

Isolation and Cultivation of Porcine Endothelial Cells, Pericytes and Astrocytes to Develop an In Vitro Blood–Brain Barrier Model for Drug Permeation Testing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
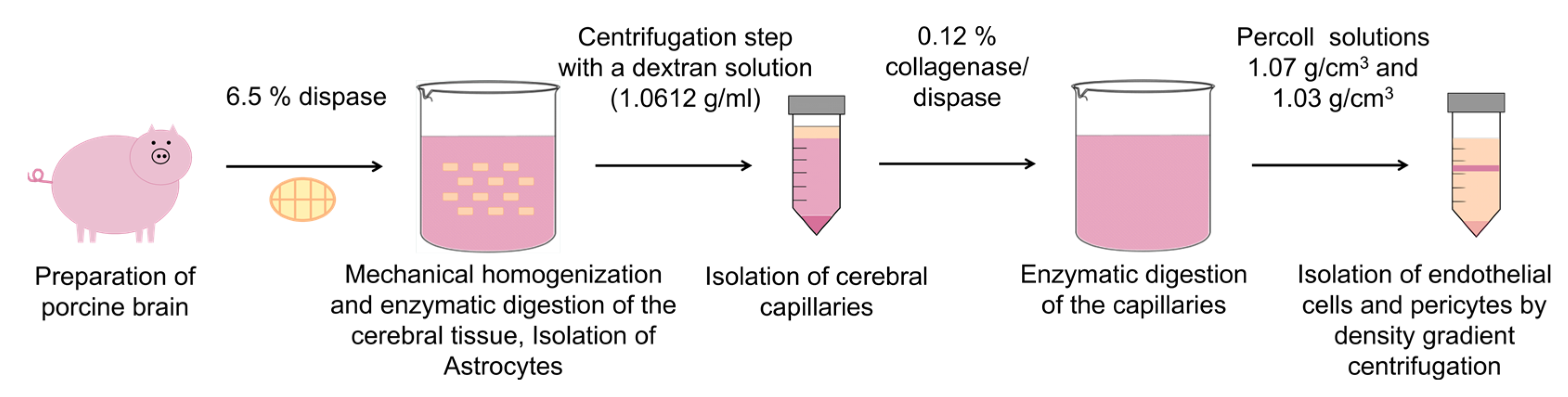
2.2. Cell Isolation
2.2.1. Endothelial Cells
2.2.2. Pericytes
2.2.3. Astrocytes
2.3. Construction of the BBB Model
2.4. Immunostaining
2.5. Transendothelial Electrical Resistance Measurements
2.6. Permeation Studies
2.7. Statistics
3. Results
3.1. Characterization of the Isolated Cells
3.2. Purity of Isolated Cells
3.3. Static Primary Coculture Model
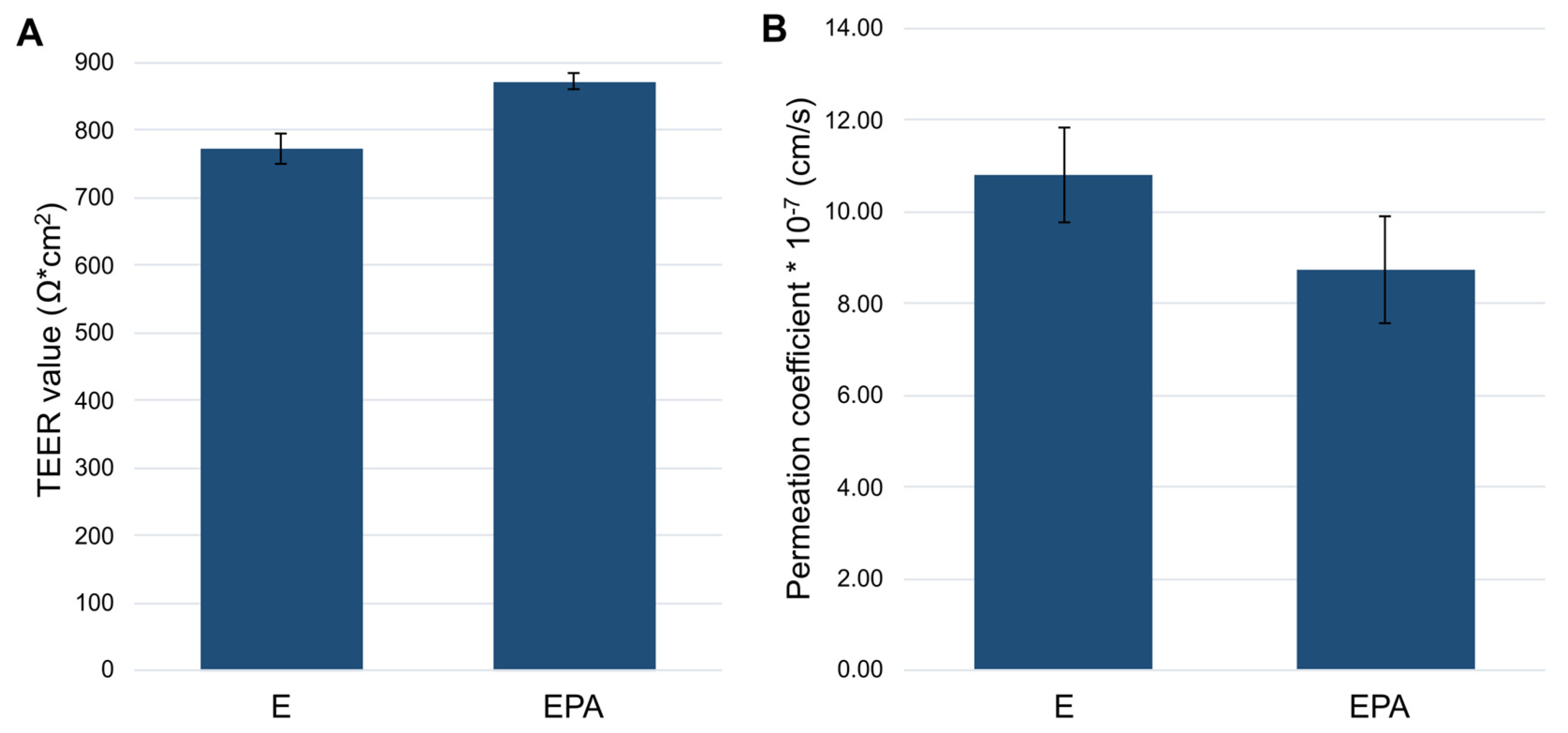
3.3.1. Influence of the Seeding Orientation of the Triple Coculture on the Barrier Integrity
3.3.2. Permeation Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Hughes, C.C.; Revest, P.A.; Greenwood, J. Development and characterisation of a rat brain capillary endothelial culture: Towards an in vitro blood-brain barrier. J. Cell Sci. 1992, 103 Pt 1, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Bowman, P.D.; Betz, A.L.; Ar, D.; Wolinsky, J.S.; Penney, J.B.; Shivers, R.R.; Goldstein, G.W. Primary culture of capillary endothelium from rat brain. In Vitro 1981, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Rutten, M.J.; Hoover, R.L.; Karnovsky, M.J. Electrical resistance and macromolecular permeability of brain endothelial monolayer cultures. Brain Res. 1987, 425, 301–310. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Yao, Y.; Tsirka, S.E.; Cao, Y. Cell-culture models of the blood-brain barrier. Stroke 2014, 45, 2514–2526. [Google Scholar] [CrossRef] [Green Version]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.A.; Shusta, E.V.; et al. In vitro models of the blood-brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- Haseloff, R.F.; Blasig, I.E.; Bauer, H.C.; Bauer, H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell. Mol. Neurobiol. 2005, 25, 25–39. [Google Scholar] [CrossRef]
- Deli, M.A.; Abrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability studies on in vitro blood-brain barrier models: Physiology, pathology, and pharmacology. Cell. Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Deli, M.A.; Nakao, S.; Honda, M.; Hayashi, K.; Nakaoke, R.; Kataoka, Y.; Niwa, M. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell. Mol. Neurobiol. 2007, 27, 687–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, I.; Fazakas, C.; Krizbai, I.A. In Vitro models of the blood-brain barrier. Acta Neurobiol. Exp. 2011, 71, 113–128. [Google Scholar]
- Sivandzade, F.; Cucullo, L. In-vitro blood-brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 38, 1667–1681. [Google Scholar] [CrossRef]
- Bicker, J.; Alves, G.; Fortuna, A.; Falcão, A. Blood–brain barrier models and their relevance for a successful development of CNS drug delivery systems: A review. Eur. J. Pharm. Biopharm. 2014, 87, 409–432. [Google Scholar] [CrossRef]
- Hinkel, S.; Mattern, K.; Dietzel, A.; Reichl, S.; Müller-Goymann, C.C. Parametric investigation of static and dynamic cell culture conditions and their impact on hCMEC/D3 barrier properties. Int. J. Pharm. 2019, 566, 434–444. [Google Scholar] [CrossRef]
- Lipps, C.; Klein, F.; Wahlicht, T.; Seiffert, V.; Butueva, M.; Zauers, J.; Truschel, T.; Luckner, M.; Köster, M.; MacLeod, R.; et al. Expansion of functional personalized cells with specific transgene combinations. Nat. Commun. 2018, 9, 994. [Google Scholar] [CrossRef] [Green Version]
- Bowman, P.D.; Ennis, S.R.; Rarey, K.E.; Betz, A.L.; Goldstein, G.W. Brain microvessel endothelial cells in tissue culture: A model for study of blood-brain barrier permeability. Ann. Neurol. 1983, 14, 396–402. [Google Scholar] [CrossRef] [Green Version]
- Franke, H.; Galla, H.; Beuckmann, C.T. Primary cultures of brain microvessel endothelial cells: A valid and flexible model to study drug transport through the blood-brain barrier In Vitro. Brain Res. Protoc. 2000, 5, 248–256. [Google Scholar] [CrossRef]
- Perriere, N.; Demeuse, P.; Garcia, E.; Regina, A.; Debray, M.; Andreux, J.-P.; Couvreur, P.; Scherrmann, J.-M.; Temsamani, J.; Couraud, P.-O.; et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J. Neurochem. 2005, 93, 279–289. [Google Scholar] [CrossRef]
- Hoheisel, D.; Nitz, T.; Franke, H.; Wegener, J.; Hakvoort, A.; Tilling, T.; Galla, H.J. Hydrocortisone reinforces the blood-brain barrier properties in a serum free cell culture system. Biochem. Biophys. Res. Commun. 1998, 244, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Patabendige, A.; Skinner, R.A.; Abbott, N.J. Establishment of a simplified in vitro porcine blood-brain barrier model with high transendothelial electrical resistance. Brain Res. 2013, 1521, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, M.; Reichl, S. Development of a serum-free human cornea construct for in vitro drug absorption studies: The influence of varying cultivation parameters on barrier characteristics. Int. J. Pharm. 2011, 416, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Bobilya, D.J. Isolation and cultivation of porcine astrocytes. Methods Mol. Biol. 2012, 814, 127–135. [Google Scholar]
- Jana, M.; Jana, A.; Pal, U.; Pahan, K. A simplified method for isolating highly purified neurons, oligodendrocytes, astrocytes, and microglia from the same human fetal brain tissue. Neurochem. Res. 2007, 32, 2015–2022. [Google Scholar] [CrossRef]
- Schildge, S.; Bohrer, C.; Beck, K.; Schachtrup, C. Isolation and culture of mouse cortical astrocytes. JoVE J. Vis. Exp. 2013, 71, e50079. [Google Scholar]
- Xue, Q.; Liu, Y.; Qi, H.; Ma, Q.; Xu, L.; Chen, W.; Chen, G.; Xu, X. A novel brain neurovascular unit model with neurons, astrocytes and microvascular endothelial cells of rat. Int. J. Biol. Sci. 2013, 9, 174–189. [Google Scholar] [CrossRef]
- Broman, M.T.; Kouklis, P.; Gao, X.; Ramchandran, R.; Neamu, R.F.; Minshall, R.D.; Malik, A.B. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ. Res. 2006, 98, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Vestweber, D. VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Piehl, C.; Piontek, J.; Cording, J.; Wolburg, H.; Blasig, I.E. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell. Mol. Life Sci. 2010, 67, 2131–2140. [Google Scholar] [CrossRef]
- Balda, M.S.; Whitney, J.A.; Flores, C.; González, S.; Cereijido, M.; Matter, K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J. Cell Biol. 1996, 134, 1031–1049. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, P.J.; de Boer, A.G. Relationship between permeability status of the blood–brain barrier and in vitro permeability coefficient of a drug. Eur. J. Pharm. Sci. 2000, 12, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kashi Malina, K.; Cooper, I.; Teichberg, V.I. Closing the gap between the in-vivo and in-vitro blood-brain barrier tightness. Brain Research 2009, 1284, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Omidi, Y.; Gumbleton, M. Primary porcine brain microvascular endothelial cells: Biochemical and functional characterisation as a model for drug transport and targeting. J. Drug Target. 2007, 15, 253–268. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledwig, V.; Reichl, S. Isolation and Cultivation of Porcine Endothelial Cells, Pericytes and Astrocytes to Develop an In Vitro Blood–Brain Barrier Model for Drug Permeation Testing. Pharmaceutics 2023, 15, 1688. https://doi.org/10.3390/pharmaceutics15061688
Ledwig V, Reichl S. Isolation and Cultivation of Porcine Endothelial Cells, Pericytes and Astrocytes to Develop an In Vitro Blood–Brain Barrier Model for Drug Permeation Testing. Pharmaceutics. 2023; 15(6):1688. https://doi.org/10.3390/pharmaceutics15061688
Chicago/Turabian StyleLedwig, Verena, and Stephan Reichl. 2023. "Isolation and Cultivation of Porcine Endothelial Cells, Pericytes and Astrocytes to Develop an In Vitro Blood–Brain Barrier Model for Drug Permeation Testing" Pharmaceutics 15, no. 6: 1688. https://doi.org/10.3390/pharmaceutics15061688
APA StyleLedwig, V., & Reichl, S. (2023). Isolation and Cultivation of Porcine Endothelial Cells, Pericytes and Astrocytes to Develop an In Vitro Blood–Brain Barrier Model for Drug Permeation Testing. Pharmaceutics, 15(6), 1688. https://doi.org/10.3390/pharmaceutics15061688