
Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450

 ,
,  , ,
, ,  and
and 
Abstract
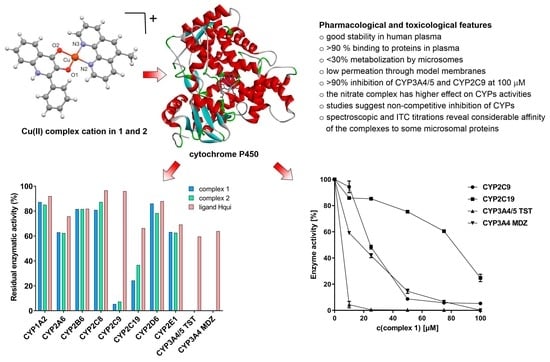
:
1. Introduction
2. Materials and Methods
2.1. In Vitro Pharmacological Properties
2.2. Chemical Stability (Stability in PBS) and Stability in Human Plasma
2.3. Microsomal Stability Assay
2.4. Plasma Protein Binding (PPB)
2.5. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.6. Cytochrome P450 Activities
2.7. Spectroscopic Study of Interactions of the Studied Complexes with Human Liver Microsomes
2.8. Isothermal Titration Calorimetry (ITC)
2.9. DFT Geometry Optimisation (Computational Details)
3. Results and Discussion
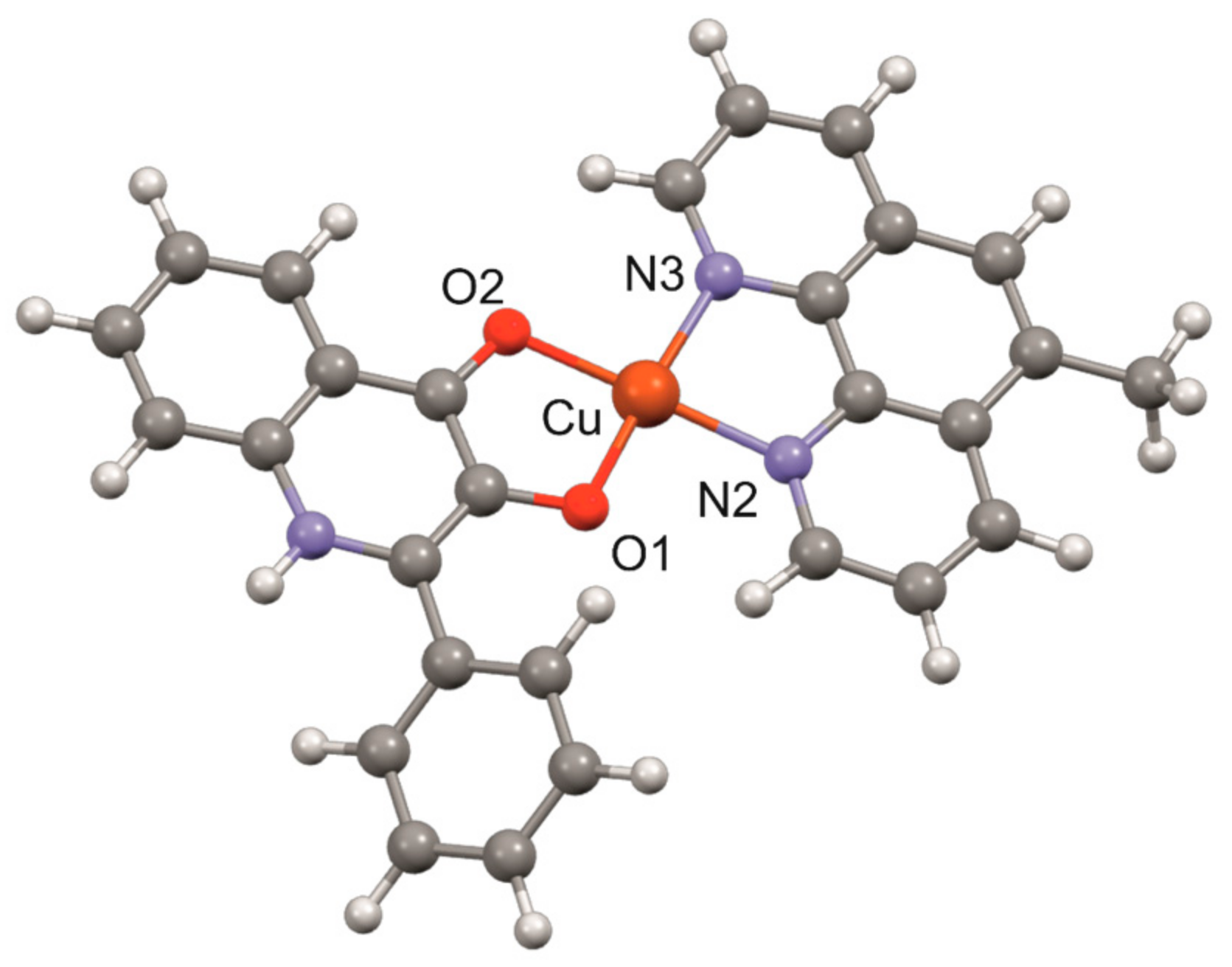
3.1. Geometry Optimisation of the [Cu(mphen)(qui)]+ Cation
3.2. Pharmacokinetic Properties of the Studied Compounds
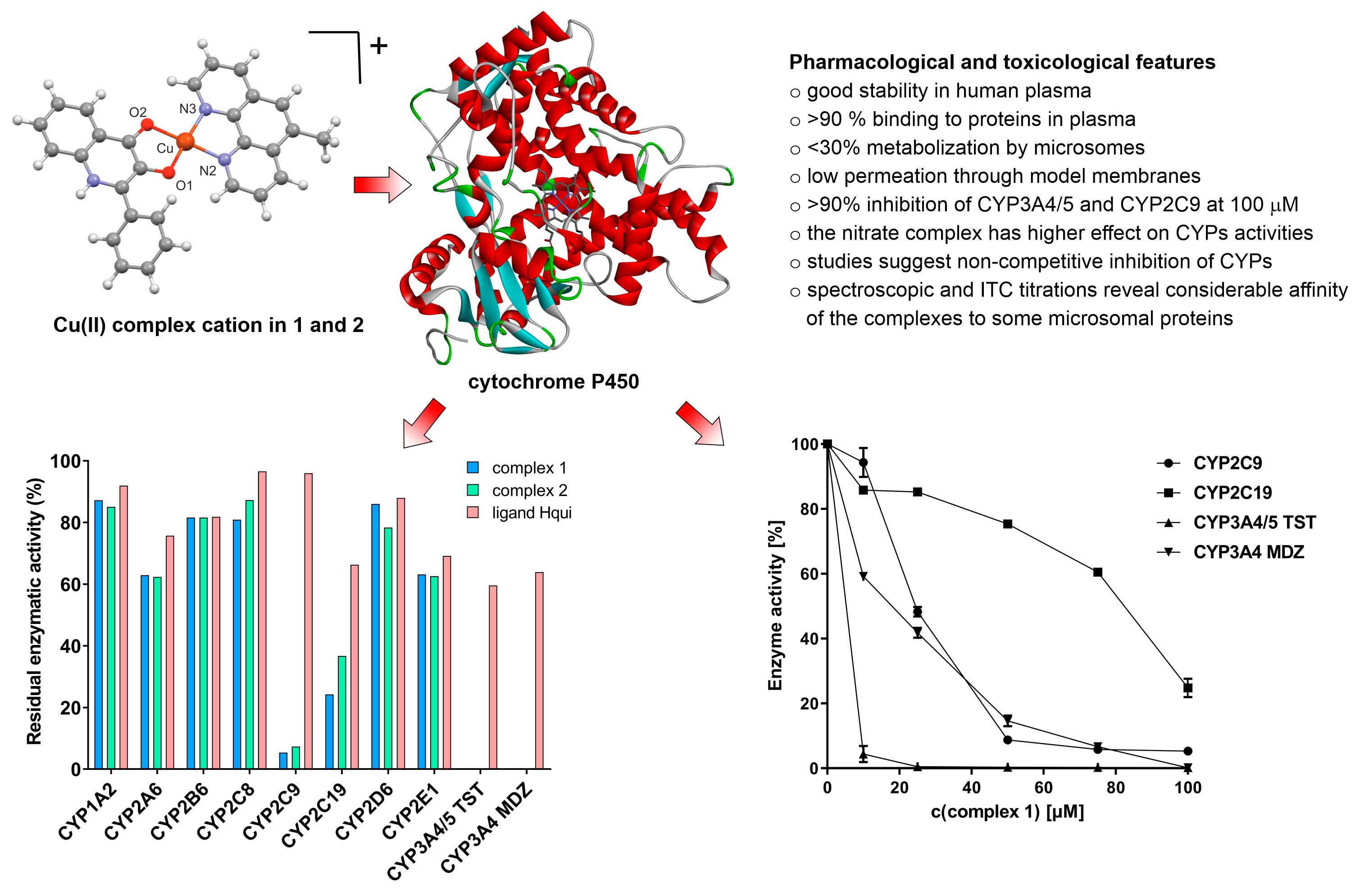
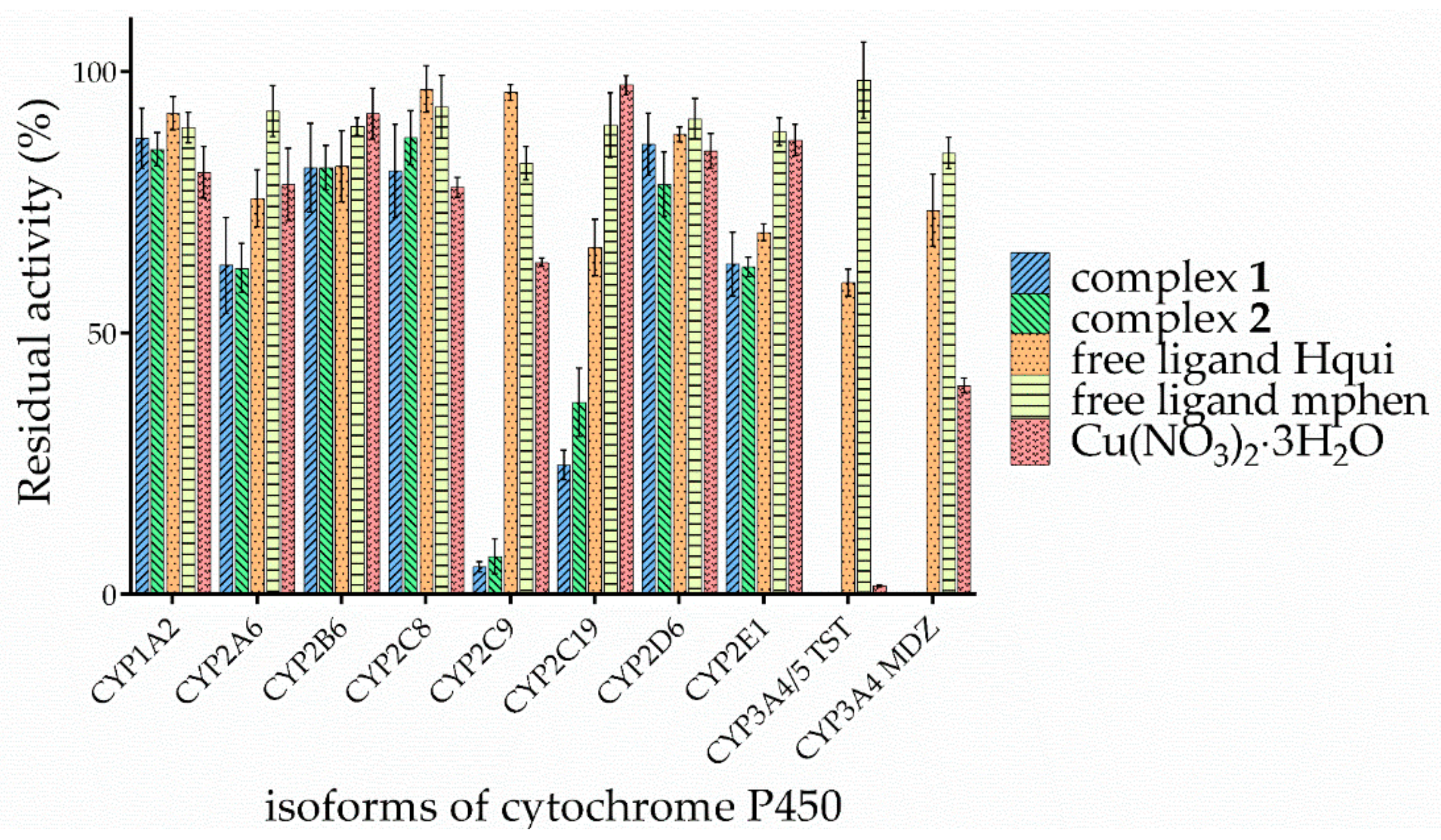
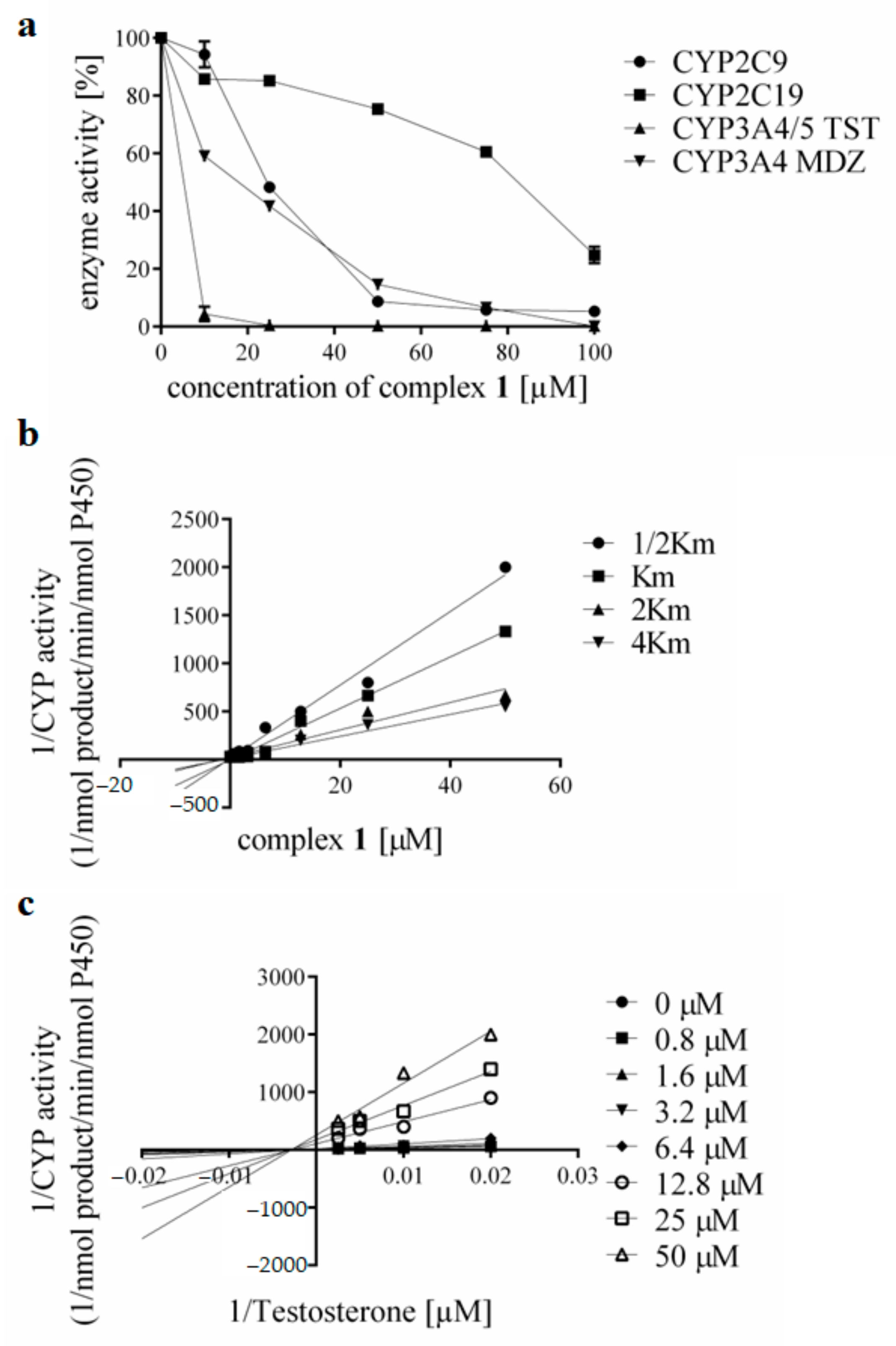
3.3. Effects of the Complexes and Free Hqui and mphen Ligands on Catalytic Activities Cytochrome P450 in Human Liver Microsomes
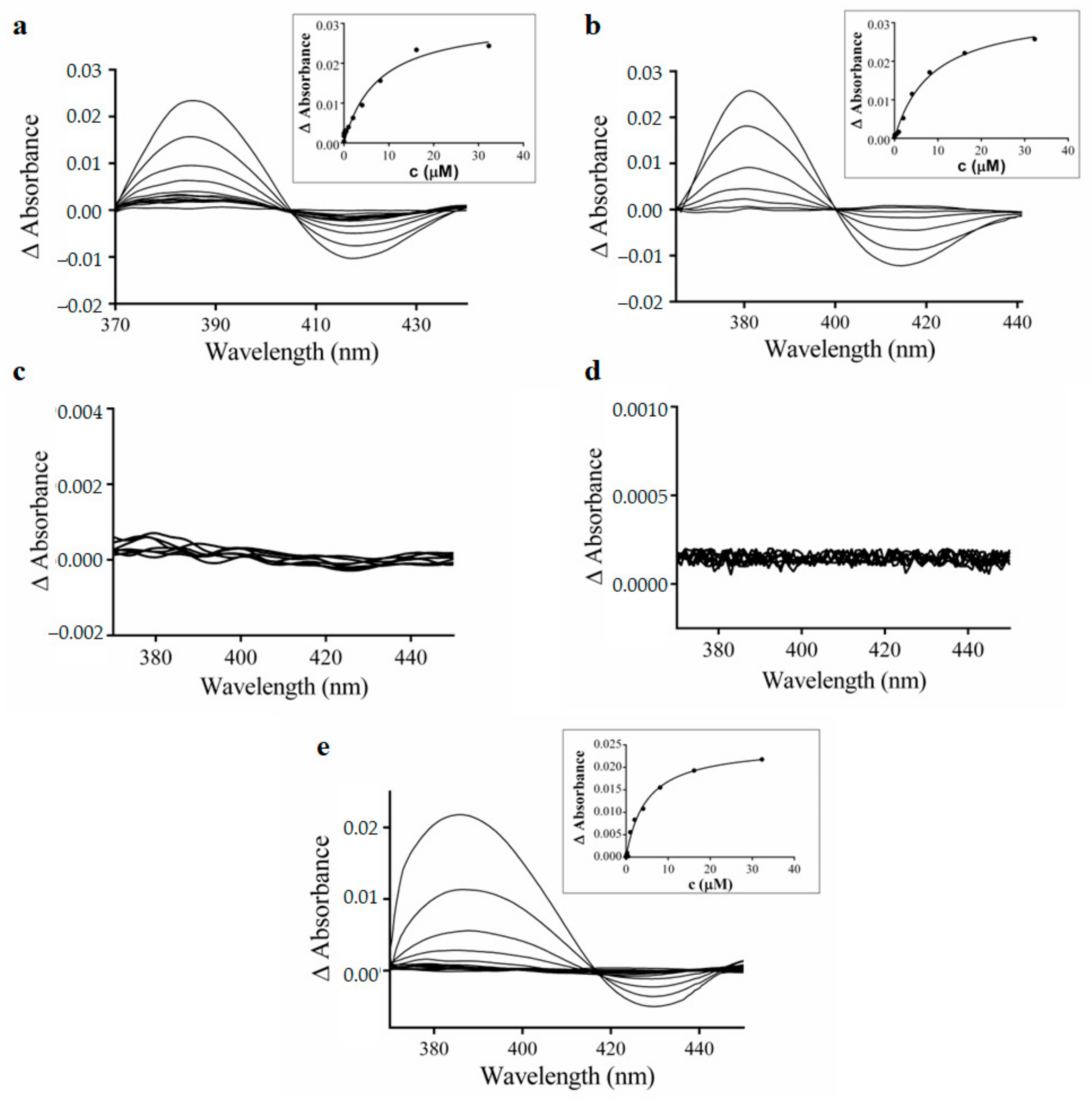
3.4. Spectral Interaction Studies of the Complexes, Free Hqui and mphen Ligands, and Copper(II) Nitrate Trihydrate with Human Liver Microsomal CYPs
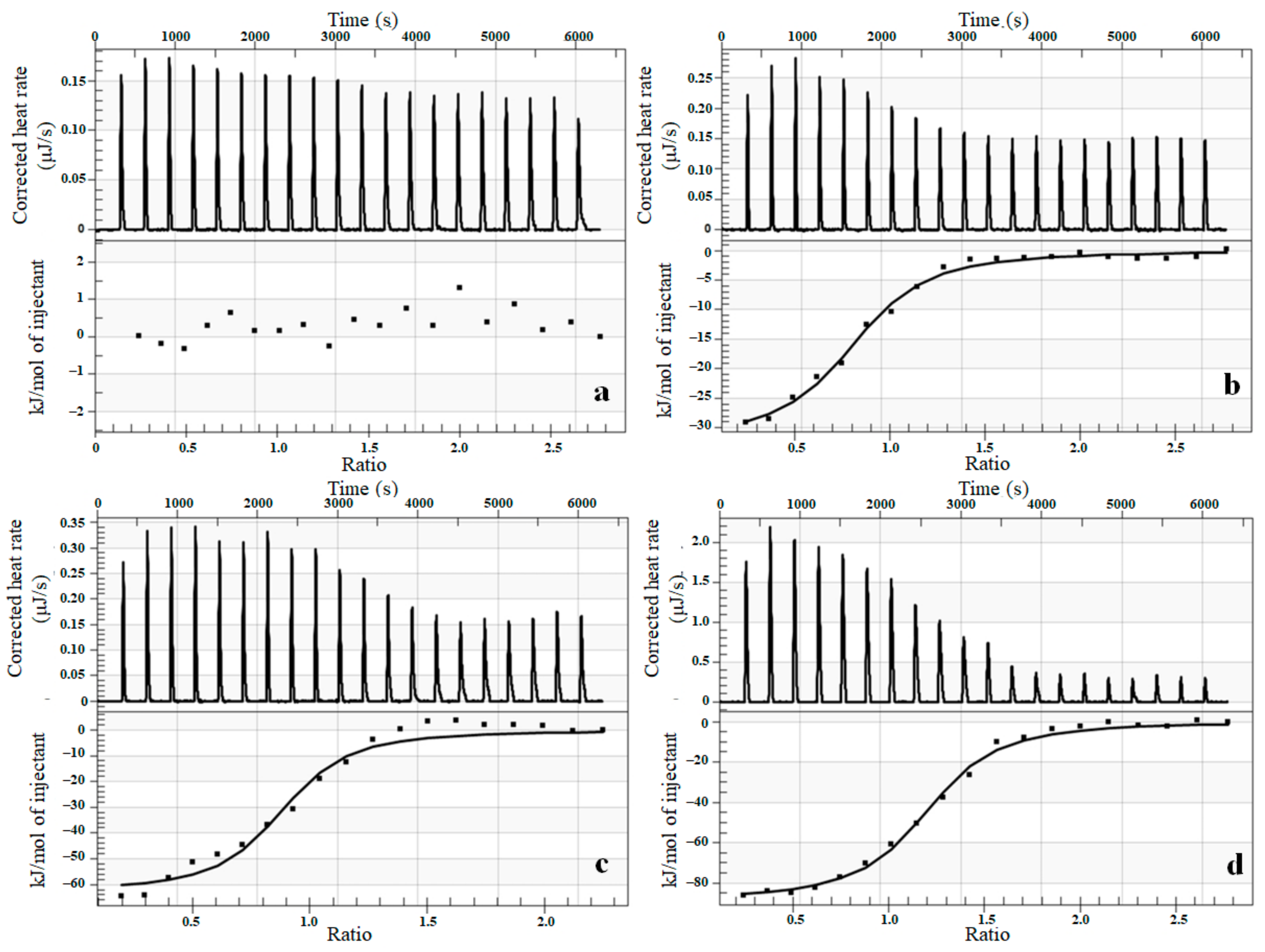
3.5. Thermodynamic Characterization of Interaction between Complexes and Cytochrome P450 Using Isothermal Titration Calorimetry (ITC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenberg, B.; Van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J.M. Understanding and Improving Platinum Anticancer Drugs—Phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug. Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, G.; Vessieres, A.; Top, S. Ferrocifen type anticancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Tabti, R.; Tounsi, N.; Gaiddon, C.; Bentouhami, E.; Désaubry, L. Progress in Copper Complexes as Anticancer Agents. Med. Chem. 2017, 7, 875–879. [Google Scholar] [CrossRef]
- Munteanu, C.R.; Suntharalingam, K. Advances in cobalt complexes as anticancer agents. Dalton Trans. 2015, 44, 13796–13808. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an Old Drug for a Golden New Age. Drugs R&D 2015, 15, 13–20. [Google Scholar] [CrossRef]
- Pragti, K.; Bidyut, K.; Mukhopadhyay, S. Target based chemotherapeutic advancement of ruthenium complexes. Coord. Chem. Rev. 2021, 448, 214169. [Google Scholar] [CrossRef]
- Leon, I.E.; Cadavid-Vargas, J.F.; di Virgilio, A.L.; Etcheverry, S.B. Vanadium, Ruthenium and Copper Compounds: A New Class of Nonplatinum Metallodrugs with Anticancer Activity. Curr. Med. Chem. 2017, 24, 112–148. [Google Scholar] [CrossRef]
- Lazarević, T.; Rilak, A.; Bugarčić, Ž.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Kar, K.; Ghost, D.; Kabi, B.; Chandra, A. A concise review on cobalt Schiff base complexes as anticancer agents. Polyhedron 2022, 222, 115890. [Google Scholar] [CrossRef]
- Sainath, B.A.; Poonam, R.I.; Mrunalini, K.; Makarand, V.P.; Srushti, P.; Vasudev, B. Silver Complexes of N-Heterocyclic Carbenes as Anticancer Agents: A Review. Int. J. Life Sci. Pharma Res. 2022, 12, L123–L129. [Google Scholar] [CrossRef]
- Omondi, R.O.; Ojwach, S.O.; Jaganyi, D. Review of comparative studies of cytotoxic activities of Pt(II), Pd(II), Ru(II)/(III) and Au(III) complexes, their kinetics of ligand substitution reactions and DNA/BSA interactions. Inorganica Chim. Acta 2020, 512, 119883. [Google Scholar] [CrossRef]
- Gasparin, C.B.; Pilger, D.A. 8-hydroxyquinoline, derivatives and metal-complexes: A review of antileukemia activities. ChemistrySelect 2023, 8, e202204219. [Google Scholar] [CrossRef]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Ping Dou, Q. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef]
- Stern, B.R.; Solioz, M.; Krewski, D.; Aggett, P.; Aw, T.C.; Baker, S.; Crump, K.; Dourson, M.; Haber, L.; Hertzberg, R.; et al. Copper and Human Health: Biochemistry, Genetics, and Strategies for Modeling Dose-response Relationships. J. Toxicol. Environ. Health B 2007, 10, 157–222. [Google Scholar] [CrossRef]
- Ng, C.H.; Kong, K.C.; Von, S.T.; Balraj, P.; Jensen, P.; Thirthagiri, E.; Hamada, H.; Chikira, M. Synthesis, characterization, DNA-binding study and anticancer properties of ternary metal(ii) complexes of edda and an intercalating ligand. Dalton Trans. 2008, 8, 447–454. [Google Scholar] [CrossRef]
- Balsa, L.M.; Baran, E.J.; León, I.E. Copper complexes as antitumor agents: In vitro and in vivo evidences. Curr. Med. Chem. 2022, 30, 510–557. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: Copper That Cancer. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Azuara, L. Process to Obtain New Mixed Copper Aminoacidate Complexes from Phenylate Phenathrolines to Be Used as Anticancerigenic Agents. European Patent EP0434444, 26 June 1991. [Google Scholar]
- Ruiz-Azuara, L. Copper Amino Acidate Diimine Nitrate Compounds and Their Methyl Derivatives and a Process for Preparing Them. U.S. Patent 5,576,326, 19 November 1996. [Google Scholar]
- Ruiz-Azuara, L. Casiopeina Parenteral Composition and Uses of the Same. Mexican Patent MX2017016444A, 17 June 2019. [Google Scholar]
- De Vizcaya-Ruiz, A.; Rivero-Muller, A.; Ruiz-Ramirez, L.; Kass, G.E.N.; Kelland, L.R.; Orr, R.M.; Dobrota, M. Induction of apoptosis by a novel copper-based anticancer compound, Casiopeina II, in L1210 murine leukaemia and CH1 human ovarian carcinoma cells. Toxicol. In Vitro 2000, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
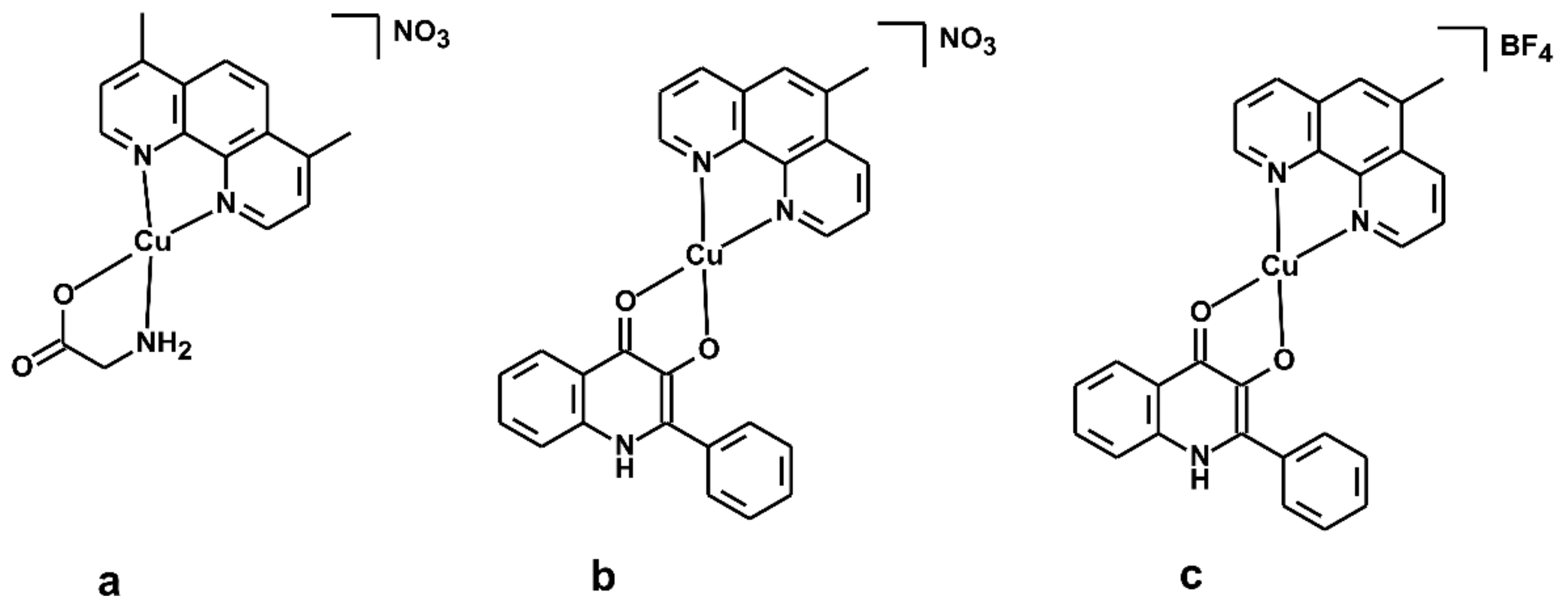
- Buchtík, R.; Trávníček, Z.; Vančo, J.; Herchel, R.; Dvořák, Z. Synthesis, characterization, DNA interaction and cleavage, and in vitro cytotoxicity of copper(ii) mixed-ligand complexes with 2-phenyl-3-hydroxy-4(1H)-quinolinone. Dalton Trans. 2011, 40, 9404. [Google Scholar] [CrossRef]
- Buchtík, R.; Trávníček, Z.; Vančo, J. In vitro cytotoxicity, DNA cleavage and SOD-mimic activity of copper(II) mixed-ligand quinolinonato complexes. J. Inorg. Biochem. 2012, 116, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Trávníček, Z.; Vančo, J.; Hošek, J.; Buchtík, R.; Dvořák, Z. Cellular responses induced by Cu(II) quinolinonato complexes in human tumor and hepatic cells. Chem. Cent. J. 2012, 6, 160. [Google Scholar] [CrossRef]
- Vančo, J.; Trávníček, Z.; Hošek, J.; Dvořák, Z. Heteroleptic copper(II) complexes of prenylated flavonoid osajin behave as selective and effective antiproliferative and anti-inflammatory agents. J. Inorg. Biochem 2022, 226, 111639. [Google Scholar] [CrossRef]
- Vančo, J.; Trávníček, Z.; Hošek, J.; Malina, T.; Dvořák, Z. Copper(II) complexes containing natural flavonoid pomiferin show considerable in vitro cytotoxicity and anti-inflammatory effects. Int. J. Mol. Sci. 2021, 22, 7626. [Google Scholar] [CrossRef]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Gul, N.S.; Khan, T.M.; Chen, M.; Huang, K.B.; Hou, C.; Choudhary, M.I.; Liang, H.; Chen, Z.F. New copper complexes inducing bimodal death through apoptosis and autophagy in A549 cancer cells. J. Inorg. Biochem. 2020, 213, 111260. [Google Scholar] [CrossRef]
- Trejo-Solís, C.; Jimenez-Farfan, D.; Rodriguez-Enriquez, S.; Fernandez-Valverde, F.; Cruz-Salgado, A.; Ruiz-Azuara, L.; Sotelo, J. Copper compound induces autophagy and apoptosis of glioma cells by reactive oxygen species and jnk activation. BMC Cancer 2012, 12, 156. Available online: http://www.biomedcentral.com/1471-2407/12/156 (accessed on 28 September 2022). [CrossRef]
- Marín-Hernández, A.; Gallardo-Pérez, J.C.; López-Ramírez, S.Y.; García-García, J.D.; Rodríguez-Zavala, J.S.; Ruiz-Ramírez, L.; Gracia-Mora, I.; Zentella-Dehesa, A.; Sosa-Garrocho, M.; Macías-Silva, M.; et al. Casiopeina II-gly and bromo-pyruvate inhibition of tumor hexokinase, glycolysis, and oxidative phosphorylation. Arch. Toxicol. 2012, 86, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef]
- Grüner, B.; Brynda, J.; Das, V.; Šicha, V.; Štěpánková, J.; Nekvinda, J.; Holub, J.; Pospisilova, K.; Fábry, M.; Pachl, P.; et al. Metallacarborane Sulfamides: Unconventional, Specific, and Highly Selective Inhibitors of Carbonic Anhydrase IX. J. Med. Chem. 2019, 62, 9560–9575. [Google Scholar] [CrossRef] [PubMed]
- Borkova, L.; Frydrych, I.; Jakubcová, N.; Adamek, R.; Lišková, B.; Gurská, S.; Medvedíková, M.; Hajduch, M.; Urban, M. Synthesis and biological evaluation of triterpenoid thiazoles derived from betulonic acid, dihydrobetulonic acid, and ursonic acid. Eur. J. Med. Chem. 2020, 185, 111806. [Google Scholar] [CrossRef] [PubMed]
- Phillips, I.; Shephard, E.; Ortiz De Montellano, P. Cytochrome P450 Protocols, 3rd ed.; Methods in Molecular Biology (Clifton, NJ, USA); Humana: New York, NY, USA, 2013; Volume 987, ISBN 9781627033213. [Google Scholar]
- Kumar Singh, J.; Olanki, A. Rapid Equilibrium Dialysis (RED): An In-vitro High-Throughput Screening Technique for Plasma Protein Binding using Human and Rat Plasma. J. Bioequivalence Bioavailab. 2012, S14, 1–4. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E. Profiling drug-like properties in discovery research. Curr. Opin. Chem. Biol. 2003, 7, 402–408. [Google Scholar] [CrossRef]
- Schenkman, J.; Jansson, I. Spectral Analyses of Cytochromes P450. Methods Mol. Biol. 2005, 320, 11–18. [Google Scholar] [CrossRef]
- Spartan 14, version 1.1.4; Wavefunction, Inc.: Irvine, CA, USA, 2013.
- Nassar, A.F.; Hollenberg, P.F.; Scatina, J. Drug Metabolism Handbook: Concepts and Applications; Wiley: Hoboken, NJ, USA, 2009; ISBN 9780470118030. [Google Scholar]
- Skolnik, S.; Lin, X.; Wang, J.; Chen, X.; He, T.; Zhang, B. Towards Prediction of In Vivo Intestinal Absorption Using a 96-Well Caco-2 Assay. J. Pharm. Sci. 2010, 99, 3246–3265. [Google Scholar] [CrossRef]
- Daly, A.; Rettie, A.; Fowler, D.; Miners, J. Pharmacogenomics of CYP2C9: Functional and Clinical Considerations. J. Pers. Med. 2018, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Bu, H.Z. A Literature Review of Enzyme Kinetic Parameters for CYP3A4-Mediated Metabolic Reactions of 113 Drugs in Human Liver Microsomes: Structure- Kinetics Relationship Assessment. Curr. Drug. Met. 2006, 7, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Hendrychová, T.; Anzenbacherová, E.; Hudeček, J.; Skopalík, J.; Lange, R.; Hildebrandt, P.; Otyepka, M.; Anzenbacher, P. Flexibility of human cytochrome P450 enzymes: Molecular dynamics and spectroscopy reveal important function-related variations. Biochim. Biophys. Acta 2011, 1814, 58–68. [Google Scholar] [CrossRef]
- Campero-Peredo, C.; Bravo-Gómez, M.E.; Hernández-Ojeda, S.L.; del Rosario Olguin-Reyes, S.; Espinosa-Aguirre, J.J.; Ruiz-Azuara, L. Effect of [Cu(4,7-dimethyl-1,10-phenanthroline)(acetylacetonato)]NO3, Casiopeína III-Ea, on the activity of cytochrome P450. Toxicol. In Vitro 2016, 33, 16–22. [Google Scholar] [CrossRef]
- Jefcoate, C.R. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 1978, 52, 258–279. [Google Scholar] [CrossRef] [PubMed]
- Kastritis, P.; Bonvin, A. On the binding affinity of macromolecular interactions: Daring to ask why proteins interact. J. R. Soc. Interface 2013, 10, 20120835. [Google Scholar] [CrossRef]
- Jelesarov, I.; Bosshard, H.R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999, 12, 3–18. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYP450 Form | CYP450 Activity Assays | CYP (pmol) | Substrate | Km (μM) | Incubation Time (min) |
|---|---|---|---|---|---|
| CYP1A2 | 7-Ethoxyresorufin O-deethylation | 35 | Ethoxyresorufin | 1.56 | 15 |
| CYP2A6 | Coumarin 7-hydroxylation | 35 | Coumarin | 14.00 | 15 |
| CYP2B6 | 7-Ethoxy-4-trifluoromethylcoumarin 7-deethylation | 35 | 7-Ethoxy-4-trifluoromethylcoumarin | 15.25 | 15 |
| CYP2C8 | Paclitaxel 6-hydroxylation | 70 | Paclitaxel | 18.41 | 15 |
| CYP2C9 | Diclofenac 4′-hydroxylation | 35 | Diclofenac | 16.00 | 25 |
| CYP2C19 | (S)-Mephenytoin 4′-hydroxylation | 50 | (S)-Mephenytoin | 28.00 | 25 |
| CYP2D6 | Bufuralol 1′-hydroxylation | 70 | Bufuralol | 14.30 | 20 |
| CYP2E1 | Chlorzoxazone 6-hydroxylation | 160 | Chlorzoxazone | 56.00 | 20 |
| CYP3A4/5 | Testosterone 6β-hydroxylation | 100 | Testosterone | 100.00 | 20 |
| CYP3A4 | Midazolam 1′-hydroxylation | 13 | Midazolam | 2.20 | 8 |
| B3LYP | BP | ϖB97X-D | ||||||
|---|---|---|---|---|---|---|---|---|
| LANL2DZ | LACVP | LANL2DZ | LACVP | LANL2DZ | LACVP | LACVP** | X-ray * | |
| Cu–O1 | 1.912 | 1.918 | 1.955 | 1.960 | 1.894 | 1.901 | 1.879 | 1.892(2) |
| Cu–O2 | 1.944 | 1.949 | 1.976 | 1.982 | 1.935 | 1.941 | 1.923 | 1.916(2) |
| Cu–N2 | 2.024 | 2.017 | 2.022 | 2.015 | 2.011 | 2.003 | 2.024 | 1.978(2) |
| Cu–N3 | 2.021 | 2.014 | 2.022 | 2.015 | 2.013 | 2.005 | 2.025 | 1.988(2) |
| O1–Cu–N3 | 175.76 | 175.86 | 166.48 | 166.66 | 176.00 | 176.10 | 175.52 | 176.22(8) |
| O2–Cu–N2 | 175.57 | 175.70 | 166.19 | 166.34 | 177.95 | 177.93 | 177.50 | 177.98(8) |
| % Compound Remaining | ||||||||
|---|---|---|---|---|---|---|---|---|
| Chemical Stability | Plasma Stability | |||||||
| Compound | 15 min | 30 | 60 | 120 | 15 min | 30 | 60 | 120 |
| complex 1 | 99.6 ± 3.6 | 99.9 ± 1.5 | 97.8 ± 3.4 | 96.6 ± 3.4 | 98.9 ± 2.5 | 94.0 ± 3.3 | 94.2 ± 2.3 | 91.2 ± 1.2 |
| complex 2 | 99.6 ± 3.0 | 96.8 ± 2.4 | 94.0 ± 3.3 | 96.6 ± 2.4 | 98.3 ± 2.4 | 94.1 ± 2.4 | 96.6 ± 3.4 | 92.6 ± 2.2 |
| % Compound remaining | ||||||||
| Microsomal stability | Plasma protein binding | PAMPA | ||||||
| Compound | 15 min | 30 | 60 | % Fraction bound | log Papp | Category a | ||
| complex 1 | 99.4 ± 3.5 | 80.7 ± 2.8 | 78.1 ± 1.7 | 95.1 ± 1.0 | −8.93 ± 0.58 | Low | ||
| complex 2 | 76.5 ± 2.7 | 76.2 ± 2.7 | 69.7 ± 2.4 | 92.9 ± 1.5 | −8.69 ± 0.66 | Low | ||
| Ka | ΔH | n | Kd | ΔS | ΔG | ||
|---|---|---|---|---|---|---|---|
| (1/M) | (kJ/mol) | (nM) | (J/mol.K) | (J/mol) | |||
| complex 1 | CYP1A2B | no interaction | |||||
| CYP2A6B | 1.04 × 106 | −31.81 | 0.801 | 962.0 | 27.64 | −40,050.9 | |
| CYP3A4B | 1.92 × 107 | −80.02 | 0.911 | 52.1 | −129 | −41,558.7 | |
| CYP3A4 | 1.30 × 107 | −88.06 | 1.171 | 76.9 | −178.3 | −34,899.9 | |
| complex 2 | CYP1A2B | no interaction | |||||
| CYP2A6B | 3.12 × 106 | −23.04 | 1.071 | 321.0 | 47.06 | −37,070.9 | |
| CYP3A4B | 2.17 × 107 | −62.71 | 0.859 | 46.1 | −69.86 | −41,881.2 | |
| CYP3A4 | 1.59 × 107 | −51.77 | 1.095 | 62.9 | −154.9 | −5586.6 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medvedíková, M.; Ranc, V.; Vančo, J.; Trávníček, Z.; Anzenbacher, P. Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450. Pharmaceutics 2023, 15, 1314. https://doi.org/10.3390/pharmaceutics15041314
Medvedíková M, Ranc V, Vančo J, Trávníček Z, Anzenbacher P. Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450. Pharmaceutics. 2023; 15(4):1314. https://doi.org/10.3390/pharmaceutics15041314
Chicago/Turabian StyleMedvedíková, Martina, Václav Ranc, Ján Vančo, Zdeněk Trávníček, and Pavel Anzenbacher. 2023. "Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450" Pharmaceutics 15, no. 4: 1314. https://doi.org/10.3390/pharmaceutics15041314
APA StyleMedvedíková, M., Ranc, V., Vančo, J., Trávníček, Z., & Anzenbacher, P. (2023). Highly Cytotoxic Copper(II) Mixed-Ligand Quinolinonato Complexes: Pharmacokinetic Properties and Interactions with Drug Metabolizing Cytochromes P450. Pharmaceutics, 15(4), 1314. https://doi.org/10.3390/pharmaceutics15041314