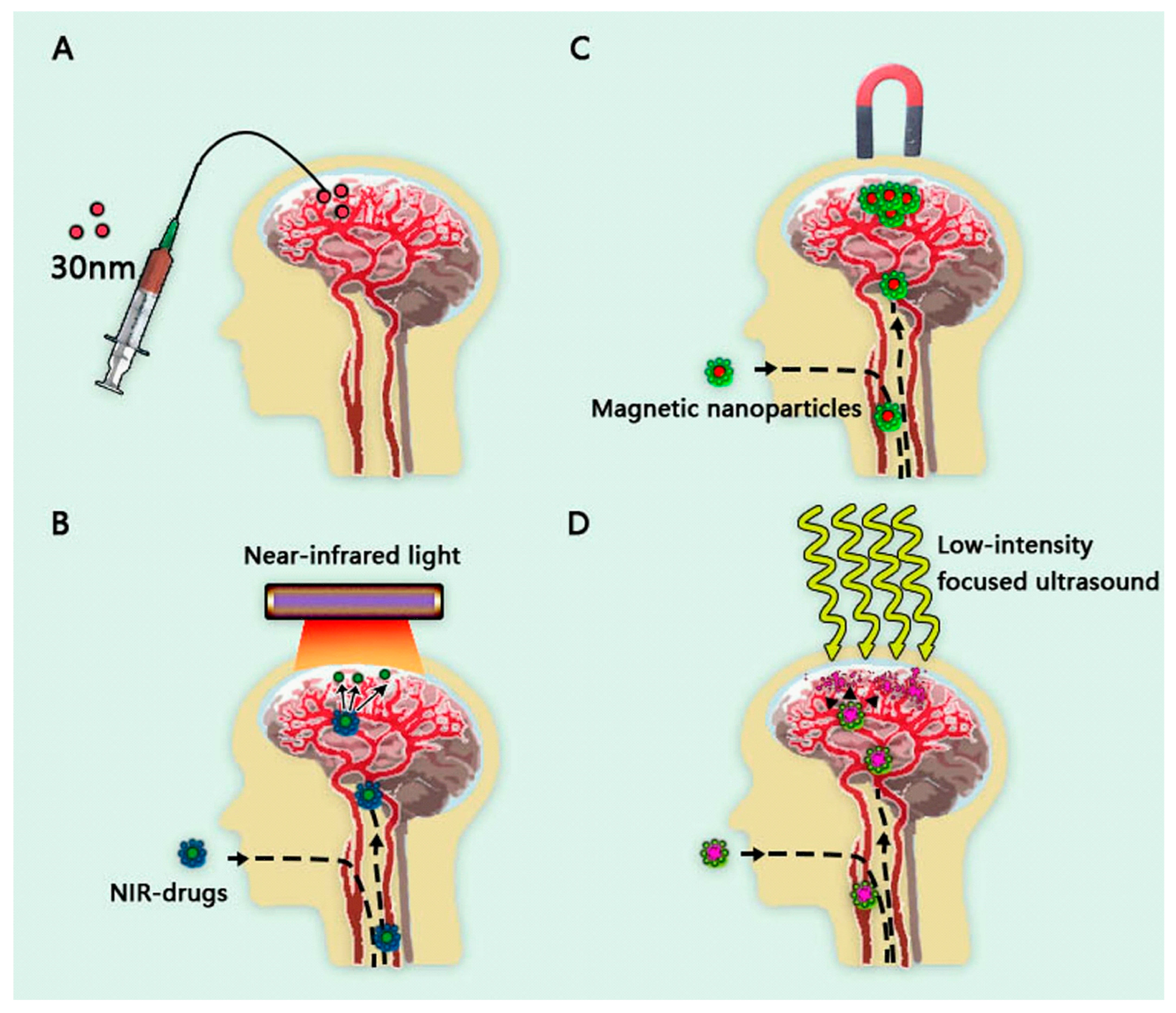
5.2. Magnetic Nanoparticles
5.3. Near-Infrared Photosensitive Nanomedicines
5.4. Combination of Focused Ultrasound and Nanomedicines

5.5. Extracellular Vesicles
5.6. BBB Shuttle Peptide
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Chen, H.; Zhao, H.; Yang, W.; Song, Y.; Li, X.; Wang, Y.; Du, D.; Liao, H.; Pan, W.; et al. New insight into brain disease therapy: Nanomedicines-crossing blood-brain barrier and extracellular space for drug delivery. Expert. Opin. Drug. Deliv. 2022, 19, 1618–1635. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, B.C.; Wu, A.J.; Li, M.; Cheung, K.H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell. Res. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Guo, J.; Ye, X.Y.; Xie, Y.; Xie, T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res. Rev. 2022, 77, 101619. [Google Scholar] [CrossRef]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug. Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Kong, C.; Yang, E.J.; Shin, J.; Park, J.; Kim, S.H.; Park, S.W.; Chang, W.S.; Lee, C.H.; Kim, H.; Kim, H.S.; et al. Enhanced delivery of a low dose of aducanumab via FUS in 5×FAD mice, an AD model. Transl. Neurodegener. 2022, 11, 57. [Google Scholar] [CrossRef]
- Shamsi, A.; Shahwan, M.; Alhumaydhi, F.A.; Alwashmi, A.S.S.; Aljasir, M.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Hassan, M.I.; Islam, A. Spectroscopic, calorimetric and in silico insight into the molecular interactions of Memantine with human transferrin: Implications of Alzheimer’s drugs. Int. J. Biol. Macromol. 2021, 190, 660–666. [Google Scholar] [CrossRef]
- Khan, M.S.; Shahwan, M.; Shamsi, A.; Alhumaydhi, F.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Abdullaev, B.; Yadav, D.K. Elucidating the Interactions of Fluoxetine with Human Transferrin Employing Spectroscopic, Calorimetric, and In Silico Approaches: Implications of a Potent Alzheimer’s Drug. ACS Omega 2022, 7, 9015–9023. [Google Scholar] [CrossRef]
- Andrade, S.; Loureiro, J.A.; Pereira, M.C. Transferrin-Functionalized Liposomes for the Delivery of Gallic Acid: A Therapeutic Approach for Alzheimer’s Disease. Pharmaceutics 2022, 14, 2163. [Google Scholar] [CrossRef]
- Nicholson, C.; Hrabětová, S. Brain Extracellular Space: The Final Frontier of Neuroscience. Biophys. J. 2017, 113, 2133–2142. [Google Scholar] [CrossRef] [Green Version]
- Hrabetova, S.; Cognet, L.; Rusakov, D.A.; Nägerl, U.V. Unveiling the Extracellular Space of the Brain: From Super-resolved Microstructure to In Vivo Function. J. Neurosci. 2018, 38, 9355–9363. [Google Scholar] [CrossRef] [Green Version]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Wang, R.; Cui, D.; Huang, X.; Yuan, L.; Liu, H.; Fu, Y.; Liang, L.; Wang, W.; He, Q.; et al. The Drainage of Interstitial Fluid in the Deep Brain is Controlled by the Integrity of Myelination. Aging Dis. 2019, 10, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Maley, J.; Yu, P.H. Potential inplications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef]
- Yue, X.; Mei, Y.; Zhang, Y.; Tong, Z.; Cui, D.; Yang, J.; Wang, A.; Wang, R.; Fei, X.; Ai, L.; et al. New insight into Alzheimer’s disease: Light reverses Aβ-obstructed interstitial fluid flow and ameliorates memory decline in APP/PS1 mice. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 671–684. [Google Scholar] [CrossRef]
- Hong, S.; Quintero-Monzon, O.; Ostaszewski, B.L.; Podlisny, D.R.; Cavanaugh, W.T.; Yang, T.; Holtzman, D.M.; Cirrito, J.R.; Selkoe, D.J. Dynamic analysis of amyloid β-protein in behaving mice reveals opposing changes in ISF versus parenchymal Aβ during age-related plaque formation. J. Neurosci. 2011, 31, 15861–15869. [Google Scholar] [CrossRef] [Green Version]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Conti Filho, C.E.; Loss, L.B.; Marcolongo-Pereira, C.; Rossoni Junior, J.V.; Barcelos, R.M.; Chiarelli-Neto, O.; da Silva, B.S.; Passamani Ambrosio, R.; Castro, F.; Teixeira, S.F.; et al. Advances in Alzheimer’s disease’s pharmacological treatment. Front. Pharmacol. 2023, 14, 1101452. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Aducanumab: First Approval. Drugs 2021, 81, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Lecanemab: First Approval. Drugs 2023, 83, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P.; Ippati, S.; Watling, M.; Balducci, C. Accelerating Alzheimer’s disease drug discovery and development: What’s the way forward? Expert. Opin. Drug. Discov. 2021, 16, 727–735. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Shin, H.; Hong, S.; Kim, Y. Physiological Roles of Monomeric Amyloid-β and Implications for Alzheimer’s Disease Therapeutics. Exp. Neurobiol. 2022, 31, 65–88. [Google Scholar] [CrossRef]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 peptides form interlaced amyloid fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Yamin, G. NMDA receptor-dependent signaling pathways that underlie amyloid beta-protein disruption of LTP in the hippocampus. J. Neurosci. Res. 2009, 87, 1729–1736. [Google Scholar] [CrossRef]
- Beckman, D.; Ott, S.; Donis-Cox, K.; Janssen, W.G.; Bliss-Moreau, E.; Rudebeck, P.H.; Baxter, M.G.; Morrison, J.H. Oligomeric Aβ in the monkey brain impacts synaptic integrity and induces accelerated cortical aging. Proc. Natl. Acad. Sci. USA 2019, 116, 26239–26246. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Yuan, Q.; Xian, Y.F.; Huang, Y.F.; Wu, W.; Song, Y.Q.; Lin, Z.X. Intracisternal injection of beta-amyloid seeds promotes cerebral amyloid angiopathy. Brain Behav. Immun. 2020, 89, 628–640. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Aman, Y.; Ng, C.T.; Chau, W.H.; Zhang, Z.; Yue, M.; Bohm, C.; Jia, Y.; Li, S.; et al. Amyloid-β toxicity modulates tau phosphorylation through the PAX6 signalling pathway. Brain 2021, 144, 2759–2770. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M.; et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef]
- Sur, C.; Kost, J.; Scott, D.; Adamczuk, K.; Fox, N.C.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; Vellas, B.; Voss, T.; et al. BACE inhibition causes rapid, regional, and non-progressive volume reduction in Alzheimer’s disease brain. Brain 2020, 143, 3816–3826. [Google Scholar] [CrossRef]
- Wang, H.; Li, R.; Shen, Y. β-Secretase: Its biology as a therapeutic target in diseases. Trends Pharmacol. Sci. 2013, 34, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; Salloway, S.; van Dyck, C.H.; Dubois, B.; Andreasen, N.; Brody, M.; Curtis, C.; Soininen, H.; Thein, S.; Shiovitz, T.; et al. Targeting Prodromal Alzheimer Disease with Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penninkilampi, R.; Brothers, H.M.; Eslick, G.D. Pharmacological Agents Targeting γ-Secretase Increase Risk of Cancer and Cognitive Decline in Alzheimer’s Disease Patients: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2016, 53, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.K.; Black, S.E.; Hendrix, S.B.; Zavitz, K.H.; Swabb, E.A.; Laughlin, M.A. Efficacy and safety of tarenflurbil in mild to moderate Alzheimer’s disease: A randomised phase II trial. Lancet Neurol. 2008, 7, 483–493. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: A randomized controlled trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Vingtdeux, V.; Marambaud, P. Identification and biology of α-secretase. J. Neurochem. 2012, 120 (Suppl. 1), 34–45. [Google Scholar] [CrossRef]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Reinhardt, S.; Stoye, N.; Luderer, M.; Kiefer, F.; Schmitt, U.; Lieb, K.; Endres, K. Identification of disulfiram as a secretase-modulating compound with beneficial effects on Alzheimer’s disease hallmarks. Sci. Rep. 2018, 8, 1329. [Google Scholar] [CrossRef] [Green Version]
- Etcheberrigaray, R.; Tan, M.; Dewachter, I.; Kuipéri, C.; Van der Auwera, I.; Wera, S.; Qiao, L.; Bank, B.; Nelson, T.J.; Kozikowski, A.P.; et al. Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 11141–11146. [Google Scholar] [CrossRef] [Green Version]
- Nelson, T.J.; Sun, M.K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin Effects on Cognitive Function and PKCɛ in Alzheimer’s Disease Phase IIa and Expanded Access Trials. J. Alzheimers Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef] [Green Version]
- Endres, K.; Fahrenholz, F.; Lotz, J.; Hiemke, C.; Teipel, S.; Lieb, K.; Tüscher, O.; Fellgiebel, A. Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 2014, 83, 1930–1935. [Google Scholar] [CrossRef]
- Ettcheto, M.; Cano, A.; Manzine, P.R.; Busquets, O.; Verdaguer, E.; Castro-Torres, R.D.; García, M.L.; Beas-Zarate, C.; Olloquequi, J.; Auladell, C.; et al. Epigallocatechin-3-Gallate (EGCG) Improves Cognitive Deficits Aggravated by an Obesogenic Diet Through Modulation of Unfolded Protein Response in APPswe/PS1dE9 Mice. Mol. Neurobiol. 2020, 57, 1814–1827. [Google Scholar] [CrossRef]
- Obregon, D.F.; Rezai-Zadeh, K.; Bai, Y.; Sun, N.; Hou, H.; Ehrhart, J.; Zeng, J.; Mori, T.; Arendash, G.W.; Shytle, D.; et al. ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 2006, 281, 16419–16427. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Liu, W.; Zhou, H.Y.; Gui, Y.R.; Yang, Y.H.; Wu, M.J.; Xiao, Y.F.; Shang, J.T.; Long, G.F.; Shu, X.J. Epigallocatechin-3-gallate Alleviates Cognitive Deficits in APP/PS1 Mice. Curr. Med. Sci. 2020, 40, 18–27. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Ohishi, T.; Tanabe, H.; Miyoshi, N.; Nakamura, Y. Beneficial Effects of Green Tea Catechins on Neurodegenerative Diseases. Molecules 2018, 23, 1297. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Situ, B.; Tan, X.; Zheng, S.; Zhang, F.; Yan, P.; Liu, P. Cryptotanshinione upregulates alpha-secretase by activation PI3K pathway in cortical neurons. Brain Res. 2010, 1348, 165–173. [Google Scholar] [CrossRef]
- Durairajan, S.S.; Liu, L.F.; Lu, J.H.; Koo, I.; Maruyama, K.; Chung, S.K.; Huang, J.D.; Li, M. Stimulation of non-amyloidogenic processing of amyloid-β protein precursor by cryptotanshinone involves activation and translocation of ADAM10 and PKC-α. J. Alzheimers Dis. 2011, 25, 245–262. [Google Scholar] [CrossRef] [Green Version]
- Kuang, X.; Zhou, H.J.; Thorne, A.H.; Chen, X.N.; Li, L.J.; Du, J.R. Neuroprotective Effect of Ligustilide through Induction of α-Secretase Processing of Both APP and Klotho in a Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 353. [Google Scholar] [CrossRef]
- Shi, C.; Zheng, D.D.; Wu, F.M.; Liu, J.; Xu, J. The phosphatidyl inositol 3 kinase-glycogen synthase kinase 3β pathway mediates bilobalide-induced reduction in amyloid β-peptide. Neurochem. Res. 2012, 37, 298–306. [Google Scholar] [CrossRef]
- Yin, Y.; Ren, Y.; Wu, W.; Wang, Y.; Cao, M.; Zhu, Z.; Wang, M.; Li, W. Protective effects of bilobalide on Aβ(25–35) induced learning and memory impairments in male rats. Pharmacol. Biochem. Behav. 2013, 106, 77–84. [Google Scholar] [CrossRef]
- Narasingappa, R.B.; Javagal, M.R.; Pullabhatla, S.; Htoo, H.H.; Rao, J.K.; Hernandez, J.F.; Govitrapong, P.; Vincent, B. Activation of α-secretase by curcumin-aminoacid conjugates. Biochem. Biophys. Res. Commun. 2012, 424, 691–696. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid-β in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef] [PubMed]
- Lopez Lopez, C.; Tariot, P.N.; Caputo, A.; Langbaum, J.B.; Liu, F.; Riviere, M.E.; Langlois, C.; Rouzade-Dominguez, M.L.; Zalesak, M.; Hendrix, S.; et al. The Alzheimer’s Prevention Initiative Generation Program: Study design of two randomized controlled trials for individuals at risk for clinical onset of Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.; Ufer, M.; Jacobson, L.H.; Rouzade-Dominguez, M.L.; Huledal, G.; Kolly, C.; Lüönd, R.M.; Machauer, R.; Veenstra, S.J.; Hurth, K.; et al. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 2018, 10, e9316. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yan, R. Inhibition of BACE1 for therapeutic use in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2010, 3, 618–628. [Google Scholar]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing β-secretase inhibitors for treatment of Alzheimer’s disease. J. Neurochem. 2012, 120 (Suppl. 1), 71–83. [Google Scholar] [CrossRef] [Green Version]
- May, P.C.; Dean, R.A.; Lowe, S.L.; Martenyi, F.; Sheehan, S.M.; Boggs, L.N.; Monk, S.A.; Mathes, B.M.; Mergott, D.J.; Watson, B.M.; et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011, 31, 16507–16516. [Google Scholar] [CrossRef] [Green Version]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [Green Version]
- Quartino, A.; Huledal, G.; Sparve, E.; Lüttgen, M.; Bueters, T.; Karlsson, P.; Olsson, T.; Paraskos, J.; Maltby, J.; Claeson-Bohnstedt, K.; et al. Population pharmacokinetic and pharmacodynamic analysis of plasma Aβ40 and Aβ42 following single oral doses of the BACE1 inhibitor AZD3839 to healthy volunteers. Clin. Pharmacol. Drug. Dev. 2014, 3, 396–405. [Google Scholar] [CrossRef]
- Forman, M.; Tseng, J.; Palcza, J.; Leempoels, J.; Ramael, S.; Krishna, G.; Ma, L.; Wagner, J.; Troyer, M. The Novel BACE Inhibitor MK-8931 Dramatically Lowers CSF Aβ Peptides in Healthy Subjects: Results from a Rising Single Dose Study (PL02.004). Neurology 2012, 78, PL02.004. [Google Scholar] [CrossRef]
- Wessels, A.M.; Tariot, P.N.; Zimmer, J.A.; Selzler, K.J.; Bragg, S.M.; Andersen, S.W.; Landry, J.; Krull, J.H.; Downing, A.M.; Willis, B.A.; et al. Efficacy and Safety of Lanabecestat for Treatment of Early and Mild Alzheimer Disease: The AMARANTH and DAYBREAK-ALZ Randomized Clinical Trials. JAMA Neurol. 2020, 77, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.; Albala, B.; Kaplow, J.M.; Aluri, J.; Yen, M.; Satlin, A. O1-06-05: First-in-human study of E2609, a novel BACE1 inhibitor, demonstrates prolonged reductions in plasma beta-amyloid levels after single dosing. Alzheimer’s Dement. 2012, 8, P96. [Google Scholar] [CrossRef]
- Albala, B.; Kaplow, J.M.; Lai, R.; Matijevic, M.; Aluri, J.; Satlin, A. S4-04-01: CSF amyloid lowering in human volunteers after 14 days’ oral administration of the novel BACE1 inhibitor E2609. Alzheimer’s Dement. 2012, 8, S743. [Google Scholar] [CrossRef]
- Timmers, M.; Van Broeck, B.; Ramael, S.; Slemmon, J.; De Waepenaert, K.; Russu, A.; Bogert, J.; Stieltjes, H.; Shaw, L.M.; Engelborghs, S.; et al. Profiling the dynamics of CSF and plasma Aβ reduction after treatment with JNJ-54861911, a potent oral BACE inhibitor. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 202–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, M.; Streffer, J.R.; Russu, A.; Tominaga, Y.; Shimizu, H.; Shiraishi, A.; Tatikola, K.; Smekens, P.; Börjesson-Hanson, A.; Andreasen, N.; et al. Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimers Res. Ther. 2018, 10, 85. [Google Scholar] [CrossRef]
- Sperling, R.; Henley, D.; Aisen, P.S.; Raman, R.; Donohue, M.C.; Ernstrom, K.; Rafii, M.S.; Streffer, J.; Shi, Y.; Karcher, K.; et al. Findings of Efficacy, Safety, and Biomarker Outcomes of Atabecestat in Preclinical Alzheimer Disease: A Truncated Randomized Phase 2b/3 Clinical Trial. JAMA Neurol. 2021, 78, 293–301. [Google Scholar] [CrossRef]
- Novak, G.; Streffer, J.R.; Timmers, M.; Henley, D.; Brashear, H.R.; Bogert, J.; Russu, A.; Janssens, L.; Tesseur, I.; Tritsmans, L.; et al. Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer’s disease spectrum patients: A randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimers Res. Ther. 2020, 12, 58. [Google Scholar] [CrossRef]
- Koriyama, Y.; Hori, A.; Ito, H.; Yonezawa, S.; Baba, Y.; Tanimoto, N.; Ueno, T.; Yamamoto, S.; Yamamoto, T.; Asada, N.; et al. Discovery of Atabecestat (JNJ-54861911): A Thiazine-Based β-Amyloid Precursor Protein Cleaving Enzyme 1 Inhibitor Advanced to the Phase 2b/3 EARLY Clinical Trial. J. Med. Chem. 2021, 64, 1873–1888. [Google Scholar] [CrossRef]
- Willis, B.A.; Lowe, S.L.; Monk, S.A.; Cocke, P.J.; Aluise, C.D.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; Dean, R.A.; Green, S.J.; et al. Robust Pharmacodynamic Effect of LY3202626, a Central Nervous System Penetrant, Low Dose BACE1 Inhibitor, in Humans and Nonclinical Species. J. Alzheimers Dis. Rep. 2022, 6, 1–15. [Google Scholar] [CrossRef]
- Lo, A.C.; Evans, C.D.; Mancini, M.; Wang, H.; Shcherbinin, S.; Lu, M.; Natanegara, F.; Willis, B.A. Phase II (NAVIGATE-AD study) Results of LY3202626 Effects on Patients with Mild Alzheimer’s Disease Dementia. J. Alzheimers Dis. Rep. 2021, 5, 321–336. [Google Scholar] [CrossRef]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef] [Green Version]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.J.; Castro, S.V.; Wang, F.; Pozdnyakov, N.; Ballard, T.E.; Sisodia, S.S.; Bales, K.R.; Johnson, D.S.; Li, Y.M. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 2012, 51, 7209–7211. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.E.; Carrieri, C.; Dela Cruz, F.; Fullerton, T.; Hajos-Korcsok, E.; He, P.; Kantaridis, C.; Leurent, C.; Liu, R.; Mancuso, J.; et al. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin. Pharmacol. Ther. 2020, 107, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.; Sharma, S.; Winston, J.; Nunez, M.; Bottini, G.; Franceschi, M.; Scarpini, E.; Frigerio, E.; Fiorentini, F.; Fernandez, M.; et al. CHF5074 reduces biomarkers of neuroinflammation in patients with mild cognitive impairment: A 12-week, double-blind, placebo-controlled study. Curr. Alzheimer Res. 2013, 10, 742–753. [Google Scholar] [CrossRef]
- Vellas, B.; Sol, O.; Snyder, P.J.; Ousset, P.J.; Haddad, R.; Maurin, M.; Lemarié, J.C.; Désiré, L.; Pando, M.P. EHT0202 in Alzheimer’s disease: A 3-month, randomized, placebo-controlled, double-blind study. Curr. Alzheimer Res. 2011, 8, 203–212. [Google Scholar] [CrossRef]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Abeta peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef]
- Gauthier, S.; Aisen, P.S.; Ferris, S.H.; Saumier, D.; Duong, A.; Haine, D.; Garceau, D.; Suhy, J.; Oh, J.; Lau, W.; et al. Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: Exploratory analyses of the MRI sub-group of the Alphase study. J. Nutr. Health Aging 2009, 13, 550–557. [Google Scholar] [CrossRef]
- Hey, J.A.; Kocis, P.; Hort, J.; Abushakra, S.; Power, A.; Vyhnálek, M.; Yu, J.Y.; Tolar, M. Discovery and Identification of an Endogenous Metabolite of Tramiprosate and Its Prodrug ALZ-801 that Inhibits Beta Amyloid Oligomer Formation in the Human Brain. CNS Drugs 2018, 32, 849–861. [Google Scholar] [CrossRef] [Green Version]
- Stark, T.; Lieblein, T.; Pohland, M.; Kalden, E.; Freund, P.; Zangl, R.; Grewal, R.; Heilemann, M.; Eckert, G.P.; Morgner, N.; et al. Peptidomimetics That Inhibit and Partially Reverse the Aggregation of Aβ(1–42). Biochemistry 2017, 56, 4840–4849. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 619667. [Google Scholar] [CrossRef] [PubMed]
- Du, W.J.; Guo, J.J.; Gao, M.T.; Hu, S.Q.; Dong, X.Y.; Han, Y.F.; Liu, F.F.; Jiang, S.; Sun, Y. Brazilin inhibits amyloid β-protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity. Sci. Rep. 2015, 5, 7992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.; Wang, Z.; Zhou, Y.; Xu, W.; Li, S.; Wang, L.; Wei, D.; Qiao, Z. A novel drug candidate for Alzheimer’s disease treatment: Gx-50 derived from Zanthoxylum bungeanum. J. Alzheimers Dis. 2013, 34, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Aucoin, D.; Ahmed, M.; Ziliox, M.; Van Nostrand, W.E.; Smith, S.O. Capping of aβ42 oligomers by small molecule inhibitors. Biochemistry 2014, 53, 7893–7903. [Google Scholar] [CrossRef] [Green Version]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.; VanSchouwen, B.; Jafari, N.; Ni, X.; Ortega, J.; Melacini, G. Molecular Mechanism for the (-)-Epigallocatechin Gallate-Induced Toxic to Nontoxic Remodeling of Aβ Oligomers. J. Am. Chem. Soc. 2017, 139, 13720–13734. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, Y.; Wang, X.; Zhang, Z.; Fu, Y.; Liu, L.; Wang, P.; Ma, H.; Ma, H.; Seeram, N.P.; et al. Ginnalin A Inhibits Aggregation, Reverses Fibrillogenesis, and Alleviates Cytotoxicity of Amyloid β(1–42). ACS Chem. Neurosci. 2020, 11, 638–647. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Kocis, P.; Tolar, M.; Yu, J.; Sinko, W.; Ray, S.; Blennow, K.; Fillit, H.; Hey, J.A. Elucidating the Aβ42 Anti-Aggregation Mechanism of Action of Tramiprosate in Alzheimer’s Disease: Integrating Molecular Analytical Methods, Pharmacokinetic and Clinical Data. CNS Drugs 2017, 31, 495–509. [Google Scholar] [CrossRef]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Pfeifer, M.; Boncristiano, S.; Bondolfi, L.; Stalder, A.; Deller, T.; Staufenbiel, M.; Mathews, P.M.; Jucker, M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 2002, 298, 1379. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Avgerinos, K.I.; Ferrucci, L.; Kapogiannis, D. Effects of monoclonal antibodies against amyloid-β on clinical and biomarker outcomes and adverse event risks: A systematic review and meta-analysis of phase III RCTs in Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101339. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients with Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Dhadda, S.; Kanekiyo, M.; Li, D.; Swanson, C.J.; Irizarry, M.; Berry, S.; Kramer, L.D.; Berry, D.A. Consistency of efficacy results across various clinical measures and statistical methods in the lecanemab phase 2 trial of early Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 182. [Google Scholar] [CrossRef]
- McDade, E.; Cummings, J.L.; Dhadda, S.; Swanson, C.J.; Reyderman, L.; Kanekiyo, M.; Koyama, A.; Irizarry, M.; Kramer, L.D.; Bateman, R.J. Lecanemab in patients with early Alzheimer’s disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res. Ther. 2022, 14, 191. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Bittner, T.; Sink, K.M.; Mackey, H.; Rabe, C.; Honig, L.S.; Cassetta, E.; Woodward, M.; Boada, M.; van Dyck, C.H.; et al. Evaluating the Safety and Efficacy of Crenezumab vs Placebo in Adults with Early Alzheimer Disease: Two Phase 3 Randomized Placebo-Controlled Trials. JAMA Neurol. 2022, 79, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 2012, 32, 9677–9689. [Google Scholar] [CrossRef]
- Lowe, S.L.; Willis, B.A.; Hawdon, A.; Natanegara, F.; Chua, L.; Foster, J.; Shcherbinin, S.; Ardayfio, P.; Sims, J.R. Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12112. [Google Scholar] [CrossRef]
- Irizarry, M.C.; Sims, J.R.; Lowe, S.L.; Nakano, M.; Hawdon, A.; Willis, B.A.; Gonzales, C.; Liu, P.; Fujimoto, S.; Dean, R.A.; et al. O4-08-06: Safety, Pharmacokinetics (PK), and Florbetapir F-18 Positron Emission Tomography (PET) After Multiple Dose Administration of LY3002813 Aβ-amyloid plaque-specific antibody, in Alzherimer’s Disease (AD). Alzheimer’s Dement. 2016, 12, P352–P353. [Google Scholar] [CrossRef]
- Lowe, S.L.; Duggan Evans, C.; Shcherbinin, S.; Cheng, Y.J.; Willis, B.A.; Gueorguieva, I.; Lo, A.C.; Fleisher, A.S.; Dage, J.L.; Ardayfio, P.; et al. Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J. Prev. Alzheimers Dis. 2021, 8, 414–424. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Demattos, R.B.; Lu, J.; Tang, Y.; Racke, M.M.; Delong, C.A.; Tzaferis, J.A.; Hole, J.T.; Forster, B.M.; McDonnell, P.C.; Liu, F.; et al. A plaque-specific antibody clears existing β-amyloid plaques in Alzheimer’s disease mice. Neuron 2012, 76, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Lacosta, A.M.; Pascual-Lucas, M.; Pesini, P.; Casabona, D.; Pérez-Grijalba, V.; Marcos-Campos, I.; Sarasa, L.; Canudas, J.; Badi, H.; Monleón, I.; et al. Safety, tolerability and immunogenicity of an active anti-Aβ(40) vaccine (ABvac40) in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res. Ther. 2018, 10, 12. [Google Scholar] [CrossRef]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, P.N.; Chiu, M.J.; Finstad, C.L.; Lin, F.; Lynn, S.; Tai, Y.H.; De Fang, X.; Zhao, K.; Hung, C.H.; et al. UB-311, a novel UBITh(®) amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 262–272. [Google Scholar] [CrossRef]
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid-β. Sci. Rep. 2018, 8, 6412. [Google Scholar] [CrossRef] [Green Version]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (mAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of tg-ArcSwe Mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Söllvander, S.; Nikitidou, E.; Gallasch, L.; Zyśk, M.; Söderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. The Aβ protofibril selective antibody mAb158 prevents accumulation of Aβ in astrocytes and rescues neurons from Aβ-induced cell death. J. Neuroinflamm. 2018, 15, 98. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Savastano, A.; Flores, D.; Kadavath, H.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Disease-Associated Tau Phosphorylation Hinders Tubulin Assembly within Tau Condensates. Angew. Chem. Int. Ed. Engl. 2021, 60, 726–730. [Google Scholar] [CrossRef]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimers Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [Green Version]
- Tsai, R.M.; Miller, Z.; Koestler, M.; Rojas, J.C.; Ljubenkov, P.A.; Rosen, H.J.; Rabinovici, G.D.; Fagan, A.M.; Cobigo, Y.; Brown, J.A.; et al. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients With Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Schmidt, R.; Kontsekova, E.; Zilka, N.; Kovacech, B.; Skrabana, R.; Vince-Kazmerova, Z.; Katina, S.; Fialova, L.; Prcina, M.; et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017, 16, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Kovacech, B.; Katina, S.; Schmidt, R.; Scheltens, P.; Kontsekova, E.; Ropele, S.; Fialova, L.; Kramberger, M.; Paulenka-Ivanovova, N.; et al. ADAMANT: A placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer’s disease. Nature Aging 2021, 1, 521–534. [Google Scholar] [CrossRef]
- West, T.; Hu, Y.; Verghese, P.B.; Bateman, R.J.; Braunstein, J.B.; Fogelman, I.; Budur, K.; Florian, H.; Mendonca, N.; Holtzman, D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimers Dis. 2017, 4, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Nobuhara, C.K.; DeVos, S.L.; Commins, C.; Wegmann, S.; Moore, B.D.; Roe, A.D.; Costantino, I.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Tau Antibody Targeting Pathological Species Blocks Neuronal Uptake and Interneuron Propagation of Tau in Vitro. Am. J. Pathol. 2017, 187, 1399–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courade, J.P.; Angers, R.; Mairet-Coello, G.; Pacico, N.; Tyson, K.; Lightwood, D.; Munro, R.; McMillan, D.; Griffin, R.; Baker, T.; et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018, 136, 729–745. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.; Mairet-Coello, G.; Danis, C.; Lieger, S.; Caillierez, R.; Carrier, S.; Skrobala, E.; Landrieu, I.; Michel, A.; Schmitt, M.; et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019, 142, 1736–1750. [Google Scholar] [CrossRef]
- Alam, R.; Driver, D.; Wu, S.; Lozano, E.; Key, S.L.; Hole, J.T.; Hayashi, M.L.; Lu, J. Preclinical characterization of an antibody [LY3303560] targeting aggregated tau. Alzheimer’s Dement. 2017, 13, P592–P593. [Google Scholar] [CrossRef]
- Chai, X.; Wu, S.; Murray, T.K.; Kinley, R.; Cella, C.V.; Sims, H.; Buckner, N.; Hanmer, J.; Davies, P.; O’Neill, M.J.; et al. Passive immunization with anti-Tau antibodies in two transgenic models: Reduction of Tau pathology and delay of disease progression. J. Biol. Chem. 2011, 286, 34457–34467. [Google Scholar] [CrossRef] [Green Version]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res. Ther. 2014, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [Green Version]
- Guan, P.P.; Cao, L.L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Mishra, S.K.; Hidau, M.; Rai, S. Memantine and Ibuprofen pretreatment exerts anti-inflammatory effect against streptozotocin-induced astroglial inflammation via modulation of NMDA receptor-associated downstream calcium ion signaling. Inflammopharmacology 2021, 29, 183–192. [Google Scholar] [CrossRef]
- Chappell, A.S.; Gonzales, C.; Williams, J.; Witte, M.M.; Mohs, R.C.; Sperling, R. AMPA potentiator treatment of cognitive deficits in Alzheimer disease. Neurology 2007, 68, 1008–1012. [Google Scholar] [CrossRef]
- Jhee, S.S.; Chappell, A.S.; Zarotsky, V.; Moran, S.V.; Rosenthal, M.; Kim, E.; Chalon, S.; Toublanc, N.; Brandt, J.; Coutant, D.E.; et al. Multiple-dose plasma pharmacokinetic and safety study of LY450108 and LY451395 (AMPA receptor potentiators) and their concentration in cerebrospinal fluid in healthy human subjects. J. Clin. Pharmacol. 2006, 46, 424–432. [Google Scholar] [CrossRef]
- Bernard, K.; Danober, L.; Thomas, J.Y.; Lebrun, C.; Muñoz, C.; Cordi, A.; Desos, P.; Lestage, P.; Morain, P. DRUG FOCUS: S 18986: A positive allosteric modulator of AMPA-type glutamate receptors pharmacological profile of a novel cognitive enhancer. CNS Neurosci. Ther. 2010, 16, e193–e212. [Google Scholar] [CrossRef]
- Shevtsova, E.F.; Angelova, P.R.; Stelmashchuk, O.A.; Esteras, N.; Vasil’eva, N.A.; Maltsev, A.V.; Shevtsov, P.N.; Shaposhnikov, A.V.; Fisenko, V.P.; Bachurin, S.O.; et al. Pharmacological sequestration of mitochondrial calcium uptake protects against dementia and β-amyloid neurotoxicity. Sci. Rep. 2022, 12, 12766. [Google Scholar] [CrossRef]
- Malek, R.; Maj, M.; Wnorowski, A.; Jóźwiak, K.; Martin, H.; Iriepa, I.; Moraleda, I.; Chabchoub, F.; Marco-Contelles, J.; Ismaili, L. Multi-target 1,4-dihydropyridines showing calcium channel blockade and antioxidant capacity for Alzheimer’s disease therapy. Bioorg. Chem. 2019, 91, 103205. [Google Scholar] [CrossRef]
- Bhatt, S.; Puli, L.; Patil, C.R. Role of reactive oxygen species in the progression of Alzheimer’s disease. Drug. Discov. Today 2021, 26, 794–803. [Google Scholar] [CrossRef]
- Balendra, V.; Singh, S.K. Therapeutic potential of astaxanthin and superoxide dismutase in Alzheimer’s disease. Open. Biol. 2021, 11, 210013. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cai, X.; Hu, W.; Li, Z.; Kong, F.; Chen, X.; Wang, D. Investigation of the neuroprotective effects of crocin via antioxidant activities in HT22 cells and in mice with Alzheimer’s disease. Int. J. Mol. Med. 2019, 43, 956–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, C.; Shin, M.; Park, Y.; Choi, B.; Jang, S.; Lim, C.; Yun, H.S.; Lee, I.S.; Won, S.Y.; Cho, K.S. Linalool Alleviates Aβ42-Induced Neurodegeneration via Suppressing ROS Production and Inflammation in Fly and Rat Models of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2021, 2021, 8887716. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Zhang, Y.; Mei, Y.; Yue, X.; Jiang, W.; Ai, L.; Yu, Y.; Luo, H.; Li, H.; Luo, W.; et al. Degradation of FA reduces Aβ neurotoxicity and Alzheimer-related phenotypes. Mol. Psychiatry 2021, 26, 5578–5591. [Google Scholar] [CrossRef] [PubMed]
- Letellier, N.; Gutierrez, L.A.; Pilorget, C.; Artaud, F.; Descatha, A.; Ozguler, A.; Goldberg, M.; Zins, M.; Elbaz, A.; Berr, C. Association Between Occupational Exposure to Formaldehyde and Cognitive Impairment. Neurology 2022, 98, e633–e640. [Google Scholar] [CrossRef]
- Li, F.; Yujie, Q.; Gong, S.; Zhang, H.; Ding, S. Learning and memory impairment of mice caused by gaseous formaldehyde. Environ. Res. 2020, 184, 109318. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Wu, R.; Ye, M.; Zhao, Y.; Kang, J.; Ma, P.; Li, J.; Yang, X. Acute formaldehyde exposure induced early Alzheimer-like changes in mouse brain. Toxicol. Mech. Methods 2018, 28, 95–104. [Google Scholar] [CrossRef]
- Zhai, R.; Rizak, J.; Zheng, N.; He, X.; Li, Z.; Yin, Y.; Su, T.; He, Y.; He, R.; Ma, Y.; et al. Alzheimer’s Disease-Like Pathologies and Cognitive Impairments Induced by Formaldehyde in Non-Human Primates. Curr. Alzheimer Res. 2018, 15, 1304–1321. [Google Scholar] [CrossRef]
- Tong, Z.; Han, C.; Qiang, M.; Wang, W.; Lv, J.; Zhang, S.; Luo, W.; Li, H.; Luo, H.; Zhou, J.; et al. Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 100–110. [Google Scholar] [CrossRef]
- Kou, Y.; Zhao, H.; Cui, D.; Han, H.; Tong, Z. Formaldehyde toxicity in age-related neurological dementia. Ageing Res. Rev. 2022, 73, 101512. [Google Scholar] [CrossRef]
- Tong, Z.; Zhang, J.; Luo, W.; Wang, W.; Li, F.; Li, H.; Luo, H.; Lu, J.; Zhou, J.; Wan, Y.; et al. Urine formaldehyde level is inversely correlated to mini mental state examination scores in senile dementia. Neurobiol. Aging 2011, 32, 31–41. [Google Scholar] [CrossRef]
- Tong, Z.; Wang, W.; Luo, W.; Lv, J.; Li, H.; Luo, H.; Jia, J.; He, R. Urine Formaldehyde Predicts Cognitive Impairment in Post-Stroke Dementia and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 1031–1038. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, F.; Xie, F.; He, R.; Guo, Q. Correlation Between Urine Formaldehyde and Cognitive Abilities in the Clinical Spectrum of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 820385. [Google Scholar] [CrossRef]
- Boor, P.J.; Trent, M.B.; Lyles, G.A.; Tao, M.; Ansari, G.A. Methylamine metabolism to formaldehyde by vascular semicarbazide-sensitive amine oxidase. Toxicology 1992, 73, 251–258. [Google Scholar] [CrossRef]
- Li, Z.H.; He, X.P.; Li, H.; He, R.Q.; Hu, X.T. Age-associated changes in amyloid-β and formaldehyde concentrations in cerebrospinal fluid of rhesus monkeys. Zool. Res. 2020, 41, 444–448. [Google Scholar] [CrossRef]
- Zhao, Q.; Lu, J.; Yao, Z.; Wang, S.; Zhu, L.; Wang, J.; Chen, B. Upregulation of Aβ42 in the Brain and Bodily Fluids of Rhesus Monkeys with Aging. J. Mol. Neurosci. 2017, 61, 79–87. [Google Scholar] [CrossRef]
- Tao, R.; Liao, M.; Wang, Y.; Wang, H.; Tan, Y.; Qin, S.; Wei, W.; Tang, C.; Liang, X.; Han, Y.; et al. In Situ Imaging of Formaldehyde in Live Mice with High Spatiotemporal Resolution Reveals Aldehyde Dehydrogenase-2 as a Potential Target for Alzheimer’s Disease Treatment. Anal. Chem. 2022, 94, 1308–1317. [Google Scholar] [CrossRef]
- Zhai, R.; Zheng, N.; Rizak, J.; Hu, X. Evidence for Conversion of Methanol to Formaldehyde in Nonhuman Primate Brain. Anal. Cell. Pathol. 2016, 2016, 4598454. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Miao, J.; Rizak, J.; Zhai, R.; Wang, Z.; Huma, T.; Li, T.; Zheng, N.; Wu, S.; Zheng, Y.; et al. Alzheimer’s disease and methanol toxicity (part 2): Lessons from four rhesus macaques (Macaca mulatta) chronically fed methanol. J. Alzheimers Dis. 2014, 41, 1131–1147. [Google Scholar] [CrossRef]
- Del Mar Hernandez, M.; Esteban, M.; Szabo, P.; Boada, M.; Unzeta, M. Human plasma semicarbazide sensitive amine oxidase (SSAO), beta-amyloid protein and aging. Neurosci. Lett. 2005, 384, 183–187. [Google Scholar] [CrossRef]
- Zhang, J.; Yue, X.; Luo, H.; Jiang, W.; Mei, Y.; Ai, L.; Gao, G.; Wu, Y.; Yang, H.; An, J.; et al. Illumination with 630 nm Red Light Reduces Oxidative Stress and Restores Memory by Photo-Activating Catalase and Formaldehyde Dehydrogenase in SAMP8 Mice. Antioxid. Redox Signal. 2019, 30, 1432–1449. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Han, C.; Luo, W.; Wang, X.; Li, H.; Luo, H.; Zhou, J.; Qi, J.; He, R. Accumulated hippocampal formaldehyde induces age-dependent memory decline. Age 2013, 35, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Miao, J.; Su, T.; Liu, Y.; He, R. Formaldehyde induces hyperphosphorylation and polymerization of Tau protein both in vitro and in vivo. Biochim. Biophys. Acta 2013, 1830, 4102–4116. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.L.; Wang, X.S.; Liu, Y.; Perrett, S.; He, R.Q. Amyloid-like aggregates of neuronal tau induced by formaldehyde promote apoptosis of neuronal cells. BMC Neurosci. 2007, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E.S. Coenzyme Q10 attenuates beta-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J. Mol. Neurosci. 2008, 34, 165–171. [Google Scholar] [CrossRef]
- Mei, Y.; Jiang, C.; Wan, Y.; Lv, J.; Jia, J.; Wang, X.; Yang, X.; Tong, Z. Aging-associated formaldehyde-induced norepinephrine deficiency contributes to age-related memory decline. Aging Cell. 2015, 14, 659–668. [Google Scholar] [CrossRef]
- He, X.; Li, Z.; Rizak, J.D.; Wu, S.; Wang, Z.; He, R.; Su, M.; Qin, D.; Wang, J.; Hu, X. Resveratrol Attenuates Formaldehyde Induced Hyperphosphorylation of Tau Protein and Cytotoxicity in N2a Cells. Front. Neurosci. 2016, 10, 598. [Google Scholar] [CrossRef] [Green Version]
- Corpas, R.; Griñán-Ferré, C.; Rodríguez-Farré, E.; Pallàs, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef] [Green Version]
- Jhaveri, A.; Deshpande, P.; Pattni, B.; Torchilin, V. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J. Control. Release 2018, 277, 89–101. [Google Scholar] [CrossRef]
- Sun, J.; Wei, C.; Liu, Y.; Xie, W.; Xu, M.; Zhou, H.; Liu, J. Progressive release of mesoporous nano-selenium delivery system for the multi-channel synergistic treatment of Alzheimer’s disease. Biomaterials 2019, 197, 417–431. [Google Scholar] [CrossRef]
- Abozaid, O.A.R.; Sallam, M.W.; El-Sonbaty, S.; Aziza, S.; Emad, B.; Ahmed, E.S.A. Resveratrol-Selenium Nanoparticles Alleviate Neuroinflammation and Neurotoxicity in a Rat Model of Alzheimer’s Disease by Regulating Sirt1/miRNA-134/GSK3β Expression. Biol. Trace Elem. Res. 2022, 200, 5104–5114. [Google Scholar] [CrossRef]
- Takagaki, A.; Fukai, K.; Nanjo, F.; Hara, Y. Reactivity of green tea catechins with formaldehyde. J. Wood Sci. 2000, 46, 334–338. [Google Scholar] [CrossRef]
- Pervin, M.; Unno, K.; Takagaki, A.; Isemura, M.; Nakamura, Y. Function of Green Tea Catechins in the Brain: Epigallocatechin Gallate and its Metabolites. Int. J. Mol. Sci. 2019, 20, 3630. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Lu, Y.; Zhang, B.; Yang, S.; Zhang, Q.; Cui, H.; Lu, X.; Zhao, Y.; Yang, X.; Li, R. Antagonistic effect of epigallocatechin-3-gallate on neurotoxicity induced by formaldehyde. Toxicology 2019, 412, 29–36. [Google Scholar] [CrossRef]
- Cano, A.; Ettcheto, M.; Chang, J.H.; Barroso, E.; Espina, M.; Kühne, B.A.; Barenys, M.; Auladell, C.; Folch, J.; Souto, E.B.; et al. Dual-drug loaded nanoparticles of Epigallocatechin-3-gallate (EGCG)/Ascorbic acid enhance therapeutic efficacy of EGCG in a APPswe/PS1dE9 Alzheimer’s disease mice model. J. Control. Release 2019, 301, 62–75. [Google Scholar] [CrossRef]
- Rezai-Zadeh, K.; Arendash, G.W.; Hou, H.; Fernandez, F.; Jensen, M.; Runfeldt, M.; Shytle, R.D.; Tan, J. Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 2008, 1214, 177–187. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Ban, J.O.; Ha, T.Y.; Yun, Y.P.; Han, S.B.; Oh, K.W.; Hong, J.T. Green tea (-)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappaB pathways in mice. J. Nutr. 2009, 139, 1987–1993. [Google Scholar] [CrossRef] [Green Version]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [Green Version]
- Disbrow, E.; Stokes, K.Y.; Ledbetter, C.; Patterson, J.; Kelley, R.; Pardue, S.; Reekes, T.; Larmeu, L.; Batra, V.; Yuan, S.; et al. Plasma hydrogen sulfide: A biomarker of Alzheimer’s disease and related dementias. Alzheimers Dement. 2021, 17, 1391–1402. [Google Scholar] [CrossRef]
- Li, X.; Zhuang, Y.Y.; Wu, L.; Xie, M.; Gu, H.F.; Wang, B.; Tang, X.Q. Hydrogen Sulfide Ameliorates Cognitive Dysfunction in Formaldehyde-Exposed Rats: Involvement in the Upregulation of Brain-Derived Neurotrophic Factor. Neuropsychobiology 2020, 79, 119–130. [Google Scholar] [CrossRef]
- Cao, L.; Cao, X.; Zhou, Y.; Nagpure, B.V.; Wu, Z.Y.; Hu, L.F.; Yang, Y.; Sethi, G.; Moore, P.K.; Bian, J.S. Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ(1–42) synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav. Immun. 2018, 73, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Aboulhoda, B.E.; Rashed, L.A.; Ahmed, H.; Obaya, E.M.M.; Ibrahim, W.; Alkafass, M.A.L.; Abd El-Aal, S.A.; ShamsEldeen, A.M. Hydrogen sulfide and mesenchymal stem cells-extracted microvesicles attenuate LPS-induced Alzheimer’s disease. J. Cell. Physiol. 2021, 236, 5994–6010. [Google Scholar] [CrossRef] [PubMed]
- Xuan, A.; Long, D.; Li, J.; Ji, W.; Zhang, M.; Hong, L.; Liu, J. Hydrogen sulfide attenuates spatial memory impairment and hippocampal neuroinflammation in β-amyloid rat model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Zhang, Y.; Zhou, Y.; Liu, Q.; Chen, X.; Liu, X.; Grune, T.; Shi, L.; Hou, M.; Liu, Z. Effects of methionine intake on cognitive function in mild cognitive impairment patients and APP/PS1 Alzheimer’s Disease model mice: Role of the cystathionine-β-synthase/H(2)S pathway. Redox Biol. 2023, 59, 102595. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.J.; Malek, K.; Crabb, D.W. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J. Investig. Med. 1996, 44, 42–46. [Google Scholar]
- Li, R.; Zhao, Z.; Sun, M.; Luo, J.; Xiao, Y. ALDH2 gene polymorphism in different types of cancers and its clinical significance. Life Sci. 2016, 147, 59–66. [Google Scholar] [CrossRef]
- Yang, K.; Ren, J.; Li, X.; Wang, Z.; Xue, L.; Cui, S.; Sang, W.; Xu, T.; Zhang, J.; Yu, J.; et al. Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur. Heart J. 2020, 41, 2442–2453. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, Z.; Ye, T.; Lin, W.; Wang, Q.; Lin, B. Aldehyde dehydrogenase II rs671 polymorphism in essential hypertension. Clin. Chim. Acta 2018, 487, 153–160. [Google Scholar] [CrossRef]
- Tanaka, F.; Shiratori, Y.; Yokosuka, O.; Imazeki, F.; Tsukada, Y.; Omata, M. High incidence of ADH2*1/ALDH2*1 genes among Japanese alcohol dependents and patients with alcoholic liver disease. Hepatology 1996, 23, 234–239. [Google Scholar] [CrossRef]
- Dingler, F.A.; Wang, M.; Mu, A.; Millington, C.L.; Oberbeck, N.; Watcham, S.; Pontel, L.B.; Kamimae-Lanning, A.N.; Langevin, F.; Nadler, C.; et al. Two Aldehyde Clearance Systems Are Essential to Prevent Lethal Formaldehyde Accumulation in Mice and Humans. Mol. Cell. 2020, 80, 996–1012.e1019. [Google Scholar] [CrossRef]
- Chen, C.H.; Kraemer, B.R.; Mochly-Rosen, D. ALDH2 variance in disease and populations. Dis. Model. Mech. 2022, 15, dmm049601. [Google Scholar] [CrossRef]
- Jin, X.; Long, T.; Chen, H.; Zeng, Y.; Zhang, X.; Yan, L.; Wu, C. Associations of Alcohol Dehydrogenase and Aldehyde Dehydrogenase Polymorphism With Cognitive Impairment Among the Oldest-Old in China. Front. Aging Neurosci. 2021, 13, 710966. [Google Scholar] [CrossRef]
- Chen, J.; Huang, W.; Cheng, C.H.; Zhou, L.; Jiang, G.B.; Hu, Y.Y. Association Between Aldehyde dehydrogenase-2 Polymorphisms and Risk of Alzheimer’s Disease and Parkinson’s Disease: A Meta-Analysis Based on 5,315 Individuals. Front. Neurol. 2019, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, J.; Zhou, S.; Tan, S.; He, X.; Yang, Z.; Xie, Y.C.; Li, S.; Zheng, C.; Ma, X. The association of mitochondrial aldehyde dehydrogenase gene (ALDH2) polymorphism with susceptibility to late-onset Alzheimer’s disease in Chinese. J. Neurol. Sci. 2008, 268, 172–175. [Google Scholar] [CrossRef]
- Ohsawa, I.; Nishimaki, K.; Murakami, Y.; Suzuki, Y.; Ishikawa, M.; Ohta, S. Age-dependent neurodegeneration accompanying memory loss in transgenic mice defective in mitochondrial aldehyde dehydrogenase 2 activity. J. Neurosci. 2008, 28, 6239–6249. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, Y.; Elharram, A.; Soon-Shiong, R.; Andrew, R.D.; Bennett, B.M. Characterization of Aldh2 (-/-) mice as an age-related model of cognitive impairment and Alzheimer’s disease. Mol. Brain 2015, 8, 27. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, W.; Wang, X.; Ge, W. Impact of mitochondrial aldehyde dehydrogenase 2 on cognitive impairment in the AD model mouse. Acta Biochim. Biophys. Sin. 2021, 53, 837–847. [Google Scholar] [CrossRef]
- Joshi, A.U.; Van Wassenhove, L.D.; Logas, K.R.; Minhas, P.S.; Andreasson, K.I.; Weinberg, K.I.; Chen, C.H.; Mochly-Rosen, D. Aldehyde dehydrogenase 2 activity and aldehydic load contribute to neuroinflammation and Alzheimer’s disease related pathology. Acta Neuropathol. Commun. 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Merlini, M.; Meyer, E.P.; Ulmann-Schuler, A.; Nitsch, R.M. Vascular β-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 2011, 122, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, K.C.; Learman, C.R.; Dunbar, G.L.; Maiti, P.; Jang, W.C.; Cha, H.C.; Song, M.S. Characterization of Impaired Cerebrovascular Structure in APP/PS1 Mouse Brains. Neuroscience 2018, 385, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., 3rd; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Yang, L.; Yang, Z.; Yu, W.; Wang, Y.; Liu, R.; Chen, M.; Gao, H. Intelligent lesion blood-brain barrier targeting nano-missiles for Alzheimer’s disease treatment by anti-neuroinflammation and neuroprotection. Acta Pharm. Sin. B 2022, 12, 1987–1999. [Google Scholar] [CrossRef]
- Han, H.; Li, K.; Yan, J.; Zhu, K.; Fu, Y. An in vivo study with an MRI tracer method reveals the biophysical properties of interstitial fluid in the rat brain. Sci. China Life Sci. 2012, 55, 782–787. [Google Scholar] [CrossRef] [Green Version]
- Syková, E.; Nicholson, C. Diffusion in brain extracellular space. Physiol. Rev. 2008, 88, 1277–1340. [Google Scholar] [CrossRef] [Green Version]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef]
- Syková, E.; Vorísek, I.; Antonova, T.; Mazel, T.; Meyer-Luehmann, M.; Jucker, M.; Hájek, M.; Ort, M.; Bures, J. Changes in extracellular space size and geometry in APP23 transgenic mice: A model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Mueggler, T.; Meyer-Luehmann, M.; Rausch, M.; Staufenbiel, M.; Jucker, M.; Rudin, M. Restricted diffusion in the brain of transgenic mice with cerebral amyloidosis. Eur. J. Neurosci. 2004, 20, 811–817. [Google Scholar] [CrossRef]
- Xu, F.; Hongbin, H.; Yan, J.; Chen, H.; He, Q.; Xu, W.; Zhu, N.; Zhang, H.; Zhou, F.; Lee, K. Greatly improved neuroprotective efficiency of citicoline by stereotactic delivery in treatment of ischemic injury. Drug. Deliv. 2011, 18, 461–467. [Google Scholar] [CrossRef]
- Kong, S.D.; Lee, J.; Ramachandran, S.; Eliceiri, B.P.; Shubayev, V.I.; Lal, R.; Jin, S. Magnetic targeting of nanoparticles across the intact blood-brain barrier. J. Control. Release 2012, 164, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Gkountas, A.A.; Polychronopoulos, N.D.; Sofiadis, G.N.; Karvelas, E.G.; Spyrou, L.A.; Sarris, I.E. Simulation of magnetic nanoparticles crossing through a simplified blood-brain barrier model for Glioblastoma multiforme treatment. Comput. Methods Programs Biomed. 2021, 212, 106477. [Google Scholar] [CrossRef]
- Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: Opportunities and challenges. Expert. Opin. Drug. Deliv. 2014, 11, 1449–1470. [Google Scholar] [CrossRef]
- Pedram, M.Z.; Shamloo, A.; Alasty, A.; Ghafar-Zadeh, E. Optimal Magnetic Field for Crossing Super-Para-Magnetic Nanoparticles through the Brain Blood Barrier: A Computational Approach. Biosensors 2016, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zhang, B.; Xie, S.; Yang, B.; Xu, Q.; Tan, J. Superparamagnetic Iron Oxide Nanoparticles Modified with Tween 80 Pass through the Intact Blood-Brain Barrier in Rats under Magnetic Field. ACS Appl. Mater. Interfaces 2016, 8, 11336–11341. [Google Scholar] [CrossRef]
- Chen, J.; Yuan, M.; Madison, C.A.; Eitan, S.; Wang, Y. Blood-brain barrier crossing using magnetic stimulated nanoparticles. J. Control. Release 2022, 345, 557–571. [Google Scholar] [CrossRef]
- Hong, K.S.; Khan, M.N.A.; Ghafoor, U. Non-invasive transcranial electrical brain stimulation guided by functional near-infrared spectroscopy for targeted neuromodulation: A review. J. Neural Eng. 2022, 19, 4. [Google Scholar] [CrossRef]
- Cai, Y.; Wei, Z.; Song, C.; Tang, C.; Han, W.; Dong, X. Optical nano-agents in the second near-infrared window for biomedical applications. Chem. Soc. Rev. 2019, 48, 22–37. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Z.; Liu, Q.; Wang, Y.; Hao, J.; Kang, Z.; Wang, C.; Zhao, X.; Liu, Y.; Shi, L. A near-infrared light-excitable immunomodulating nano-photosensitizer for effective photoimmunotherapy. Biomater. Sci. 2021, 9, 4191–4198. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gu, Y.; Liu, X.; Fan, Y.; Zhang, Y.; Yi, C.; Cheng, C.; Yang, M. Near-Infrared Photothermally Enhanced Photo-Oxygenation for Inhibition of Amyloid-β Aggregation Based on RVG-Conjugated Porphyrinic Metal-Organic Framework and Indocyanine Green Nanoplatform. Int. J. Mol. Sci. 2022, 23, 10885. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, H.; Dong, X.; Sun, Y. Composite of gold nanoclusters and basified human serum albumin significantly boosts the inhibition of Alzheimer’s β-amyloid by photo-oxygenation. Acta Biomater. 2022, 144, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive hippocampal blood-brain barrier opening in Alzheimer’s disease with focused ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, R.I.; Carpenter, J.S.; Mehta, R.I.; Haut, M.W.; Ranjan, M.; Najib, U.; Lockman, P.; Wang, P.; D’Haese, P.F.; Rezai, A.R. Blood-Brain Barrier Opening with MRI-guided Focused Ultrasound Elicits Meningeal Venous Permeability in Humans with Early Alzheimer Disease. Radiology 2021, 298, 654–662. [Google Scholar] [CrossRef]
- Rezai, A.R.; Ranjan, M.; Haut, M.W.; Carpenter, J.; D’Haese, P.F.; Mehta, R.I.; Najib, U.; Wang, P.; Claassen, D.O.; Chazen, J.L.; et al. Focused ultrasound-mediated blood-brain barrier opening in Alzheimer’s disease: Long-term safety, imaging, and cognitive outcomes. J. Neurosurg. 2022, 1, 1–9. [Google Scholar] [CrossRef]
- Wasielewska, J.M.; Chaves, J.C.S.; Johnston, R.L.; Milton, L.A.; Hernández, D.; Chen, L.; Song, J.; Lee, W.; Leinenga, G.; Nisbet, R.M.; et al. A sporadic Alzheimer’s blood-brain barrier model for developing ultrasound-mediated delivery of Aducanumab and anti-Tau antibodies. Theranostics 2022, 12, 6826–6847. [Google Scholar] [CrossRef]
- Rich, M.C.; Sherwood, J.; Bartley, A.F.; Whitsitt, Q.A.; Lee, M.; Willoughby, W.R.; Dobrunz, L.E.; Bao, Y.; Lubin, F.D.; Bolding, M. Focused ultrasound blood brain barrier opening mediated delivery of MRI-visible albumin nanoclusters to the rat brain for localized drug delivery with temporal control. J. Control. Release 2020, 324, 172–180. [Google Scholar] [CrossRef]
- Liu, Y.; Gong, Y.; Xie, W.; Huang, A.; Yuan, X.; Zhou, H.; Zhu, X.; Chen, X.; Liu, J.; Liu, J.; et al. Microbubbles in combination with focused ultrasound for the delivery of quercetin-modified sulfur nanoparticles through the blood brain barrier into the brain parenchyma and relief of endoplasmic reticulum stress to treat Alzheimer’s disease. Nanoscale 2020, 12, 6498–6511. [Google Scholar] [CrossRef]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular vesicles through the blood-brain barrier: A review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C.V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Cui, G.H.; Guo, H.D.; Li, H.; Zhai, Y.; Gong, Z.B.; Wu, J.; Liu, J.S.; Dong, Y.R.; Hou, S.X.; Liu, J.R. RVG-modified exosomes derived from mesenchymal stem cells rescue memory deficits by regulating inflammatory responses in a mouse model of Alzheimer’s disease. Immun. Ageing 2019, 16, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; He, T.; Chai, Z.; Samulski, R.J.; Li, C. Blood-brain barrier shuttle peptides enhance AAV transduction in the brain after systemic administration. Biomaterials 2018, 176, 71–83. [Google Scholar] [CrossRef]
- Zhang, X.; Chai, Z.; Lee Dobbins, A.; Itano, M.S.; Askew, C.; Miao, Z.; Niu, H.; Samulski, R.J.; Li, C. Customized blood-brain barrier shuttle peptide to increase AAV9 vector crossing the BBB and augment transduction in the brain. Biomaterials 2022, 281, 121340. [Google Scholar] [CrossRef]
- Hersh, D.S.; Anastasiadis, P.; Mohammadabadi, A.; Nguyen, B.A.; Guo, S.; Winkles, J.A.; Kim, A.J.; Gullapalli, R.; Keller, A.; Frenkel, V.; et al. MR-guided transcranial focused ultrasound safely enhances interstitial dispersion of large polymeric nanoparticles in the living brain. PLoS ONE 2018, 13, e0192240. [Google Scholar] [CrossRef] [Green Version]


| Drug Name | Principle | Phase | Effect in Clinical Trials | Status | Refs. |
|---|---|---|---|---|---|
| PBT2 | Reduction in Aβ aggregation | Phase 2 (NCT01590888) | The higher dose reportedly reduced Aβ42 levels in CSF * | Completed | [99,100] |
| Resveratrol | Anti-oxidant capacity; prevention of amyloid deposition | Phase 3 (NCT01504854) | Reduce cognitive impairment and Aβ42 in CSF *; increased Aβ40 levels in CSF * and plasma; increased brain volume loss | Withdraw | [101,102] |
| Alzhemed™ (Tramiprosate) | Inhibit the interaction of Aβ with endogenous glycosaminoglycans | Phase 3 (NCT00314912) | Slowed cognitive decline in ApoE4 homozygotes | Unknown | [89,103] |
| Epigallocatechin Gallate | Remodel toxic amyloid-beta fibrils | Phase 2/3 (NCT00951834) | No public information | Completed | [47] |
| Drug Name | Principle | Phase | Effect in Clinical Trials | Status | Refs. |
|---|---|---|---|---|---|
| Aducanumab (BIIB037) | Passive immunity | Phase 3 (NCT02484547; NCT02477800; NCT01677572) | Bound to soluble monomeric Aβ and reduce brain Aβ; reduced cognitive impairment only at the highest dose; adverse reactions: ARIA * | Approved | [106,107,108] |
| Lecanemab (BAN2401) | Passive immunity | Phase 3 (NCT04468659; NCT03887455) | Reduced markers of amyloid in early AD * Alleviated cognitive and functional decline; adverse reactions: ARIA *, infusion-related reactions | Approved | [20,109,110] |
| Remternetug (LY3372993) | Passive immunity | Phase 3 (NCT05463731) | No public information | Recruiting | https://www.clinicaltrials.gov/ (accessed on 28 March 2023) |
| Gantenerumab (RO4909832) | Passive immunity | Phase 3 (NCT04339413; NCT04339413; NCT02051608) | No reduction in cognitive impairment; adverse reactions: ARIA * | Terminated | [111,112] |
| Solanezumab (LY2062430) | Passive immunity | Phase 3 (NCT02760602; NCT01900665; NCT01127633) | Did not significantly affect cognitive decline | Terminated | [112,113] |
| Crenezumab (MABT5102A) | Passive immunity | Phase 3 (NCT03491150; NCT03114657; NCT03114657) | Did not reduce cognitive decline in participants with early AD * | Terminated | [114,115] |
| Donanemab (LY30028123) | Passive immunity | Phase 2 (NCT03367403) | Improved cognition and daily living ability in early AD patients; reduce amyloid plaque levels and overall tau load | Recruiting | [116,117,118,119,120] |
| ABvac40 | Active immunity | Phase 1 (NCT03113812) | Good safety and tolerance; triggered a consistent and specific immune response | Unknown | [121] |
| ACI-24 | Active immunity | Phase 2 (2018-000445-39) | Produced a low IgG antibody response, increased CSF * Aβ40 and Aβ42 levels but caused no change in amyloid-PET. | Completed | https://www.clinicaltrialsregister.eu/ (accessed on 28 March 2023) |
| Amilomotide (CAD106) | Active immunity | Phase 2 Phase 3 (NCT00795418) | Unexpected changes in cognitive function, brain volume loss, and body weight loss | Terminated | [122] |
| UB-311 | Active immunity | Phase 2 (NCT03531710; NCT03531710) | A slower rate of increase in ADAS-Cog in mild AD * patients; 100% responder rate | Completed | [123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Xu, J.; Xu, H.; Luo, T.; Li, Y.; Jiang, K.; Shentu, Y.; Tong, Z. New Insights into Alzheimer’s Disease: Novel Pathogenesis, Drug Target and Delivery. Pharmaceutics 2023, 15, 1133. https://doi.org/10.3390/pharmaceutics15041133
Chen H, Xu J, Xu H, Luo T, Li Y, Jiang K, Shentu Y, Tong Z. New Insights into Alzheimer’s Disease: Novel Pathogenesis, Drug Target and Delivery. Pharmaceutics. 2023; 15(4):1133. https://doi.org/10.3390/pharmaceutics15041133
Chicago/Turabian StyleChen, Haishu, Jinan Xu, Hanyuan Xu, Tiancheng Luo, Yihao Li, Ke Jiang, Yangping Shentu, and Zhiqian Tong. 2023. "New Insights into Alzheimer’s Disease: Novel Pathogenesis, Drug Target and Delivery" Pharmaceutics 15, no. 4: 1133. https://doi.org/10.3390/pharmaceutics15041133
APA StyleChen, H., Xu, J., Xu, H., Luo, T., Li, Y., Jiang, K., Shentu, Y., & Tong, Z. (2023). New Insights into Alzheimer’s Disease: Novel Pathogenesis, Drug Target and Delivery. Pharmaceutics, 15(4), 1133. https://doi.org/10.3390/pharmaceutics15041133