
Smuggling on the Nanoscale—Fusogenic Liposomes Enable Efficient RNA-Transfer with Negligible Immune Response In Vitro and In Vivo

 , , ,
, , ,  , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Ethics Statement for Neuron Isolation
2.3. Preparation of Fusion- and Lipofection-Based Reagents
2.4. Fluorescence Microscopy
2.5. Flow Cytometry
2.6. Live/Dead Stain Assay
2.7. RNA Isolation and cDNA Synthesis for In Vitro and In Vivo Studies
2.8. qRT-PCR Assays for In Vitro and In Vivo Studies
2.9. Inhibition of Endocytosis
2.10. Live Cell Calcium Imaging
2.11. Neuronal Meshwork Synchronization Analysis
2.12. In Vivo Experiments with Zebrafish
2.12.1. Ethics Statement
2.12.2. Fish and Transgenic Lines
2.12.3. Injection into the Adult Zebrafish Brain
2.12.4. Injection into the Zebrafish Embryo
2.12.5. Immunohistochemistry of Fish Brains
2.12.6. Fluorescence Microscopy of Zebrafish
2.12.7. Quantification of eGFP-Positive Cells in Zebrafish Telencephalon-Slices
2.13. Statistical Analysis
3. Results
3.1. Quantity of Transferred Nucleic Acids and Transfer Mechanism Determine Biocompatibility in Neuronal Cells
3.2. The Reduced Influence of Endosomal Uptake Mechanisms for Fusion-Based RNA Transfer Allows High Biocompatibility and Low Cytokine Responses
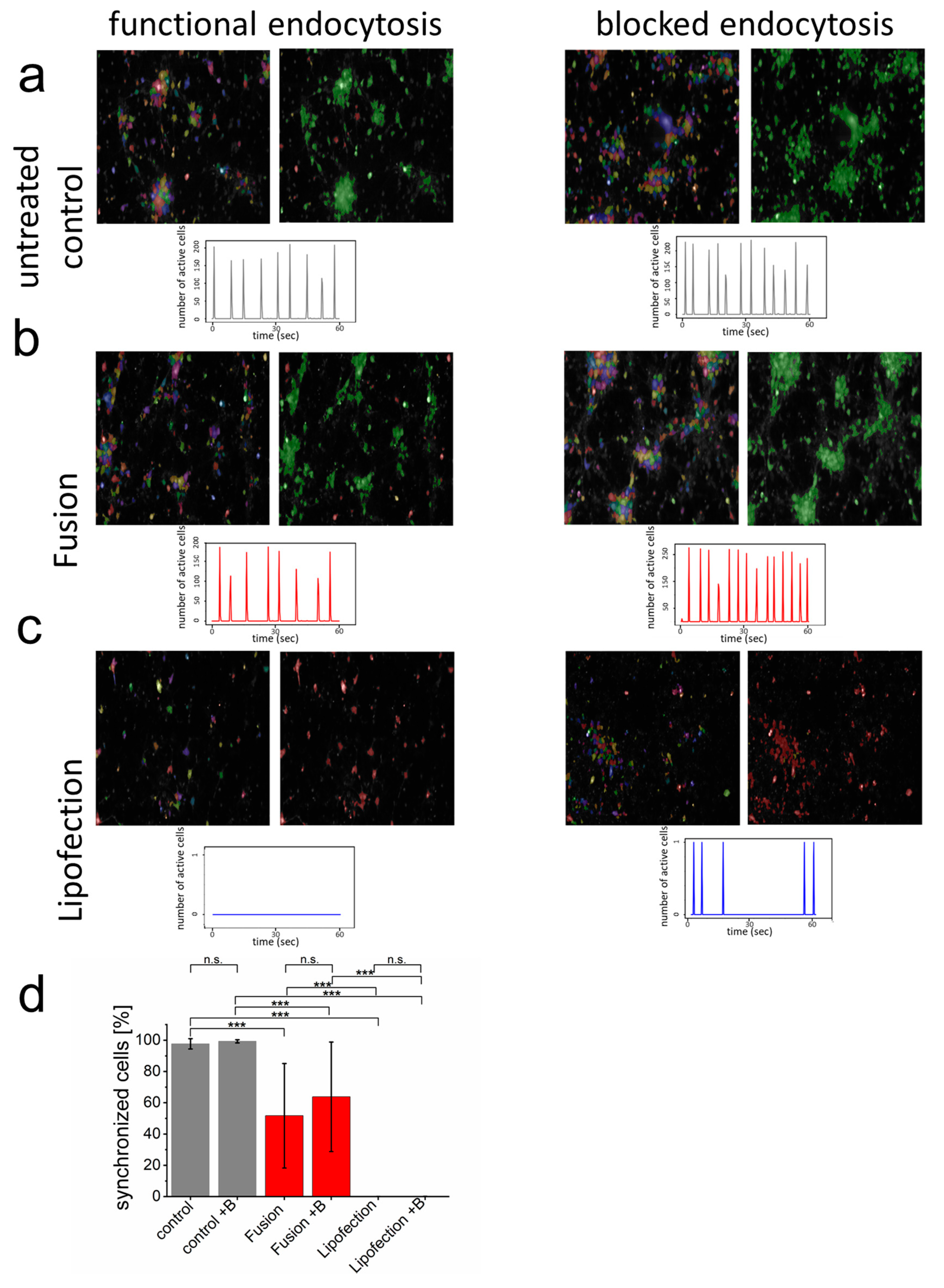
3.3. Endocytosis-Dependent RNA Transfer Blocks Functionality of Neuronal Networks
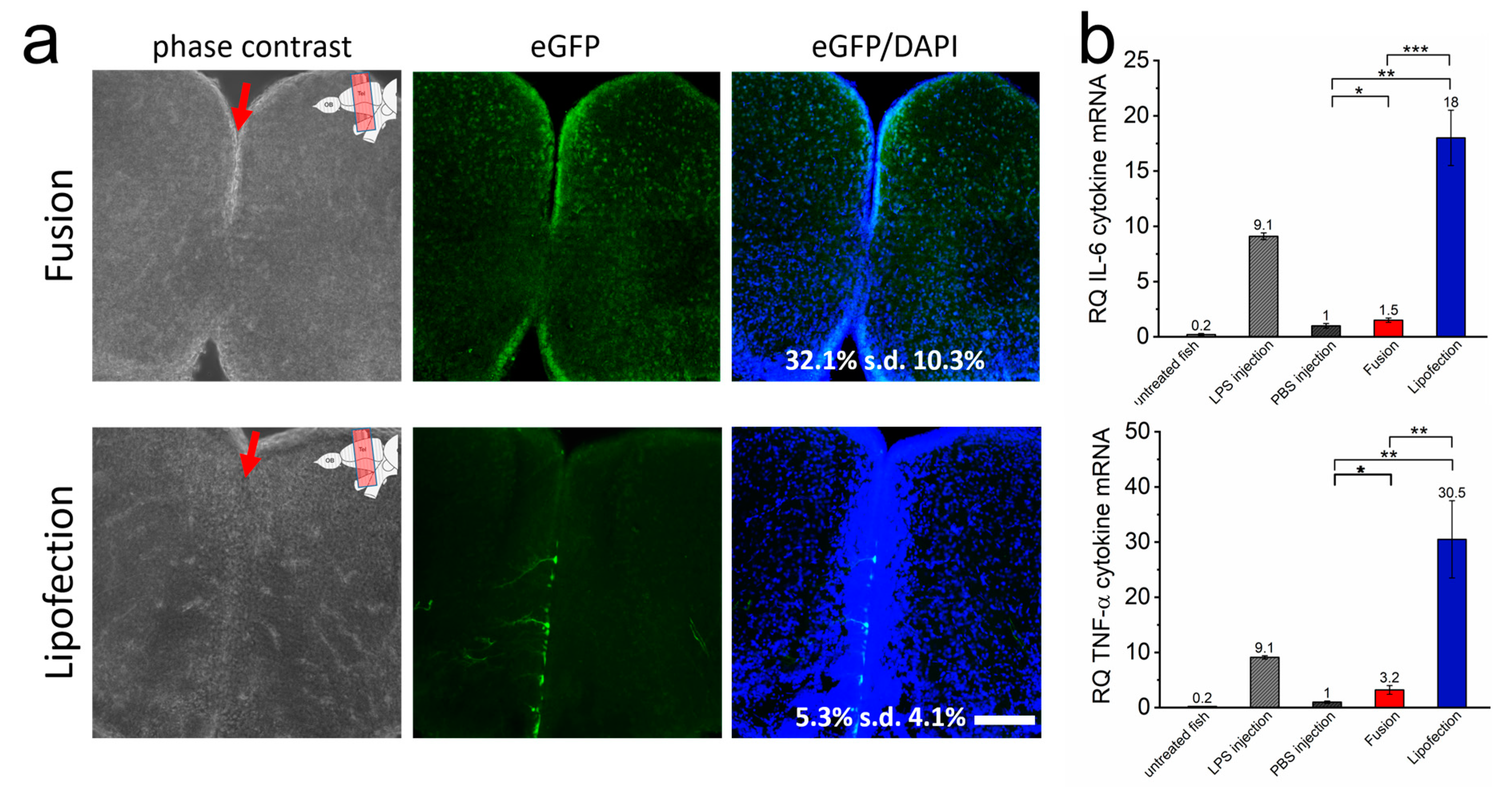
3.4. Inflammatory Responses Are Also Initiated In Vivo by Endosomal Transfer Mechanisms of Nucleic Acids
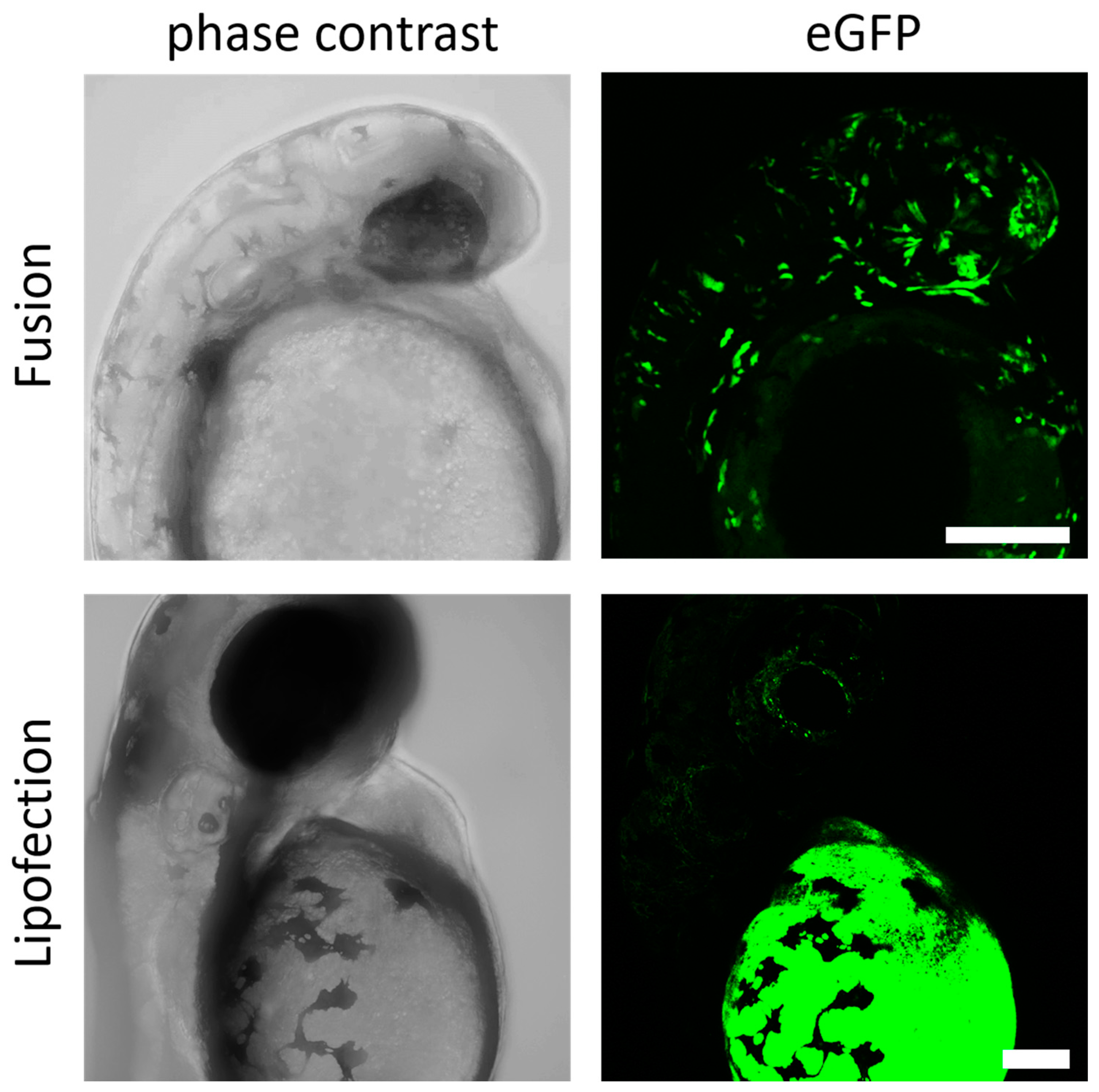
3.5. Endosome-Independent Transfer Mechanisms Are Highly Efficient without Detectable Teratological Effects in Early Embryogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beermann, J.; Piccoli, M.-T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [Green Version]
- Egli, M.; Manoharan, M. Re-Engineering RNA Molecules into Therapeutic Agents. Acc. Chem. Res. 2019, 52, 1036–1047. [Google Scholar] [CrossRef]
- Chung, Y.H.; Beiss, V.; Fiering, S.N.; Steinmetz, N.F. COVID-19 Vaccine Frontrunners and Their Nanotechnology Design. ACS Nano 2020, 14, 12522–12537. [Google Scholar] [CrossRef]
- Walsh, E.E. RNA-Based COVID-19 Vaccine BNT162b2 Selected for a Pivotal Efficacy Study. medRxiv 2020. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Kreiter, S. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Zaks, T.Z.; Wang, R.-F.; Irvine, K.R.; Kammula, U.S.; Marincola, F.M.; Leitner, W.; Restifo, N.P. Cancer therapy using a self-replicating RNA vaccine. Nat. Med. 1999, 5, 823–827. [Google Scholar] [CrossRef]
- Oh, Y.K.; Park, T.G. siRNA delivery systems for cancer treatment. Adv. Drug Deliv. Rev. 2009, 61, 850–862. [Google Scholar] [CrossRef]
- Schott, J.W.; Galla, M.; Godinho, T.; Baum, C.; Schambach, A. Viral and non-viral approaches for transient delivery of mRNA and proteins. Curr. Gene Ther. 2011, 11, 382–398. [Google Scholar] [CrossRef]
- Xue, H.Y.; Guo, P.; Wen, W.-C.; Wong, H.L. Lipid-Based Nanocarriers for RNA Delivery. Curr. Pharm. Des. 2015, 21, 3140–3147. [Google Scholar] [CrossRef]
- Grigsby, C.L.; Leong, K.W. Balancing protection and release of DNA: Tools to address a bottleneck of non-viral gene delivery. J. R. Soc. Interface 2010, 7 (Suppl. S1), S67–S82. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Q.; Jang, Y.; Kang, S.H.; Moon, J.; Kim, W.J.; Park, H. Modulation of immune responses with nanoparticles and reduction of their immunotoxicity. Biomater. Sci. 2020, 8, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Huysmans, H.; Zhong, Z.; De Temmerman, J.; Mui, B.L.; Tam, Y.K.; Mc Cafferty, S.; Gitsels, A.; Vanrompay, D.; Sanders, N.N. Expression Kinetics and Innate Immune Response after Electroporation and LNP-Mediated Delivery of a Self-Amplifying mRNA in the Skin. Mol. Ther. Nucleic Acids 2019, 17, 867–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardarelli, F.; Digiacomo, L.; Marchini, C.; Amici, A.; Salomone, F.; Fiume, G.; Rossetta, A.; Gratton, E.; Pozzi, D.; Caracciolo, G. The intracellular trafficking mechanism of Lipofectamine-based transfection reagents and its implication for gene delivery. Sci. Rep. 2016, 6, 25879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S. Pattern recognition receptors: Doubling up for the innate immune response. Cell 2002, 111, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, J.M. Replication of HIV-1 in vivo and in vitro. Ultrastruct. Pathol. 2007, 31, 151–167. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Slaats, J. IL-1beta/IL-6/CRP and IL-18/ferritin: Distinct Inflammatory Programs in Infections. PLoS Pathog. 2016, 12, e1005973. [Google Scholar] [CrossRef] [Green Version]
- Shechter, R.; Schwartz, M. CNS sterile injury: Just another wound healing? Trends Mol. Med. 2013, 19, 135–143. [Google Scholar] [CrossRef]
- Kaslin, J.; Ganz, J.; Brand, M. Proliferation, neurogenesis and regeneration in the non-mammalian vertebrate brain. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 101–122. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Ma, T.; Zhang, X.; Ying, Q.; Han, M.; Zhang, M.; Yang, R.; Li, Y.; Wang, F.; Liu, R.; et al. Incorporation of CD40 ligand or granulocyte-macrophage colony stimulating factor into Hantaan virus (HTNV) virus-like particles significantly enhances the long-term immunity potency against HTNV infection. J. Med. Microbiol. 2019, 68, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Hinsch, K.; Zupanc, G.K. Generation and long-term persistence of new neurons in the adult zebrafish brain: A quantitative analysis. Neuroscience 2007, 146, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Strahle, U.; Scholpp, S. Neurogenesis in zebrafish—from embryo to adult. Neural Dev. 2013, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Marz, M.; Schmidt, R.; Rastegar, S.; Strähle, U. Regenerative response following stab injury in the adult zebrafish telencephalon. Dev. Dyn. 2011, 240, 2221–2231. [Google Scholar] [CrossRef]
- Rodriguez Viales, R.; Diotel, N.; Ferg, M.; Armant, O.; Eich, J.; Alunni, A.; März, M.; Bally-Cuif, L.; Rastegar, S.; Strähle, U. The helix-loop-helix protein id1 controls stem cell proliferation during regenerative neurogenesis in the adult zebrafish telencephalon. Stem Cells 2015, 33, 892–903. [Google Scholar] [CrossRef]
- Iismaa, S.E.; Kaidonis, X.; Nicks, A.M.; Bogush, N.; Kikuchi, K.; Naqvi, N.; Harvey, R.P.; Husain, A.; Graham, R.M. Comparative regenerative mechanisms across different mammalian tissues. NPJ Regen. Med. 2018, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Modo, M. Bioscaffold-Induced Brain Tissue Regeneration. Front. Neurosci. 2019, 13, 1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Atobe, K.; Barichello, J.M.; Ishida, T.; Kiwada, H. Complex formation with plasmid DNA increases the cytotoxicity of cationic liposomes. Biol. Pharm. Bull. 2007, 30, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Hersch, N.; Dieluweit, S.; Biehl, R.; Merkel, R.; Hoffmann, B. Novel fusogenic liposomes for fluorescent cell labeling and membrane modification. Bioconj. Chem. 2010, 21, 537–543. [Google Scholar] [CrossRef]
- Kube, S.; Hersch, N.; Naumovska, E.; Gensch, T.; Hendriks, J.; Franzen, A.; Landvogt, L.; Siebrasse, J.-P.; Kubitscheck, U.; Hoffmann, B.; et al. Fusogenic Liposomes as Nanocarriers for the Delivery of Intracellular Proteins. Langmuir 2017, 33, 1051–1059. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hersch, N.; Gerlach, S.; Dreissen, G.; Springer, R.; Merkel, R.; Csiszár, A.; Hoffmann, B. Complex Size and Surface Charge Determine Nucleic Acid Transfer by Fusogenic Liposomes. Int. J. Mol. Sci. 2020, 21, 2244. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Hersch, N.; Merkel, R.; Csiszar, A.; Hoffmann, B. Changing the Way of Entrance: Highly Efficient Transfer of mRNA and siRNA via Fusogenic Nano-Carriers. J. Biomed. Nanotechnol. 2019, 15, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Kleusch, C.; Naumovska, E.; Merkel, R.; Csiszár, A. A bioanalytical assay to distinguish cellular uptake routes for liposomes. Cytom. A 2016, 89, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Fricke, R.; Zentis, P.D.; Rajappa, L.T.; Hofmann, B.; Banzet, M.; Offenhäusser, A.; Meffert, S.H. Axon guidance of rat cortical neurons by microcontact printed gradients. Biomaterials 2011, 32, 2070–2076. [Google Scholar] [CrossRef]
- Iwasa, A.; Akita, H.; Khalil, I.; Kogure, K.; Futaki, S.; Harashima, H. Cellular uptake and subsequent intracellular trafficking of R8-liposomes introduced at low temperature. Biochim. Biophys. Acta 2006, 1758, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Eilers PH, B.H. Baseline correction with asymmetric least squares smoothing. Leiden Univ. Med. Centre Rep. 2005, 1, 5. [Google Scholar]
- Alestrom, P.; D’Angelo, L.; Midtlyng, P.J.; Schorderet, D.F.; Schulte-Merker, S.; Sohm, F.; Warner, S. Zebrafish: Housing and husbandry recommendations. Lab. Anim. 2020, 54, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio); Institute of Neuroscience, University of Oregon: Eugene, OR, USA, 2000. [Google Scholar]
- Ando, H.; Okamoto, H. Efficient transfection strategy for the spatiotemporal control of gene expression in zebrafish. Mar. Biotechnol. 2006, 8, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Cubbage, C.C.; Mabee, P.M. Development of the cranium and paired fins in the zebrafish Danio rerio (Ostariophysi, Cyprinidae). J. Morphol. 1996, 229, 121–160. [Google Scholar] [CrossRef]
- Zuiderveld, K. Contrast Limited Adaptive Histogram Equalization. In Graphics Gems IV; Paul, S.H., Ed.; Academic Press Professional, Inc.: Cambridge, MA, USA, 1994; pp. 474–485. [Google Scholar]
- Bradley, D.; Roth, G. Adaptive Thresholding using the Integral Image. J. Gr. Tools 2007, 12, 13–21. [Google Scholar] [CrossRef]
- Carvalho, L.; Heisenberg, C.P. The yolk syncytial layer in early, zebrafish development. Trends Cell Biol. 2010, 20, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Mukalel, A.J.; Riley, R.S.; Zhang, R.; Mitchell, M.J. Nanoparticles for nucleic acid delivery: Applications in cancer immunotherapy. Cancer Lett. 2019, 458, 102–112. [Google Scholar] [CrossRef]
- Youn, H.; Chung, J.K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348. [Google Scholar]
- Pascolo, S. Messenger RNA-based vaccines. Expert Opin. Biol. Ther. 2004, 4, 1285–1294. [Google Scholar] [CrossRef]
- Michel, T.; Luft, D.; Abraham, M.-K.; Reinhardt, S.; Medina, M.L.S.; Kurz, J.; Schaller, M.; Avci-Adali, M.; Schlensak, C.; Peter, K.; et al. Cationic Nanoliposomes Meet mRNA: Efficient Delivery of Modified mRNA Using Hemocompatible and Stable Vectors for Therapeutic Applications. Mol. Ther. Nucleic Acids 2017, 8, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Zeromski, J.; Kaczmarek, M.; Boruczkowski, M.; Kierepa, A.; Kowala-Piaskowska, A.; Mozer-Lisewska, I. Significance and Role of Pattern Recognition Receptors in Malignancy. Arch. Immunol. Ther. Exp. 2019, 67, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA sensors of the innate immune system and their detection of pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Pisetsky, D.S. The origin and properties of extracellular DNA: From PAMP to DAMP. Clin. Immunol. 2012, 144, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.L.; Barton, G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014, 24, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Ohto, U. Toll-like Receptor 9 Contains Two DNA Binding Sites that Function Cooperatively to Promote Receptor Dimerization and Activation. Immunity 2018, 48, 649–658.e4. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Reich, I.C.; Pisetsky, D.S. Mechanisms of activation of the RAW264.7 macrophage cell line by transfected mammalian DNA. Cell. Immunol. 2004, 229, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Durbin, J.K.; Miller, D.K.; Niekamp, J.; Khisamutdinov, E.F. Modulating Immune Response with Nucleic Acid Nanoparticles. Molecules 2019, 24, 3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levenson, E.A.; Kiick, K.L. DNA-polymer conjugates for immune stimulation through Toll-like receptor 9 mediated pathways. Acta Biomater. 2014, 10, 1134–1145. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis E Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Saunders, L.R.; Barber, G.N. The dsRNA binding protein family: Critical roles, diverse cellular functions. FASEB J. 2003, 17, 961–983. [Google Scholar] [CrossRef]
- Moradian, H.; Roch, T.; Lendlein, A.; Gossen, M. mRNA Transfection-Induced Activation of Primary Human Monocytes and Macrophages: Dependence on Carrier System and Nucleotide Modification. Sci. Rep. 2020, 10, 4181. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, M. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef]
- Li, Y.; Cui, X.-L.; Chen, Q.-S.; Yu, J.; Zhang, H.; Gao, J.; Sun, D.-X.; Zhang, G.-Q. Cationic liposomes induce cytotoxicity in HepG2 via regulation of lipid metabolism based on whole-transcriptome sequencing analysis. BMC Pharmacol. Toxicol. 2018, 19, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11, 20140459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.F.; Leong, K.W. Emerging links between surface nanotechnology and endocytosis: Impact on nonviral gene delivery. Nano Today 2010, 5, 553–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolasinac, R.; Kleusch, C.; Braun, T.; Merkel, R.; Csiszár, A. Deciphering the Functional Composition of Fusogenic Liposomes. Int. J. Mol. Sci. 2018, 19, 346. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Brencicova, E.; Diebold, S.S. Nucleic acids and endosomal pattern recognition: How to tell friend from foe? Front. Cell. Infect. Microbiol. 2013, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Kolasinac, R.; Kleusch, C.; Braun, T.; Merkel, R.; Csiszár, A. Influence of Environmental Conditions on the Fusion of Cationic Liposomes with Living Mammalian Cells. Nanomaterials 2019, 9, 1025. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Lu, Y.; Xie, F.; Zou, H.; Fan, X.; Li, B.; Li, W.; Zhang, W.; Mei, L.; Feng, S.-S.; et al. Cationic liposomes induce cell necrosis through lysosomal dysfunction and late-stage autophagic flux inhibition. Nanomedicine 2016, 11, 3117–3137. [Google Scholar] [CrossRef]
- Kleusch, C.; Monzel, C.; Sridhar, K.; Hoffmann, B.; Csiszár, A.; Merkel, R. Fluorescence Correlation Spectroscopy Reveals Interaction of Some Microdomain-Associated Lipids with Cellular Focal Adhesion Sites. Int. J. Mol. Sci. 2020, 21, 8149. [Google Scholar] [CrossRef]
- Kariko, K. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.U.; Lee, H.J.; Kim, Y.B. Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 2013, 33, 491–504. [Google Scholar] [CrossRef]
- Jiao, Y.; Jiao, Y.; Liu, Y.-W.; Chen, W.-G. Neuroregeneration and functional recovery after stroke: Advancing neural stem cell therapy toward clinical application. Neural Regen. Res. 2021, 16, 80–92. [Google Scholar] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef]
- Morcos, P.A.; Li, Y.; Jiang, S. Vivo-Morpholinos: A non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. Biotechniques 2008, 45, 613–614, 616, 618 passim. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.P.; Dangott, L.J.; Lightfoot, J.T. Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques 2014, 56, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Asakawa, K.; Suster, M.L.; Mizusawa, K.; Nagayoshi, S.; Kotani, T.; Urasaki, A.; Kishimoto, Y.; Hibi, M.; Kawakami, K. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc. Natl. Acad. Sci. USA 2008, 105, 1255–1260. [Google Scholar] [CrossRef] [Green Version]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHOK1 | nHEK | Neuronal-Like PC-12 | Primary Embryonic Cortical Neurons | |
|---|---|---|---|---|
| Medium | DMEM F12 Glutmax (Gibco, USA), 1× PenStrep (Gibco, USA), 10% FBS (Superior, Biochrom) | Dermalife K (CellSystems, Germany) without EGF | RPMI 1640 (Gibco, USA), 1× PenStrep (Gibco, USA), 10% FBS (Superior, Biochrom) | 50 mL Gibco Neurobasal Medium + 1 mL B-27 Supplement (Thermo Scientific, USA), 2.5 mg Gentamicin Reagent Solution (Sigma, USA), 125 µL GlutaMax (Thermo Scientific, USA) |
| Lipofectamine® 2000 standard protocol (LSP 1) | 2 µL reagent in 25 µL OptiMEM + 1 µg eGFP mRNA in 25 µL OptiMEM/complexed mRNA + reagent (50 µL) add 500 µL media/incubation over night | |||
| Lipofectamine® 2000 reduced uptake protocol (RU 2) by reduced incubation time (LRT 3) | Lipoplex prepared as in L SP add 500 µL OptiMEM/incubation for 40 min (min) | / | ||
| Lipofectamine® 2000 reduced uptake protocol (RU 2) by LRT and PBS washing (LRT + PBS 4) | Lipoplex prepared as in L SP add 500 µL OptiMEM/incubation for 40 min/wash cells 3× with 1× PBS, pH 7.2, room temperature (RT) | / | ||
| Lipofectamine® 2000 reduced uptake protocol (RU 2) by LRT and PBS-H washing (LRT + PBSH 5) | Lipoplex prepared as in L SP add 500 µL OptiMEM/incubation for 40 min/wash cells 3× with 1× PBS-Heparin (20 U/mL Heparin-Natrium-25.000 (i.v./s.c.) ratiopharm GmbH), pH 7.2, RT | / | ||
| Fuse-It-mRNA standard protocol (F SP 6) | 2 µL neutralization buffer (NB) + 1 µg eGFP mRNA 10 min RT/add 2.5 µL fusogenic solution (FS) | |||
| 10 min fusion in 250 µL PBS | 8 min fusion in 250 µL PBS | 10 min fusion in 250 µL PBS | 10 min fusion in 250 µL PBS | |
| Fuse-It-mRNA reduced uptake protocol (RU 2) by PBS washing after fusion (F SP + PBS 7) | after incubation at 37 °C washed 3× with 1× PBS, pH 7.2, RT | / | ||
| Fuse-It-mRNA reduced uptake protocol (RU 2) by PBS-H washing after fusion (FSP + PBSH 8) | after incubation at 37 °C washed 3× with 1× PBS-Heparin (20 U/mL Heparin-Natrium-25.000 (i.v./s.c.) ratiopharm GmbH), pH 7.2, RT | |||
| CHOK1 | nHEK | Neuronal-Like PC-12 | Primary Embryonic Cortical Neurons | |
|---|---|---|---|---|
| endogenous control | GAPDH chinese hamster (Thermo Scientific; Cg04424039-gh GAPDH FAM) | GAPDH homo sapiens (Thermo Scientific; HS02786624-g1 GAPDH) | GAPDH rattus norvegius (Thermo Scientific; Rn01775763_g1) | |
| eGFP | eGFP mr Enhance (Thermo Scientific; Mr04097229_mr eGFP) | |||
| IL-6 | / | IL-6 Fam homo sapiens (Thermo Scientific; HS00174131_m1 IL-6) | IL-6 Fam rattus norvegius (Thermo Scientific; Rn01410330_m1) | |
| TNF-α | / | TNF-α homo sapiens (Thermo Scientific; HS00174128_m1 TNF-α) | / | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, M.; Gerlach, S.; Takamiya, M.; Tarazi, S.; Hersch, N.; Csiszár, A.; Springer, R.; Dreissen, G.; Scharr, H.; Rastegar, S.; et al. Smuggling on the Nanoscale—Fusogenic Liposomes Enable Efficient RNA-Transfer with Negligible Immune Response In Vitro and In Vivo. Pharmaceutics 2023, 15, 1210. https://doi.org/10.3390/pharmaceutics15041210
Hoffmann M, Gerlach S, Takamiya M, Tarazi S, Hersch N, Csiszár A, Springer R, Dreissen G, Scharr H, Rastegar S, et al. Smuggling on the Nanoscale—Fusogenic Liposomes Enable Efficient RNA-Transfer with Negligible Immune Response In Vitro and In Vivo. Pharmaceutics. 2023; 15(4):1210. https://doi.org/10.3390/pharmaceutics15041210
Chicago/Turabian StyleHoffmann, Marco, Sven Gerlach, Masanari Takamiya, Samar Tarazi, Nils Hersch, Agnes Csiszár, Ronald Springer, Georg Dreissen, Hanno Scharr, Sepand Rastegar, and et al. 2023. "Smuggling on the Nanoscale—Fusogenic Liposomes Enable Efficient RNA-Transfer with Negligible Immune Response In Vitro and In Vivo" Pharmaceutics 15, no. 4: 1210. https://doi.org/10.3390/pharmaceutics15041210
APA StyleHoffmann, M., Gerlach, S., Takamiya, M., Tarazi, S., Hersch, N., Csiszár, A., Springer, R., Dreissen, G., Scharr, H., Rastegar, S., Beil, T., Strähle, U., Merkel, R., & Hoffmann, B. (2023). Smuggling on the Nanoscale—Fusogenic Liposomes Enable Efficient RNA-Transfer with Negligible Immune Response In Vitro and In Vivo. Pharmaceutics, 15(4), 1210. https://doi.org/10.3390/pharmaceutics15041210