

Iminosugar-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors as Potential Anti-Pancreatic Cancer Agents




 ,
, 
Abstract
:1. Introduction
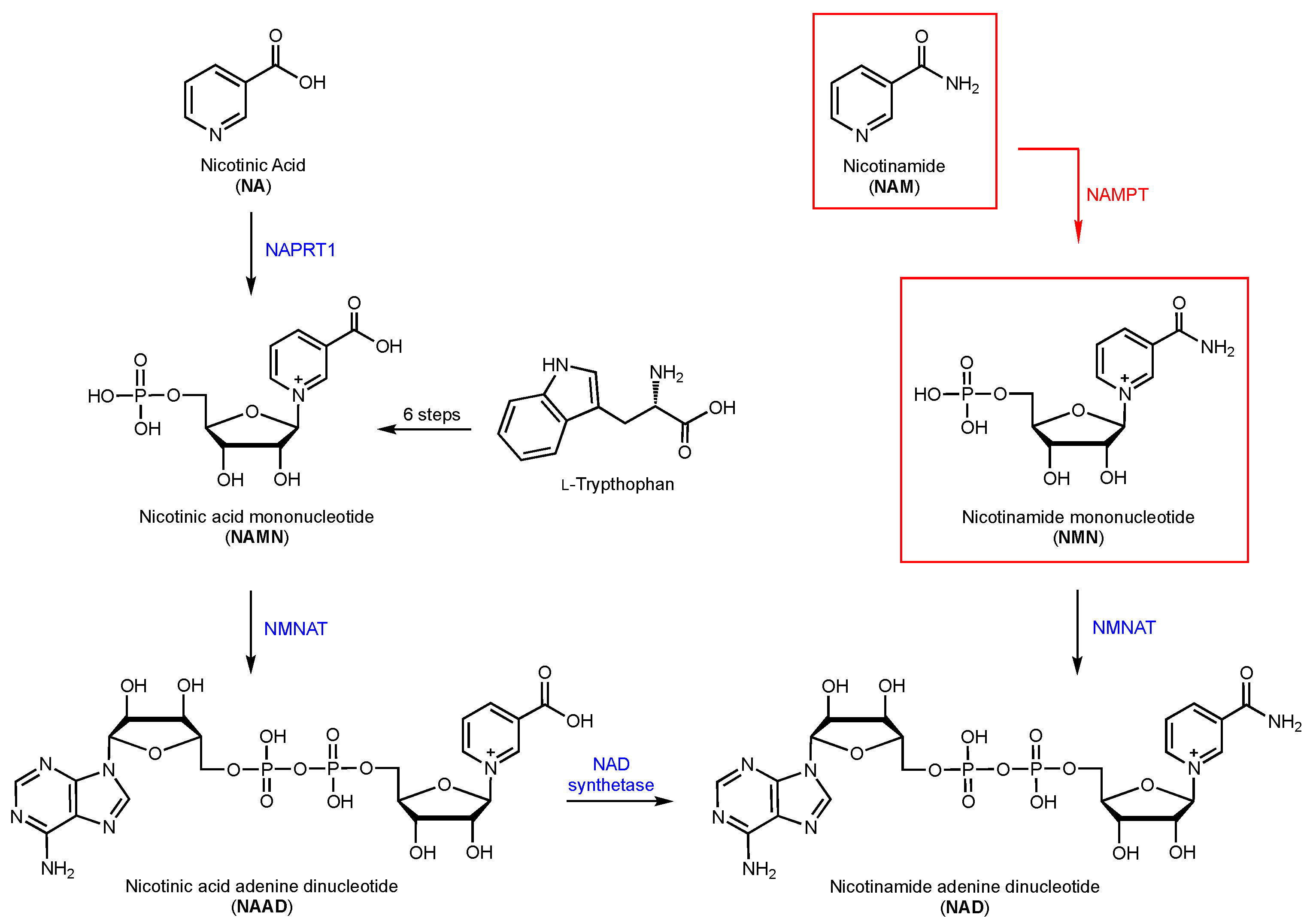
1.1. The Role of NAD Co-Enzyme in Cancer Cells
1.2. The Structure of the NAMPT Enzyme
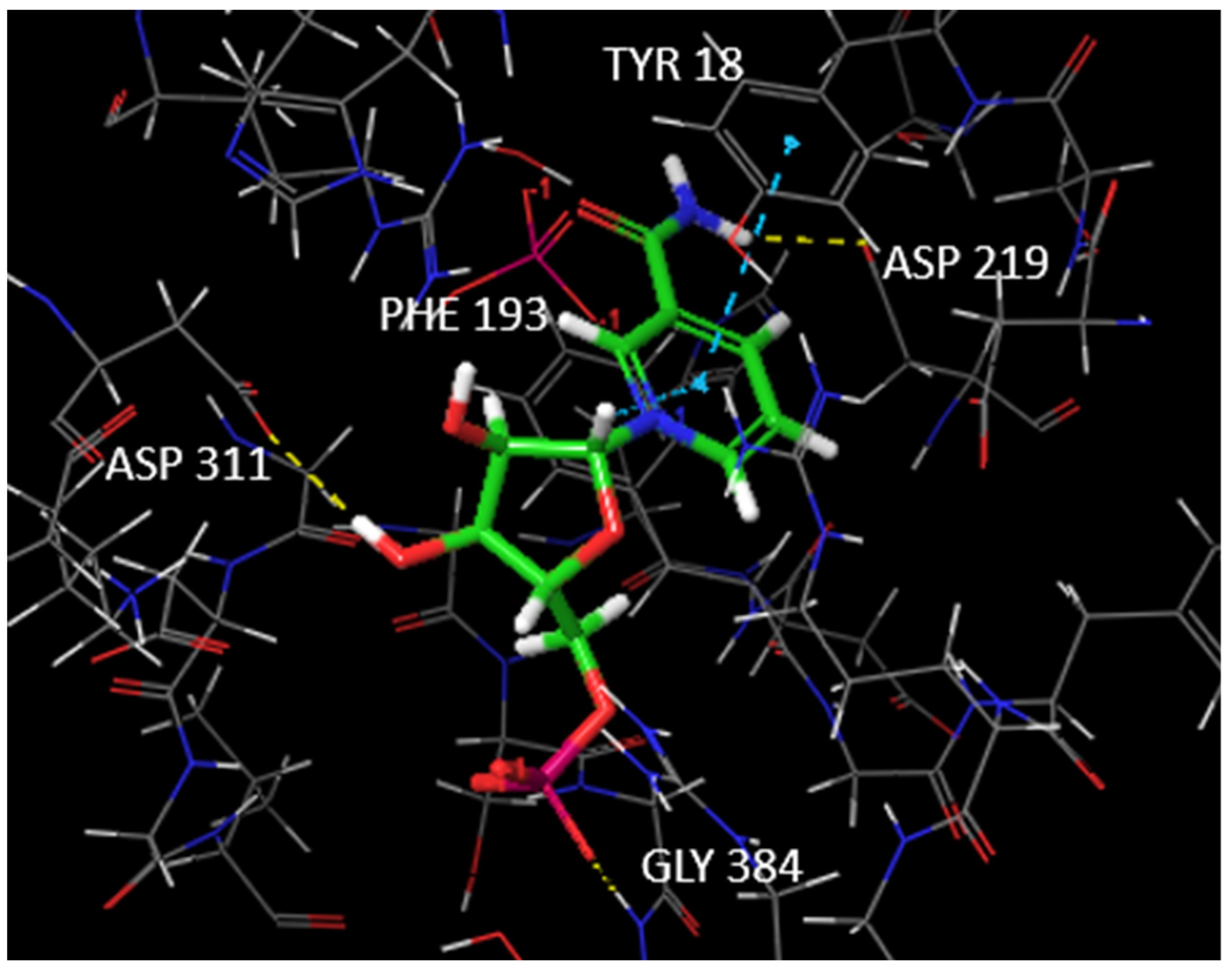
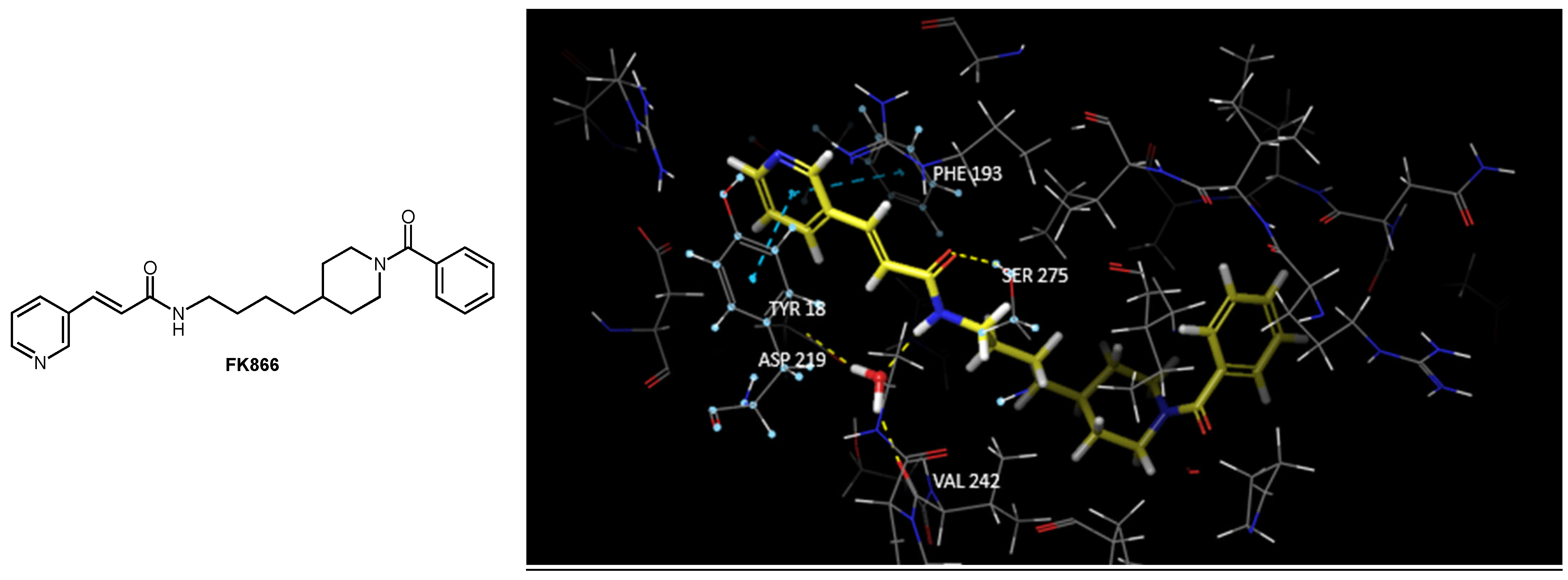
1.3. The Interactions of NAMPT with Its Inhibitors
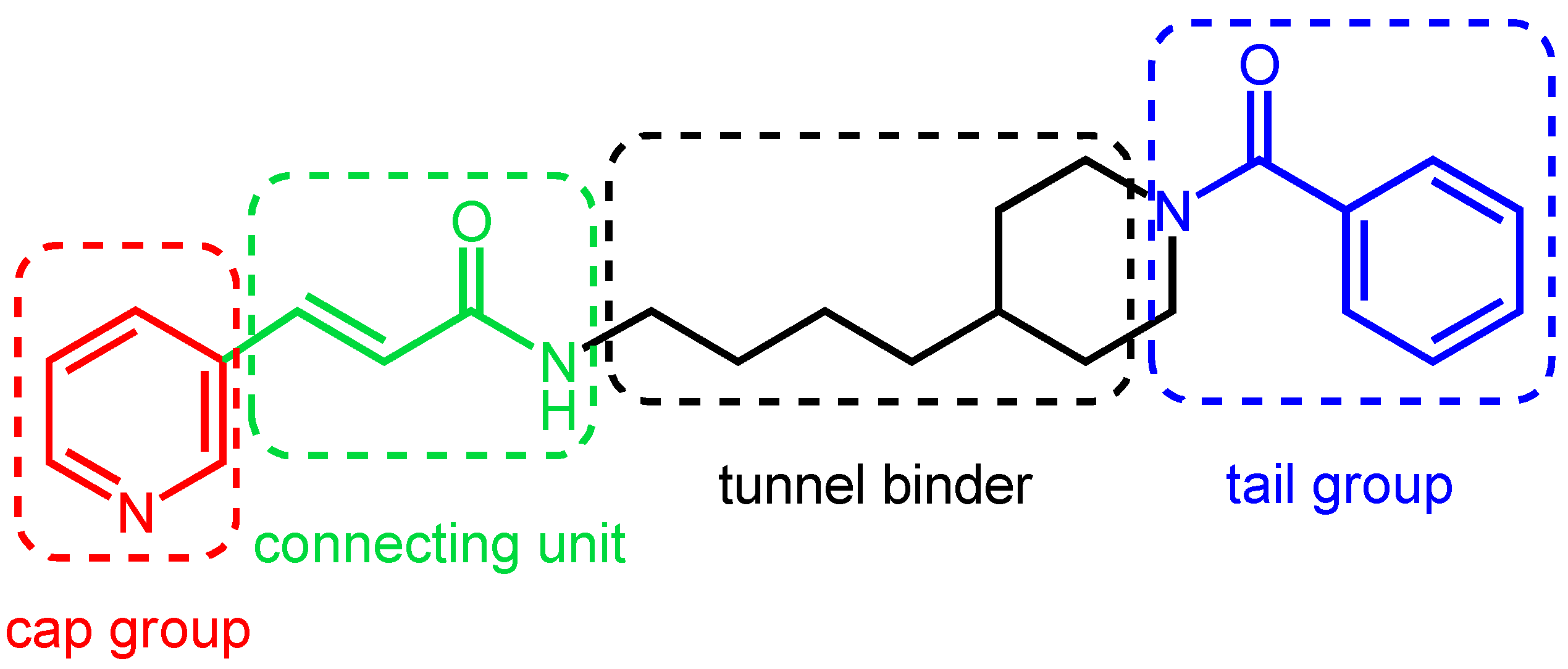
1.4. The Controversial Role of the Pyridine Moiety
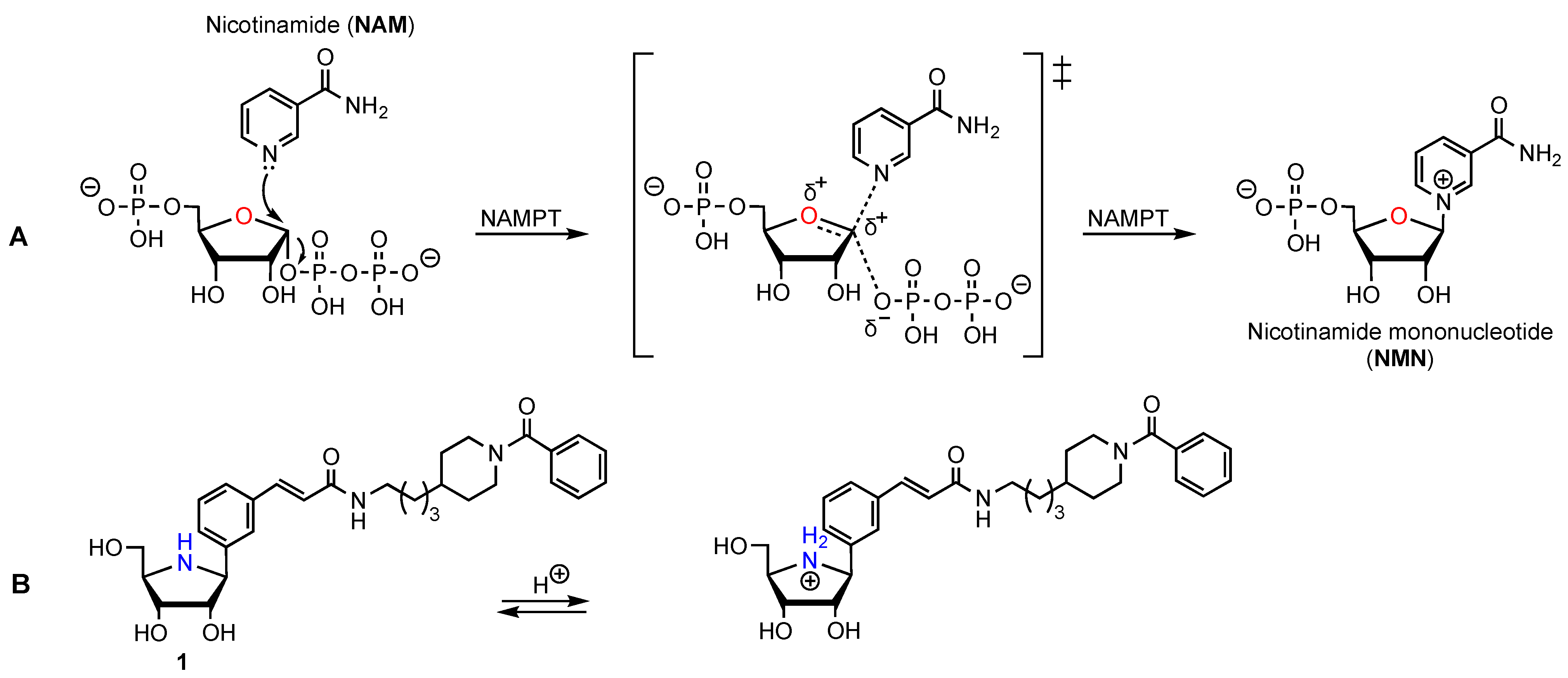
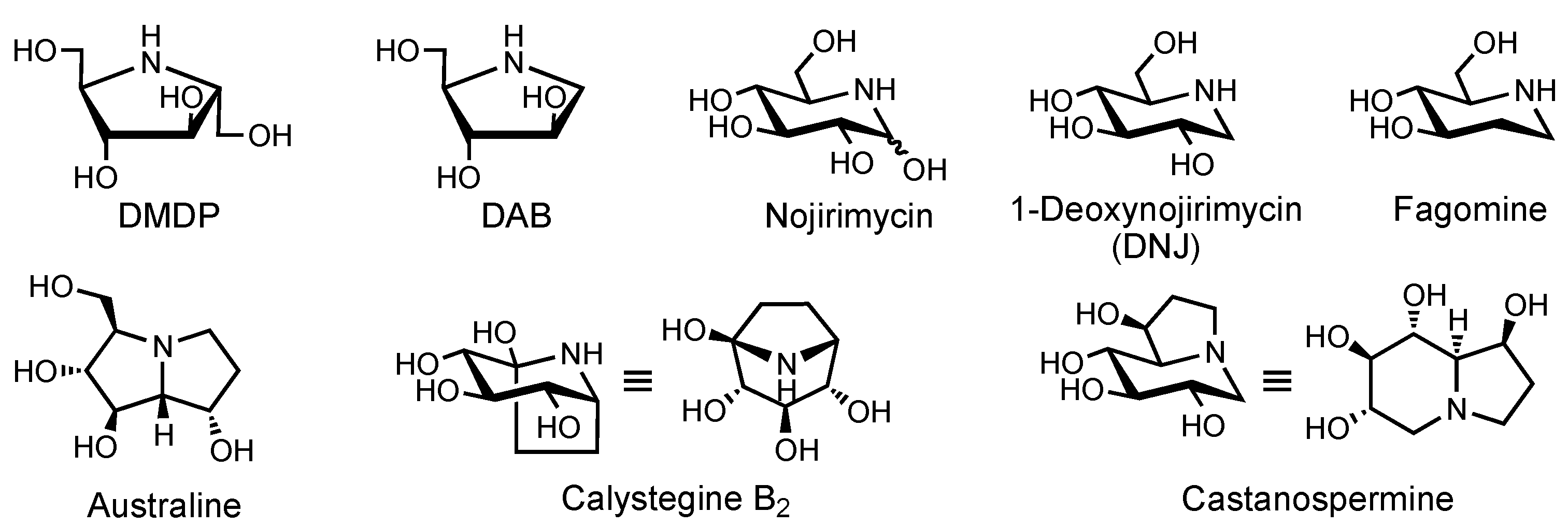
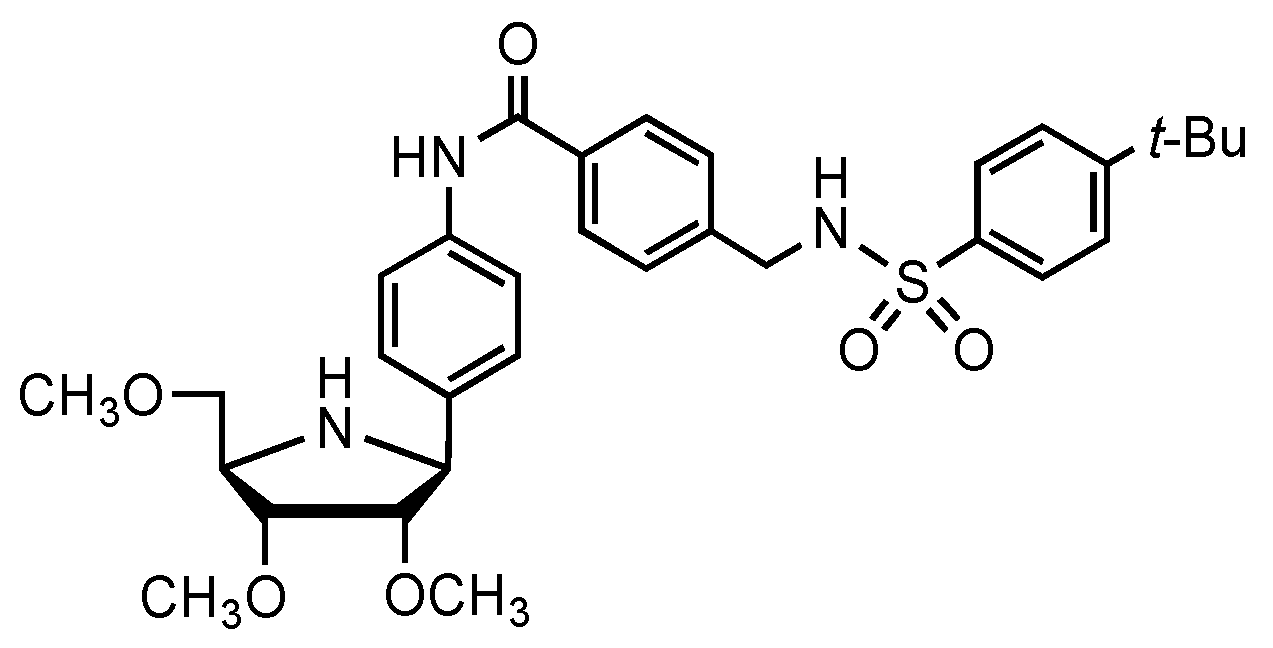
1.5. Mimics of the NAMPT Transition State as New Inhibitors
2. Materials and Method
3. Results and Discussion
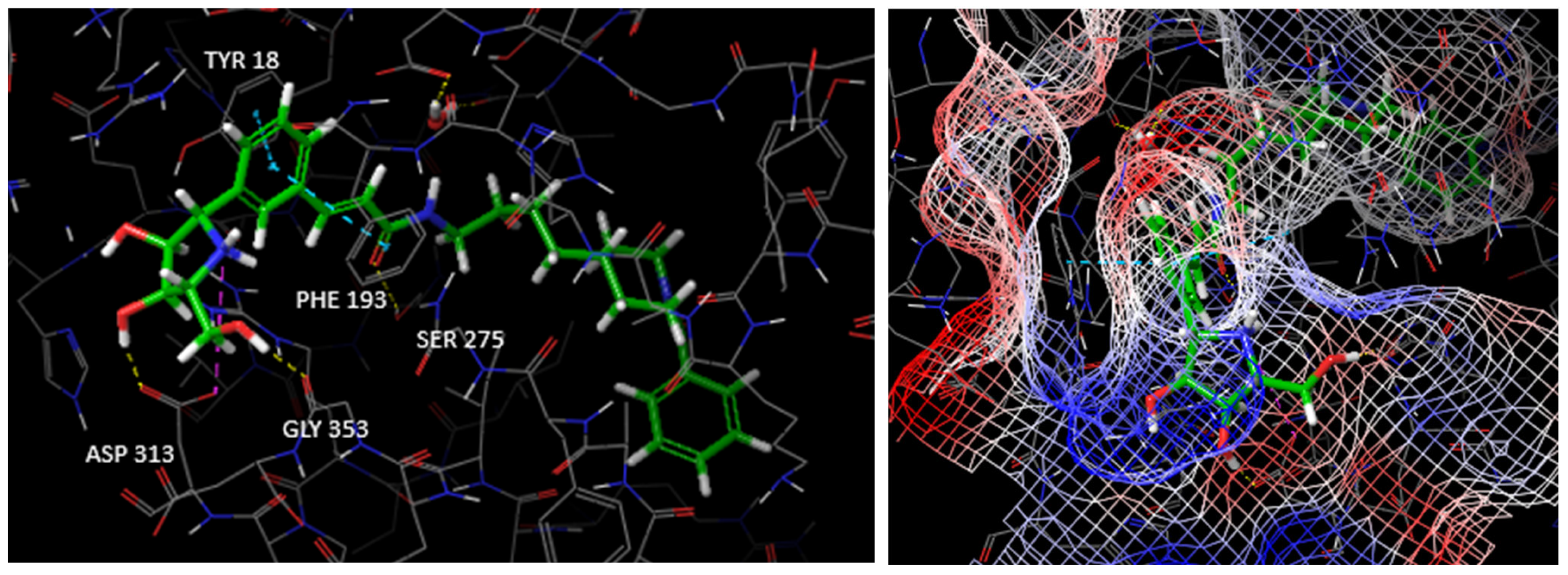
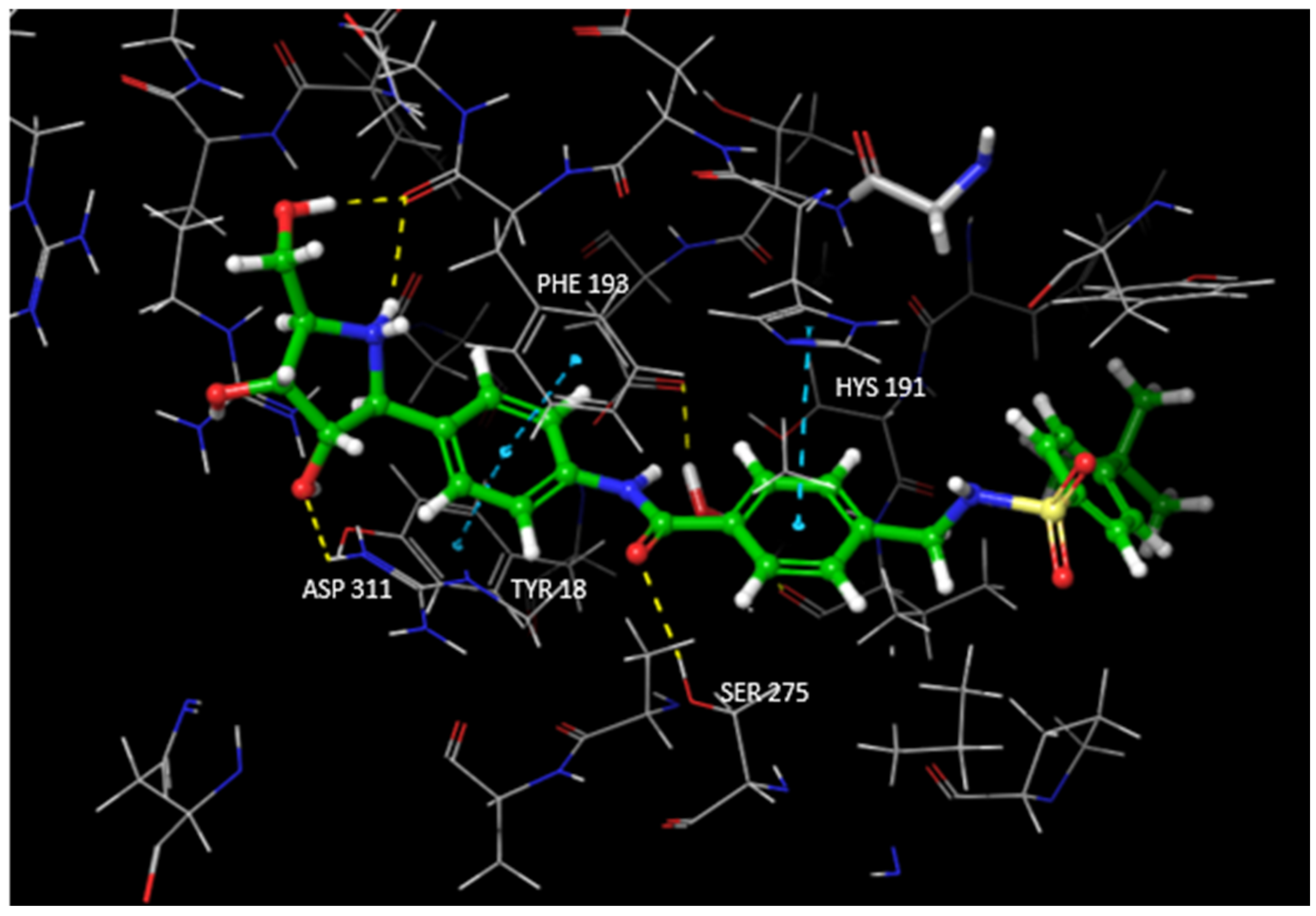
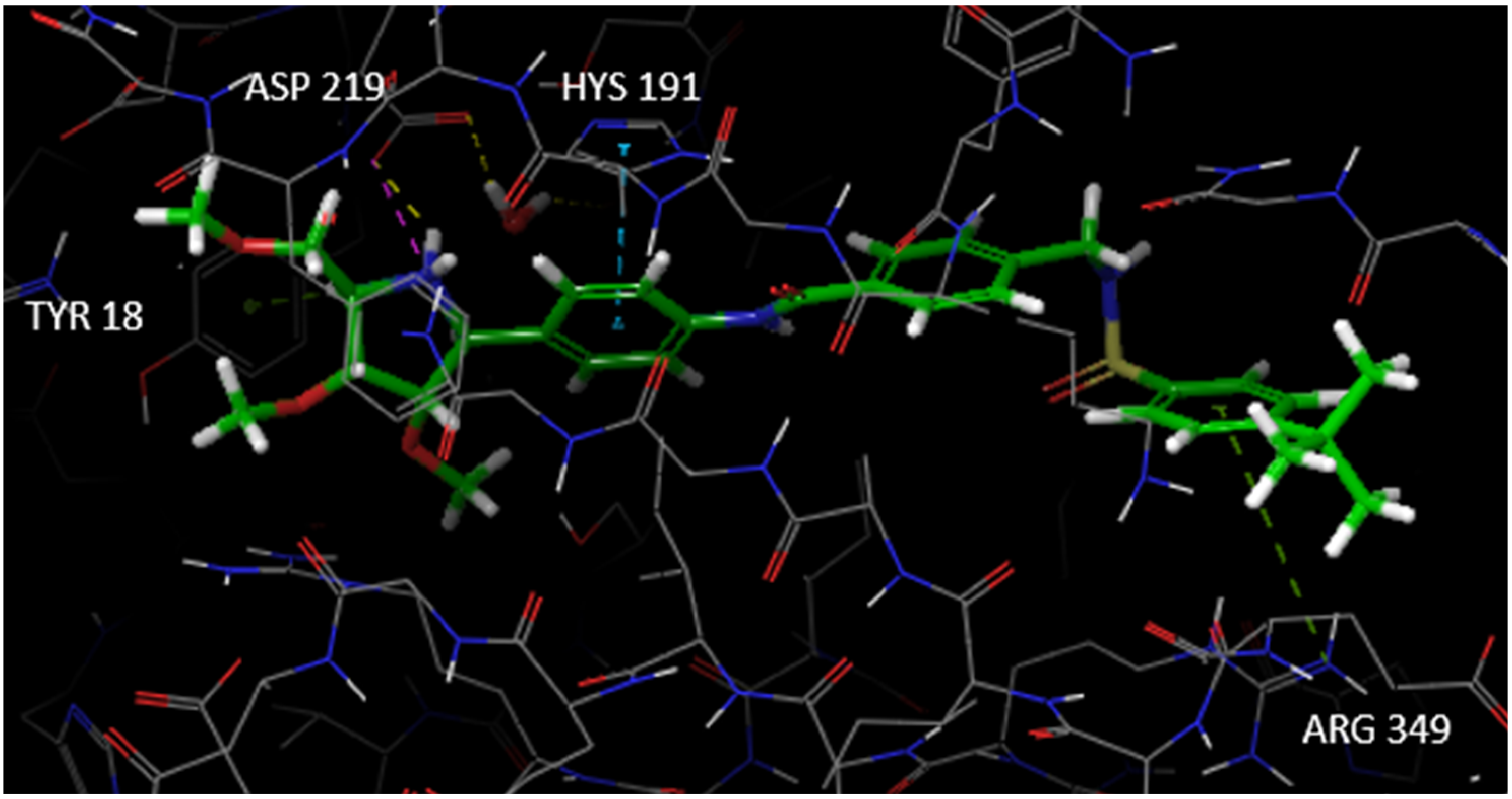
3.1. Docking Studies

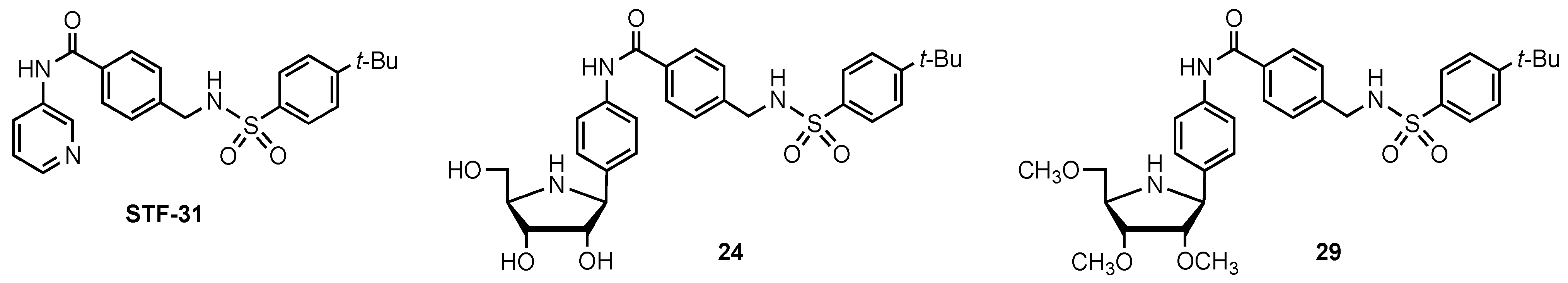
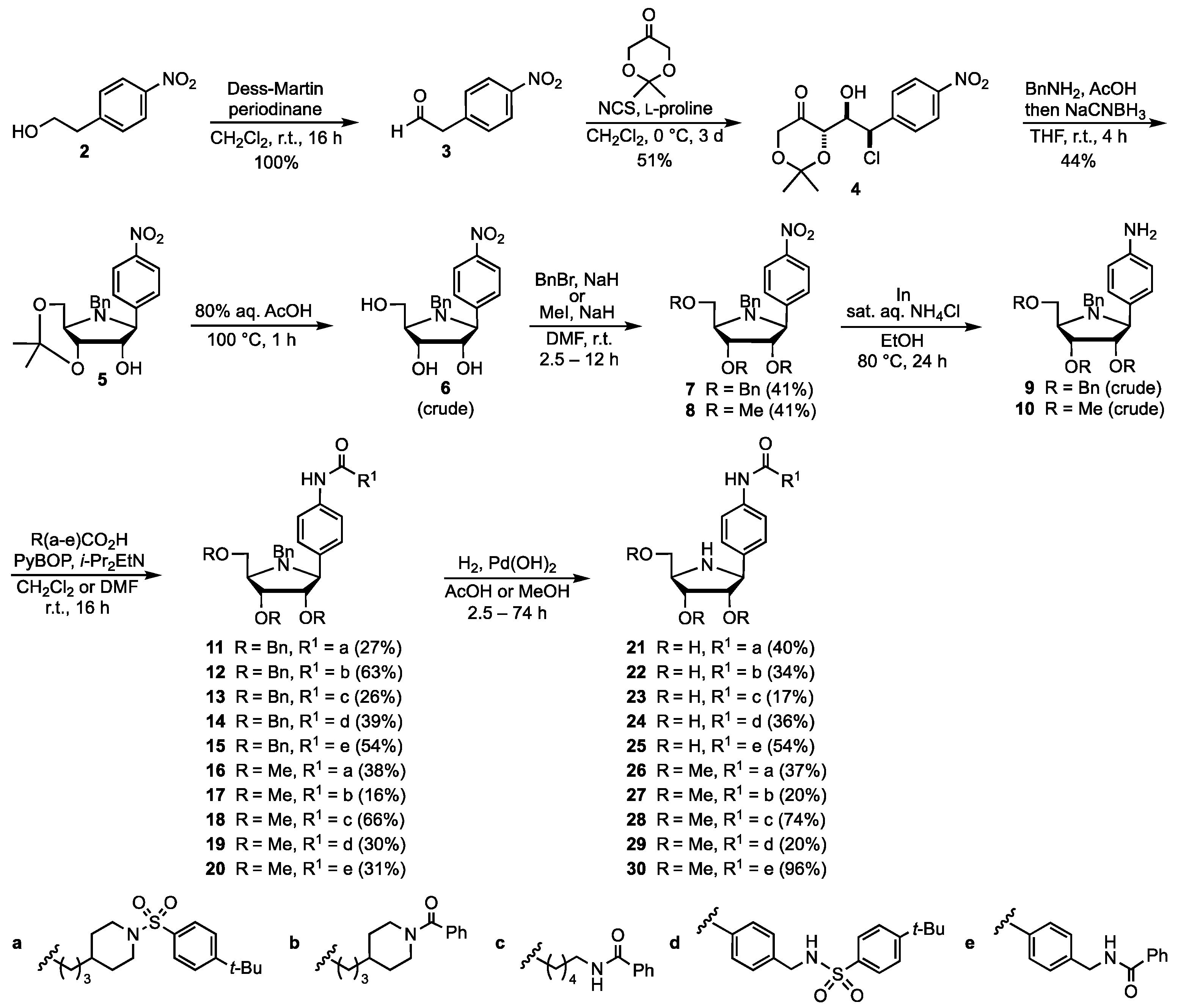
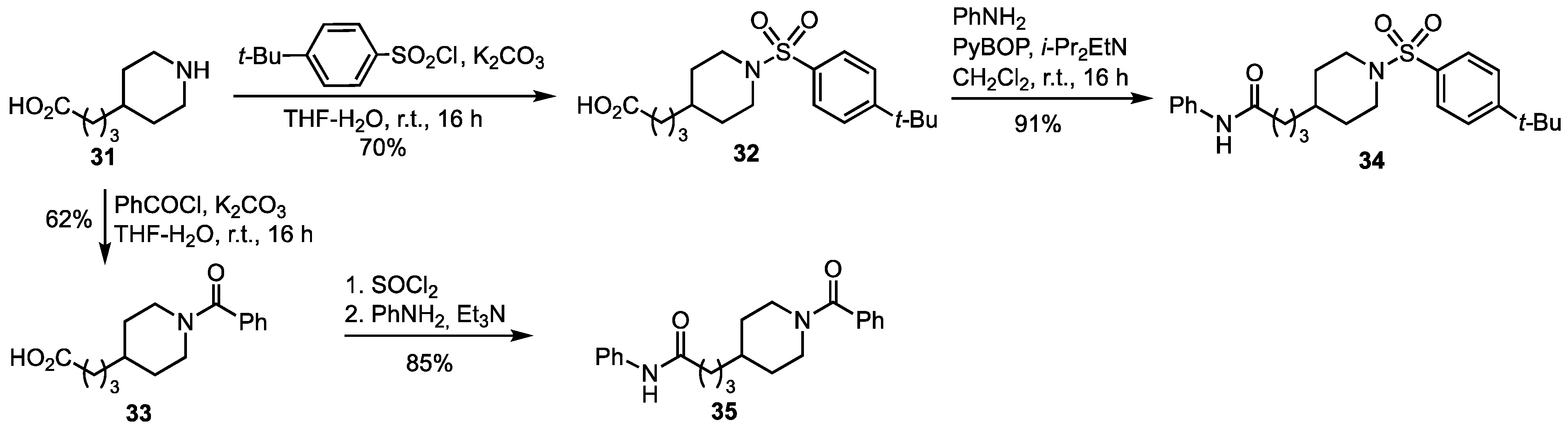
3.2. Synthesis of the Iminosugar Derivatives

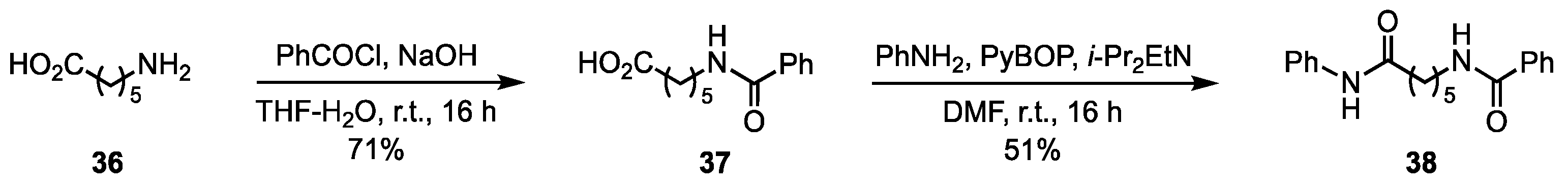
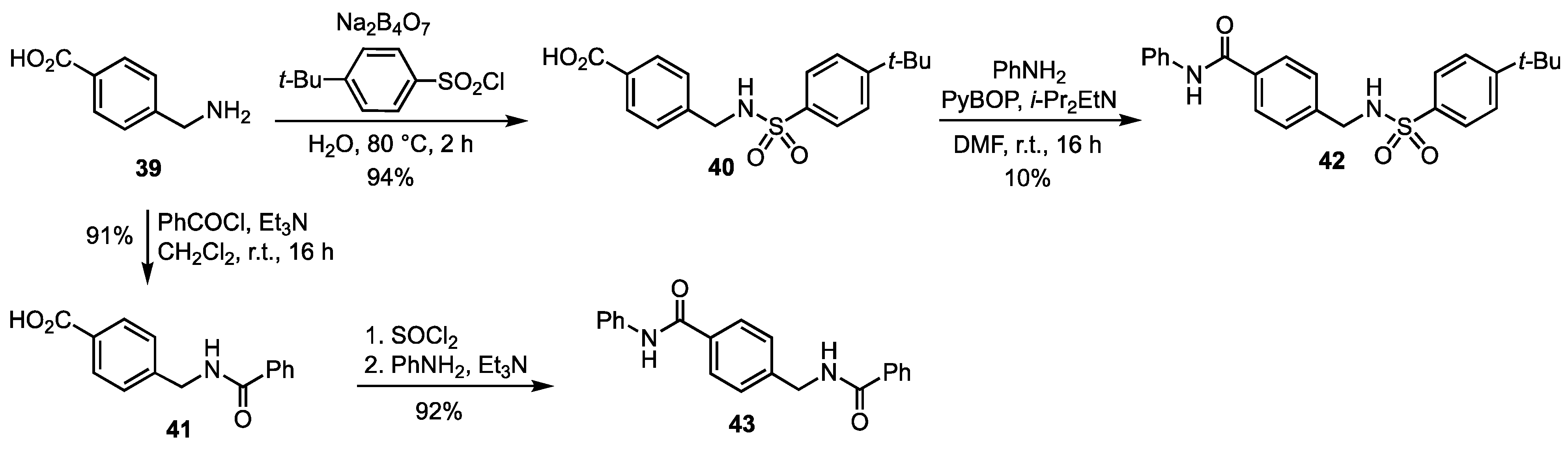
3.3. Synthesis of the Non Iminosugar-Based Inhibitors



3.4. Biological Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolla, A.A.; Travelli, C.; Genazzani, A.A.; Sethi, J.K. Extracellular nicotinamide phosphoribosyltransferase, a new cancer metabokine. Br. J. Pharmacol. 2016, 173, 2182–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Burgos, E.S.; Ho, M.-C.; Almo, S.C.; Schramm, V.L. A phosphoenzyme mimic, overlapping catalytic sites and reaction coordinate motion for human NAMPT. Proc. Natl. Acad. Sci. USA 2009, 106, 13748–13753. [Google Scholar] [CrossRef] [Green Version]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Galli, U.; Colombo, G.; Travelli, C.; Tron, G.C.; Genazzani, A.A.; Grolla, A.A. Recent Advances in NAMPT Inhibitors: A Novel Immunotherapic Strategy. Front. Pharmacol. 2020, 11, 656. [Google Scholar] [CrossRef]
- Oh, A.; Ho, Y.-C.; Zak, M.; Liu, Y.; Chen, X.; Yuen, P.; Zheng, X.; Liu, Y.; Dragovich, P.S.; Wang, W. Structural and Biochemical Analyses of the Catalysis and Potency Impact of Inhibitor Phosphoribosylation by Human Nicotinamide Phosphoribosyltransferase. ChemBioChem 2014, 15, 1121–1130. [Google Scholar] [CrossRef]
- Wilsbacher, J.L.; Cheng, M.; Cheng, D.; Trammell, S.A.J.; Shi, Y.; Guo, J.; Koeniger, S.L.; Kovar, P.J.; He, Y.; Selvaraju, S.; et al. Discovery and Characterization of Novel Nonsubstrate and Substrate NAMPT Inhibitors. Mol. Cancer Ther. 2017, 16, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Travelli, C.; Aprile, S.; Rahimian, R.; Grolla, A.A.; Rogati, F.; Bertolotti, M.; Malagnino, F.; di Paola, R.; Impellizzeri, D.; Fusco, R.; et al. Identification of Novel Triazole-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors Endowed with Antiproliferative and Antiinflammatory Activity. J. Med. Chem. 2017, 60, 1768–1792. [Google Scholar] [CrossRef]
- Zak, M.; Yuen, P.; Liu, X.; Patel, S.; Sampath, D.; Oeh, J.; Liederer, B.M.; Wang, W.; O’Brien, T.; Xiao, Y.; et al. Minimizing CYP2C9 Inhibition of Exposed-Pyridine NAMPT (Nicotinamide Phosphoribosyltransferase) Inhibitors. J. Med. Chem. 2016, 59, 8345–8368. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xiang, H.; Zhang, W. Review of various NAMPT inhibitors for the treatment of cancer. Front. Pharmacol. 2022, 13, 970553. [Google Scholar] [CrossRef] [PubMed]
- Gillig, A.; Majjigapu, S.R.; Sordat, B.; Vogel, P. Synthesis of a C-Iminoribofuranoside Analog of the Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitor FK866. Helv. Chim. Acta 2012, 95, 34–42. [Google Scholar] [CrossRef]
- Benzi, A.; Sturla, L.; Heine, M.; Fischer, A.W.; Spinelli, S.; Magnone, M.; Sociali, G.; Parodi, A.; Fenoglio, D.; Emionite, L.; et al. CD38 downregulation modulates NAD+ and NADP(H) levels in thermogenic adipose tissues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158819. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Nash, R.J.; Molyneux, R.J.; Fleet, G.W.J. Sugar-mimic glycosidase inhibitors: Natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Asymmetry 2000, 11, 1645–1680. [Google Scholar] [CrossRef]
- Winchester, B.G. Iminosugars: From botanical curiosities to licensed drugs. Tetrahedron Asymmetry 2009, 20, 645–651. [Google Scholar] [CrossRef]
- Compain, P.; Chagnault, V.; Martin, O.R. Tactics and strategies for the synthesis of iminosugar C-glycosides: A review. Tetrahedron Asymmetry 2009, 20, 672–711. [Google Scholar] [CrossRef]
- D’Alonzo, D.; Guaragna, A.; Palumbo, G. Glycomimetics at the mirror: Medicinal chemistry of l-iminosugars. Curr. Med. Chem. 2009, 16, 473–505. [Google Scholar] [CrossRef]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef]
- Compain, P. Searching for Glycomimetics That Target Protein Misfolding in Rare Diseases: Successes, Failures, and Unexpected Progress Made in Organic Synthesis. Synlett 2014, 25, 1215–1240. [Google Scholar] [CrossRef]
- Cipolla, L.; La Ferla, B.; Gregori, M. Combinatorial Approaches to Iminosugars as Glycosidase and Glycosyltransferase Inhibitors. Comb. Chem. High Throughput Screen. 2006, 9, 571–582. [Google Scholar] [PubMed]
- Nishimura, Y. Gem-diamine 1-N-iminosugars as versatile glycomimetics: Synthesis, biological activity and therapeutic potential. J. Antibiot. 2009, 62, 407–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compain, P.; Martin, O.R. Carbohydrate mimetics-based glycosyltransferase inhibitors. Bioorg. Med. Chem. 2001, 9, 3077–3092. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P. Monosaccharide and Disaccharide Mimics: New Molecular Tools for Biology and Medicine. Chimia 2001, 55, 359–365. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R. Design, Synthesis and Biological Evaluation of Iminosugar-Based Glycosyltransferase Inhibitors. Curr. Top. Med. Chem. 2003, 3, 541–560. [Google Scholar] [CrossRef]
- Zou, W. C-Glycosides and Aza-C-Glycosides as Potential Glycosidase and Glycosyltransferase Inhibitors. Curr. Top. Med. Chem. 2005, 5, 1363–1391. [Google Scholar] [CrossRef]
- Nicolas, C.; Martin, O.R. Glycoside Mimics from Glycosylamines: Recent Progress. Molecules 2018, 23, 1612. [Google Scholar] [CrossRef] [Green Version]
- Conforti, I.; Marra, A. Iminosugars as glycosyltransferase inhibitors. Org. Biomol. Chem. 2021, 19, 5439–5475. [Google Scholar] [CrossRef]
- Horenstein, B.A.; Zabinski, R.F.; Schramm, V.L. A New Class of C-Nucleoside Analogues. 1-(S)-aryl-1,4-dideoxy-1,4-imino-d-ribitols, Transition State Analogue Inhibitors of Nucleoside Hydrolase. Tetrahedron Lett. 1993, 34, 7213–7216. [Google Scholar] [CrossRef]
- Bergeron-Brlek, M.; Meanwell, M.; Britton, R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 2015, 6, 6903. [Google Scholar] [CrossRef] [Green Version]
- Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jørgensen, K.A. Direct Organocatalytic Asymmetric α-Chlorination of Aldehydes. J. Am. Chem. Soc. 2004, 126, 4790–4791. [Google Scholar] [CrossRef] [PubMed]
- Bergeron-Brlek, M.; Teoh, T.; Britton, R. A tandem organocatalytic α-chlorination-aldol reaction that proceeds with dynamic kinetic resolution: A powerful tool for carbohydrate synthesis. Org. Lett. 2013, 15, 3554–3557. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.J.; Pitts, M.R. Indium as Reducing Agent: Reduction of Aromatic Nitro Groups. Synlett 1998, 1998, 1028. [Google Scholar] [CrossRef]
- Andrianov, V.; Gailite, V.; Lola, D.; Loza, E.; Semenikhina, V.; Kalvinsh, I.; Finn, P.; Dumong Petersen, K.; Ritchie, J.W.A.; Khan, N.; et al. Novel amide derivatives as inhibitors of histone deacetylase: Design, synthesis and SAR. Eur. J. Med. Chem. 2009, 44, 1067–1085. [Google Scholar] [CrossRef] [PubMed]
- Frankowski, K.J.; Hedrick, M.P.; Gosalia, P.; Li, K.; Shi, S.; Whipple, D.; Ghosh, P.; Prisinzano, T.E.; Schoenen, F.J.; Su, Y.; et al. Discovery of Small Molecule Kappa Opioid Receptor Agonist and Antagonist Chemotypes through a HTS and Hit Refinement Strategy. ACS Chem. Neurosci. 2012, 3, 221–236. [Google Scholar] [CrossRef]
- Schmidt, J.; Rotter, M.; Weiser, T.; Wittmann, S.; Weizel, L.; Kaiser, A.; Heering, J.; Goebel, T.; Angioni, C.; Wurglics, M.; et al. A Dual Modulator of Farnesoid X Receptor and Soluble Epoxide Hydrolase to Counter Nonalcoholic Steatohepatisis. J. Med. Chem. 2017, 60, 7703–7724. [Google Scholar] [CrossRef]
- Zhang, R.-Y.; Qin, Y.; Lv, X.-Q.; Wang, P.; Xu, T.-Y.; Zhang, L.; Miao, C.-Y. A fluorometric assay for high-throughput screening targeting nicotinamide phosphoribosyltransferase. Anal. Biochem. 2011, 412, 18–25. [Google Scholar]
- Bai, J.-F.; Majjigapu, S.R.; Sordat, B.; Poty, S.; Vogel, P.; Elías-Rodríguez, P.; Moreno-Vargas, A.J.; Carmona, A.T.; Caffa, I.; Ghanem, M.; et al. Identification of new FK866 analogues with potent anticancer activity against pancreatic cancer. Eur. J. Med. Chem. 2022, 239, 114504. [Google Scholar] [CrossRef]
- Grau, B.W.; Bönisch, S.; Neuhauser, A.; Hampel, F.; Görling, A.; Tsogoeva, S.B. Facile Access to Challenging ortho-Terphenyls via Merging Two Multi-Step Domino Reactions in One-Pot: A Joint Experimental/Theoretical Study. ChemCatChem 2019, 11, 3982–3992. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | NAMPT Inhibition (IC50, μM) | MiaPaCa-2 Cells Viability (IC50, μM) | iNAD+ Depletion (%) |
|---|---|---|---|---|
| 1 | FK866 | 0.0033 | 0.0024 | 90 |
| 2 | 21 | NI 1 | NI 2 | 0 |
| 3 | 26 | 138.9 | 37 | 10 |
| 4 | 34 | 357 | 46.6 | 6 |
| 5 | 22 | 99.5 | 108 | 77 |
| 6 | 27 | NI 1 | 539 | 0 |
| 7 | 35 | NI 1 | 91.4 | 4 |
| 8 | 23 | 363 | NI 3 | 5 |
| 9 | 28 | 603 | NI 3 | 3 |
| 10 | 38 | 113 | 340 | 0 |
| 11 | 24 | 549 | NI 3 | 9 |
| 12 | 29 | 63 | 32 | 79 |
| 13 | 42 | 43 | NI 3 | 61 |
| 14 | 25 | NI 1 | NI 3 | 0 |
| 15 | 30 | NI 1 | 920 | 0 |
| 16 | 43 | 283 | 698 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conforti, I.; Benzi, A.; Caffa, I.; Bruzzone, S.; Nencioni, A.; Marra, A. Iminosugar-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors as Potential Anti-Pancreatic Cancer Agents. Pharmaceutics 2023, 15, 1472. https://doi.org/10.3390/pharmaceutics15051472
Conforti I, Benzi A, Caffa I, Bruzzone S, Nencioni A, Marra A. Iminosugar-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors as Potential Anti-Pancreatic Cancer Agents. Pharmaceutics. 2023; 15(5):1472. https://doi.org/10.3390/pharmaceutics15051472
Chicago/Turabian StyleConforti, Irene, Andrea Benzi, Irene Caffa, Santina Bruzzone, Alessio Nencioni, and Alberto Marra. 2023. "Iminosugar-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors as Potential Anti-Pancreatic Cancer Agents" Pharmaceutics 15, no. 5: 1472. https://doi.org/10.3390/pharmaceutics15051472
APA StyleConforti, I., Benzi, A., Caffa, I., Bruzzone, S., Nencioni, A., & Marra, A. (2023). Iminosugar-Based Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors as Potential Anti-Pancreatic Cancer Agents. Pharmaceutics, 15(5), 1472. https://doi.org/10.3390/pharmaceutics15051472