
Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy



Abstract
:1. Introduction
2. Traditional Methods of pDNA Delivery
2.1. Naked DNA
2.2. Electroporation
2.3. Sonoporation
2.4. Cell-Penetrating Peptides
2.5. Liposomes
2.6. Other Nanoparticles
3. Exosomes for Plasmid Delivery
3.1. Sources of Human Exosomes
3.2. Bovine Milk as an Exosome Source
4. Advantages of Bovine Milk Exosomes for Plasmid DNA Delivery
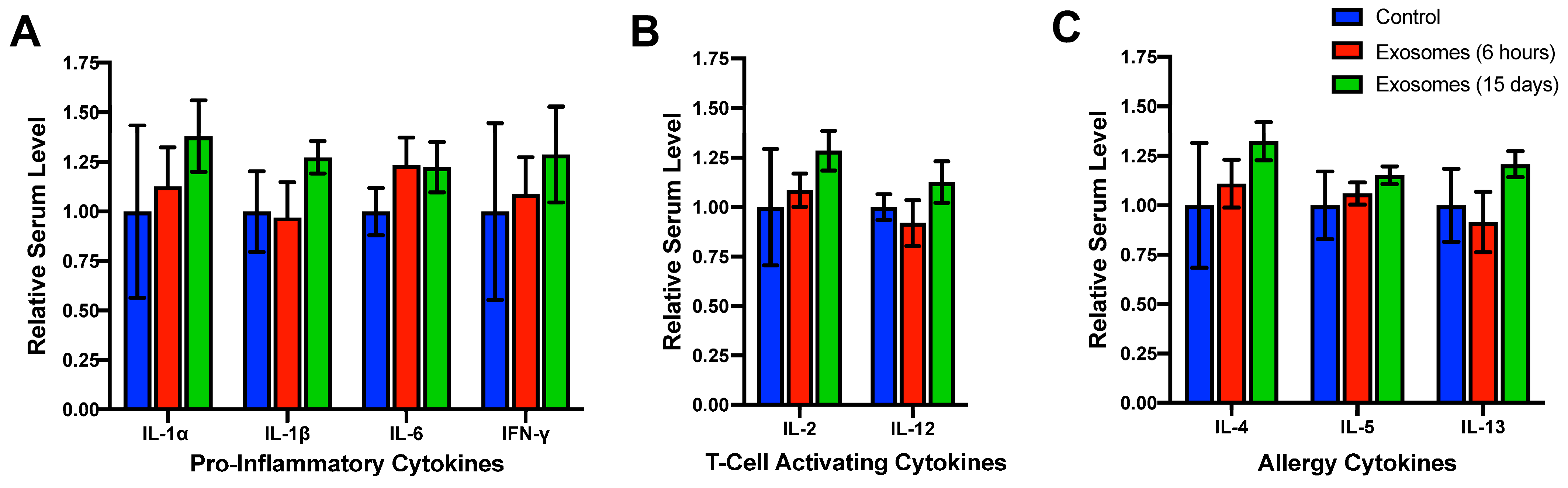
4.1. Lack of Systemic Toxicity, Immunotoxicity, and Immunogenicity
4.2. Administration by Multiple Delivery Routes
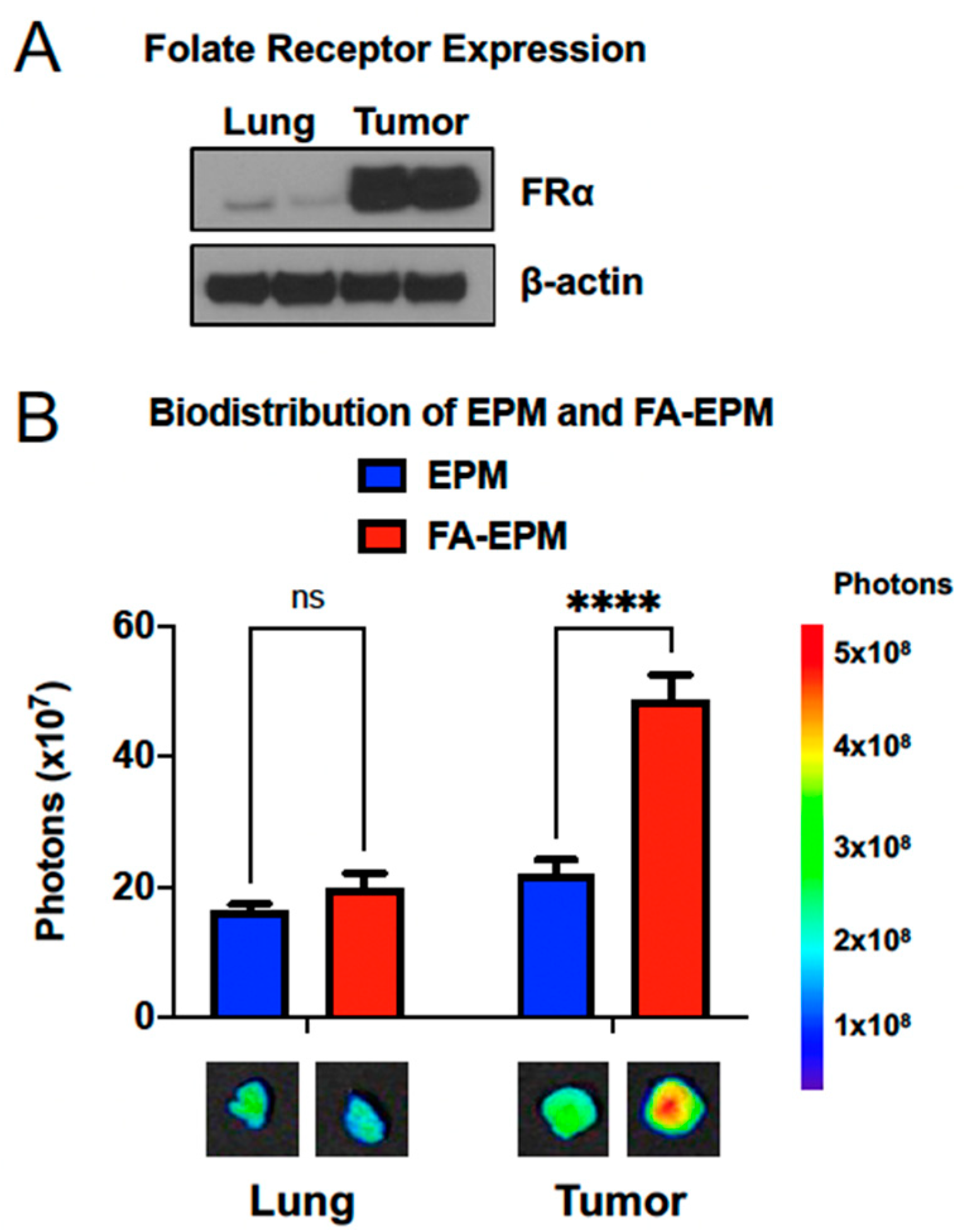
4.3. Functionalization for Targeted Delivery
5. Mechanisms of Loading Exosomes with Plasmid DNA
5.1. Electroporation
5.2. Sonication
5.3. Hybrid Exosomes
5.4. Transfection of Exosome-Producing Cells
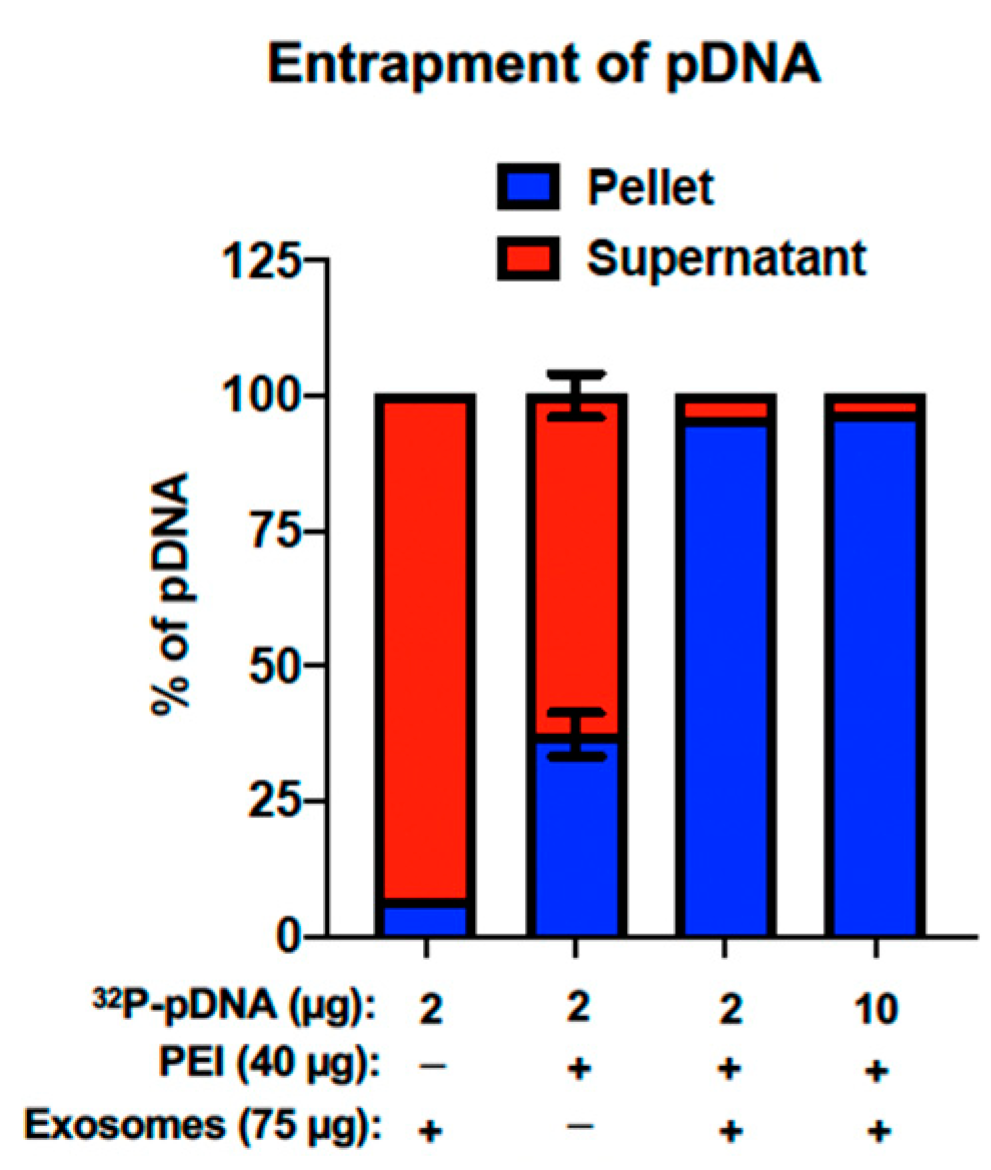
5.5. Exosome-Polyethyleneimine Matrix (EPM)
5.5.1. Entrapment of pDNA within the EPM
5.5.2. Functionalization of the EPM
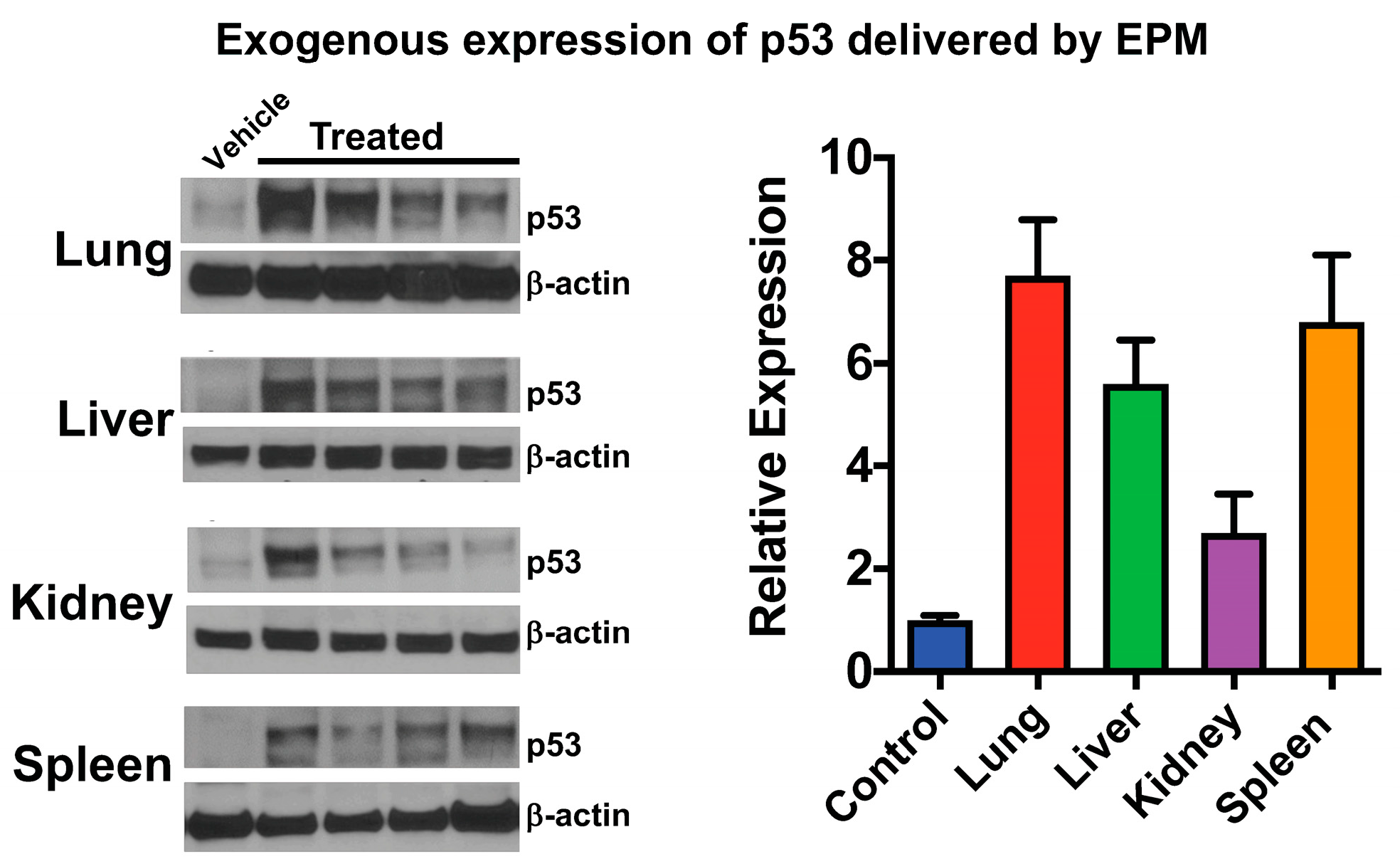
6. Therapeutic Application of pDNA via Bovine Milk/Colostrum Exosomes
7. Conclusions and Future Perspectives
8. Patents
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FDA. Long Term Follow-Up after Administration of Human Gene Therapy Products: Guidance for Industry. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy#footnote1 (accessed on 6 June 2023).
- Rubanyi, G.M. The future of human gene therapy. Mol. Asp. Med. 2001, 22, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, T. A brief history of gene therapy. Nat. Genet. 1992, 2, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Avery, O.T.; Macleod, C.M.; McCarty, M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: Induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type iii. J. Exp. Med. 1944, 79, 137–158. [Google Scholar] [CrossRef]
- Terheggen, H.G.; Lowenthal, A.; Lavinha, F.; Colombo, J.P.; Rogers, S. Unsuccessful trial of gene replacement in arginase deficiency. Eur. J. Pediatr. 1975, 119, 1–3. [Google Scholar] [CrossRef]
- Gottweis, H. Gene therapy and the public: A matter of trust. Gene Ther. 2002, 9, 667–669. [Google Scholar] [CrossRef]
- Roberts, L. Human gene transfer test approved. Science 1989, 243, 473. [Google Scholar] [CrossRef]
- Anderson, W.F. September 14, 1990: The beginning. Hum. Gene Ther. 1990, 1, 371–372. [Google Scholar] [CrossRef]
- Edelstein, M.L.; Abedi, M.R.; Wixon, J.; Edelstein, R.M. Gene Therapy Clinical Trials Worldwide 1989–2003: An Overview. Mol. Ther. 2004, 9, S375–S376. [Google Scholar] [CrossRef]
- Gene-therapy trials must proceed with caution. Nature 2016, 534, 590. [CrossRef] [Green Version]
- Somia, N.; Verma, I.M. Gene therapy: Trials and tribulations. Nat. Rev. Genet. 2000, 1, 91–99. [Google Scholar] [CrossRef]
- Verma, I.M. A Tumultuous Year for Gene Therapy. Mol. Ther. 2000, 2, 415–416. [Google Scholar] [CrossRef]
- Sibbald, B. Death but one unintended consequence of gene-therapy trial. CMAJ 2001, 164, 1612. [Google Scholar]
- Marshall, E. Gene Therapy Death Prompts Review of Adenovirus Vector. Science 1999, 286, 2244–2245. [Google Scholar] [CrossRef]
- Kaiser, J. Decades after a tragic failure, gene therapy successfully treats a rare liver disease. Science 2021. [Google Scholar] [CrossRef]
- Biotechnology, N. FDA approves hereditary blindness gene therapy. Nat. Biotechnol. 2018, 36, 6. [Google Scholar] [CrossRef]
- Darrow, J.J. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov. Today 2019, 24, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Bennett, J.; Tanabe, T.; Sun, D.; Zeng, Y.; Kjeldbye, H.; Gouras, P.; Maguire, A.M. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat. Med. 1996, 2, 649–654. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Choi, V.W.; McCarty, D.M.; Samulski, R.J. Host cell DNA repair pathways in adeno-associated viral genome processing. J. Virol. 2006, 80, 10346–10356. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008, 16, 1648–1656. [Google Scholar] [CrossRef]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial after Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Tachibana, K.; Hiraoka, K.; Aoki, M.; Yamamoto, S.; Matsumoto, K.; Nakamura, T.; Ogihara, T.; Kaneda, Y.; Morishita, R. Development of safe and efficient novel nonviral gene transfer using ultrasound: Enhancement of transfection efficiency of naked plasmid DNA in skeletal muscle. Gene Ther. 2002, 9, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardee, C.L.; Arévalo-Soliz, L.M.; Hornstein, B.D.; Zechiedrich, L. Advances in Non-Viral DNA Vectors for Gene Therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.D.; Kingston, P.A. Plasmid-mediated gene therapy for cardiovascular disease. Cardiovasc. Res. 2011, 91, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Gill, D.R.; Pringle, I.A.; Hyde, S.C. Progress and Prospects: The design and production of plasmid vectors. Gene Ther. 2009, 16, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworkin, J.P.; Lazcano, A.; Miller, S.L. The roads to and from the RNA world. J. Theor. Biol. 2003, 222, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ludtke, J.J.; Thioudellet, C.; Kleinpeter, P.; Antoniou, M.; Herweijer, H.; Braun, S.; Wolff, J.A. Intraarterial delivery of naked plasmid DNA expressing full-length mouse dystrophin in the mdx mouse model of duchenne muscular dystrophy. Hum. Gene Ther. 2004, 15, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Budker, V.; Zhang, G.; Danko, I.; Williams, P.; Wolff, J. The efficient expression of intravascularly delivered DNA in rat muscle. Gene Ther. 1998, 5, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Tada, M.; Hatano, E.; Taura, K.; Nitta, T.; Koizumi, N.; Ikai, I.; Shimahara, Y. High volume hydrodynamic injection of plasmid DNA via the hepatic artery results in a high level of gene expression in rat hepatocellular carcinoma induced by diethylnitrosamine. J. Gene Med. 2006, 8, 1018–1026. [Google Scholar] [CrossRef]
- Zhang, G.; Budker, V.; Wolff, J.A. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999, 10, 1735–1737. [Google Scholar] [CrossRef]
- Jiang, J.; Yamato, E.; Miyazaki, J.-I. Intravenous Delivery of Naked Plasmid DNA for in Vivo Cytokine Expression. Biochem. Biophys. Res. Commun. 2001, 289, 1088–1092. [Google Scholar] [CrossRef]
- Ito, T.; Fukuhara, M.; Okuda, T.; Okamoto, H. Naked pDNA/hyaluronic acid powder shows excellent long-term storage stability and gene expression in murine lungs. Int. J. Pharm. 2020, 574, 118880. [Google Scholar] [CrossRef]
- Deev, R.V.; Bozo, I.Y.; Mzhavanadze, N.D.; Voronov, D.A.; Gavrilenko, A.V.; Chervyakov, Y.V.; Staroverov, I.N.; Kalinin, R.E.; Shvalb, P.G.; Isaev, A.A. pCMV-vegf165 Intramuscular Gene Transfer is an Effective Method of Treatment for Patients with Chronic Lower Limb Ischemia. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Shahryari, A.; Saghaeian Jazi, M.; Mohammadi, S.; Razavi Nikoo, H.; Nazari, Z.; Hosseini, E.S.; Burtscher, I.; Mowla, S.J.; Lickert, H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019, 10, 868. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Huang, L. Non-Viral Vector as Vaccine Carrier. In Advances in Genetics; Academic Press: Cambridge, MA, USA, 2005; pp. 315–337. [Google Scholar]
- Al-Dosari, M.S.; Gao, X. Nonviral gene delivery: Principle, limitations, and recent progress. AAPS J. 2009, 11, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Sokołowska, E.; Błachnio-Zabielska, A.U. A Critical Review of Electroporation as A Plasmid Delivery System in Mouse Skeletal Muscle. Int. J. Mol. Sci. 2019, 20, 2776. [Google Scholar] [CrossRef] [Green Version]
- Mir, L.M.; Bureau, M.F.; Gehl, J.; Rangara, R.; Rouy, D.; Caillaud, J.-M.; Delaere, P.; Branellec, D.; Schwartz, B.; Scherman, D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. USA 1999, 96, 4262–4267. [Google Scholar] [CrossRef] [Green Version]
- Gothelf, A.; Hojman, P.; Gehl, J. Therapeutic levels of erythropoietin (EPO) achieved after gene electrotransfer to skin in mice. Gene Ther. 2010, 17, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.; Jaroszeski, M.; Atkin, A.; Moradpour, D.; Gilbert, R.; Wands, J.; Nicolau, C. In vivo gene electroinjection and expression in rat liver. FEBS Lett. 1996, 389, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujie, M.; Isaka, Y.; Nakamura, H.; Imai, E.; Hori, M. Electroporation-mediated gene transfer that targets glomeruli. J. Am. Soc. Nephrol. 2001, 12, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Dean, D.A.; Machado-Aranda, D.; Blair-Parks, K.; Yeldandi, A.V.; Young, J.L. Electroporation as a method for high-level nonviral gene transfer to the lung. Gene Ther. 2003, 10, 1608–1615. [Google Scholar] [CrossRef]
- Hojman, P.; Gissel, H.; Gehl, J. Sensitive and precise regulation of haemoglobin after gene transfer of erythropoietin to muscle tissue using electroporation. Gene Ther. 2007, 14, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Todorova, B.; Adam, L.; Culina, S.; Boisgard, R.; Martinon, F.; Cosma, A.; Ustav, M.; Kortulewski, T.; Le Grand, R.; Chapon, C. Electroporation as a vaccine delivery system and a natural adjuvant to intradermal administration of plasmid DNA in macaques. Sci. Rep. 2017, 7, 4122. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.R.F.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef]
- Morrison, C. DNA vaccines against Zika virus speed into clinical trials. Nat. Rev. Drug Discov. 2016, 15, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Tebas, P.; Kraynyak, K.A.; Patel, A.; Maslow, J.N.; Morrow, M.P.; Sylvester, A.J.; Knoblock, D.; Gillespie, E.; Amante, D.; Racine, T.; et al. Intradermal SynCon® Ebola GP DNA Vaccine Is Temperature Stable and Safely Demonstrates Cellular and Humoral Immunogenicity Advantages in Healthy Volunteers. J. Infect. Dis. 2019, 220, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Papaneri, A.B.; Johnson, R.F.; Wada, J.; Bollinger, L.; Jahrling, P.B.; Kuhn, J.H. Middle East respiratory syndrome: Obstacles and prospects for vaccine development. Expert Rev. Vaccines 2015, 14, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Riedmann, E.M. Two therapeutic HPV vaccine candidates successful in phase 1. Hum. Vaccines Immunother. 2012, 8, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newswire, P.R. INOVIO Completes Enrollment of Phase 1B Clinical Trial for its DNA Vaccine Candidate against Lassa Fever, INO-4500, in West Africa. In Health; Newswire, P., Ed.; Inovio Pharmaceuticals, Inc.: Plymouth Meeting, PA, USA, 2021. [Google Scholar]
- Vonderheide, R.H.; Kraynyak, K.A.; Shields, A.F.; McRee, A.J.; Johnson, J.M.; Sun, W.; Chintakuntlawar, A.V.; Pawlicki, J.; Sylvester, A.J.; McMullan, T. Phase 1 study of safety, tolerability and immunogenicity of the human telomerase (hTERT)-encoded DNA plasmids INO-1400 and INO-1401 with or without IL-12 DNA plasmid INO-9012 in adult patients with solid tumors. J. Immunother. Cancer 2021, 9, e003019. [Google Scholar] [CrossRef]
- Durieux, A.C.; Bonnefoy, R.; Busso, T.; Freyssenet, D. In vivo gene electrotransfer into skeletal muscle: Effects of plasmid DNA on the occurrence and extent of muscle damage. J. Gene Med. A Cross-Discip. J. Res. Sci. Gene Transf. Clin. Appl. 2004, 6, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.A.; Ford-Speelman, D.L.; Ru, L.W.; Densmore, A.L.; Roche, R.; Reed, P.W.; Bloch, R.J. Physiological and histological changes in skeletal muscle following in vivo gene transfer by electroporation. Am. J. Physiol.-Cell Physiol. 2011, 301, C1239–C1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M. Sonoporation: Gene transfer using ultrasound. World J. Methodol. 2013, 3, 39–44. [Google Scholar] [CrossRef]
- Tsunoda, S.; Mazda, O.; Oda, Y.; Iida, Y.; Akabame, S.; Kishida, T.; Shin-Ya, M.; Asada, H.; Gojo, S.; Imanishi, J.; et al. Sonoporation using microbubble BR14 promotes pDNA/siRNA transduction to murine heart. Biochem. Biophys. Res. Commun. 2005, 336, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Tachibana, K.; Kuroki, M.; Kuroki, M. Gene transfer with echo-enhanced contrast agents: Comparison between Albunex, Optison, and Levovist in mice—Initial results. Radiology 2003, 229, 423–428. [Google Scholar] [CrossRef]
- Shohet, R.V.; Chen, S.; Zhou, Y.-T.; Wang, Z.; Meidell, R.S.; Unger, R.H.; Grayburn, P.A. Echocardiographic destruction of albumin microbubbles directs gene delivery to the myocardium. Circulation 2000, 101, 2554–2556. [Google Scholar] [CrossRef] [Green Version]
- Vannan, M.; McCreery, T.; Li, P.; Han, Z.; Unger, E.; Kuersten, B.; Nabel, E.; Rajagopalan, S. Ultrasound-mediated transfection of canine myocardium by intravenous administration of cationic microbubble-linked plasmid DNA. J. Am. Soc. Echocardiogr. 2002, 15, 214–218. [Google Scholar] [CrossRef]
- Wang, W.; Li, W.; Ma, N.; Steinhoff, G. Non-viral gene delivery methods. Curr. Pharm. Biotechnol. 2013, 14, 46–60. [Google Scholar]
- Tachibana, K.; Tachibana, S. Transdermal delivery of insulin by ultrasonic vibration. J. Pharm. Pharmacol. 2011, 43, 270–271. [Google Scholar] [CrossRef]
- Panje, C.M.; Wang, D.S.; Pysz, M.A.; Paulmurugan, R.; Ren, Y.; Tranquart, F.; Tian, L.; Willmann, J.K. Ultrasound-mediated gene delivery with cationic versus neutral microbubbles: Effect of DNA and microbubble dose on in vivo transfection efficiency. Theranostics 2012, 2, 1078–1091. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Sit, W.H.; Wan, J.M.; Yu, A.C. Sonoporation induces apoptosis and cell cycle arrest in human promyelocytic leukemia cells. Ultrasound Med. Biol. 2011, 37, 2149–2159. [Google Scholar] [CrossRef]
- Nishimura, K.; Ogawa, K.; Kawaguchi, M.; Fumoto, S.; Mukai, H.; Kawakami, S. Suppression of Peritoneal Fibrosis by Sonoporation of Hepatocyte Growth Factor Gene-Encoding Plasmid DNA in Mice. Pharmaceutics 2021, 13, 115. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, G.A.; Hofmann, A.T.; Slezak, P.; Schuetzenberger, S.; Kaipel, M.; Schwartz, E.; Neef, A.; Nomikou, N.; Nau, T.; van Griensven, M.; et al. Sonoporation Increases Therapeutic Efficacy of Inducible and Constitutive BMP2/7 in Vivo Gene Delivery. Hum. Gene Ther. Methods 2014, 25, 57–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, G.; Wong, A.W.; Bez, M.; Yang, F.; Tam, S.; Even, L.; Sheyn, D.; Ben-David, S.; Tawackoli, W.; Pelled, G.; et al. Multiparameter evaluation of in vivo gene delivery using ultrasound-guided, microbubble-enhanced sonoporation. J. Control Release 2016, 223, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.E.; Zahid, M. Cell Penetrating Peptides, Novel Vectors for Gene Therapy. Pharmaceutics 2020, 12, 225. [Google Scholar] [CrossRef] [Green Version]
- Ignatovich, I.A.; Dizhe, E.B.; Pavlotskaya, A.V.; Akifiev, B.N.; Burov, S.V.; Orlov, S.V.; Perevozchikov, A.P. Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J. Biol. Chem. 2003, 278, 42625–42636. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, C.; Plank, C.; Lausier, J.; Schillinger, U.; Muller, R.H.; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J. Biol. Chem. 2003, 278, 11411–11418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehto, T.; Simonson, O.E.; Mäger, I.; Ezzat, K.; Sork, H.; Copolovici, D.-M.; Viola, J.R.; Zaghloul, E.M.; Lundin, P.; Moreno, P.M. A peptide-based vector for efficient gene transfer in vitro and in vivo. Mol. Ther. 2011, 19, 1457–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, A.M.; Trabulo, S.; Cardoso, A.L.; Maia, S.; Gomes, P.; Jurado, A.l.S.; de Lima, M.C.P. Comparison of the efficiency of complexes based on S413-PV cell-penetrating peptides in plasmid DNA and siRNA delivery. Mol. Pharm. 2013, 10, 2653–2666. [Google Scholar] [CrossRef] [Green Version]
- Alhakamy, N.A.; Ishiguro, S.; Uppalapati, D.; Berkland, C.J.; Tamura, M. AT2R Gene Delivered by Condensed Polylysine Complexes Attenuates Lewis Lung Carcinoma after Intravenous Injection or Intratracheal Spray. Mol. Cancer Ther. 2016, 15, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vij, M.; Natarajan, P.; Pattnaik, B.R.; Alam, S.; Gupta, N.; Santhiya, D.; Sharma, R.; Singh, A.; Ansari, K.M.; Gokhale, R.S. Non-invasive topical delivery of plasmid DNA to the skin using a peptide carrier. J. Control Release 2016, 222, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Mehrlatifan, S.; Mirnurollahi, S.M.; Motevalli, F.; Rahimi, P.; Soleymani, S.; Bolhassani, A. The structural HCV genes delivered by MPG cell penetrating peptide are directed to enhance immune responses in mice model. Drug Deliv. 2016, 23, 2852–2859. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Xia, X.; Teng, L.; Wang, L.; Chen, L.; Guo, X.; Belingon, B.; Li, J.; Feng, X.; Li, X.; et al. Emerging landscape of cell-penetrating peptide-mediated nucleic acid delivery and their utility in imaging, gene-editing, and RNA-sequencing. J. Control Release 2022, 341, 166–183. [Google Scholar] [CrossRef]
- Veiman, K.-L.; Künnapuu, K.; Lehto, T.; Kiisholts, K.; Pärn, K.; Langel, Ü.; Kurrikoff, K. PEG shielded MMP sensitive CPPs for efficient and tumor specific gene delivery in vivo. J. Control Release 2015, 209, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Pardridge, W.M. The Trojan Horse Liposome Technology for Nonviral Gene Transfer across the Blood-Brain Barrier. J. Drug Deliv. 2011, 2011, 296151. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lee, H.; Pardridge, W.M. Plasmid DNA gene therapy of the Niemann-Pick C1 mouse with transferrin receptor-targeted Trojan horse liposomes. Sci. Rep. 2020, 10, 13334. [Google Scholar] [CrossRef]
- Cornford, E.M.; Hyman, S.; Cornford, M.E.; Chytrova, G.; Rhee, J.; Suzuki, T.; Yamagata, T.; Yamakawa, K.; Penichet, M.L.; Pardridge, W.M. Non-invasive gene targeting to the fetal brain after intravenous administration and transplacental transfer of plasmid DNA using PEGylated immunoliposomes. J. Drug Target. 2016, 24, 58–67. [Google Scholar] [CrossRef]
- Lee, H.; Jiang, D.; Pardridge, W.M. Lyoprotectant Optimization for the Freeze-Drying of Receptor-Targeted Trojan Horse Liposomes for Plasmid DNA Delivery. Mol. Pharm. 2020, 17, 2165–2174. [Google Scholar] [CrossRef]
- Zheng, C.; Baum, B.J. Evaluation of promoters for use in tissue-specific gene delivery. Gene Ther. Protoc. 2008, 434, 205–219. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, K.; Kawakami, K.; Fujita, Y.; Kato, T.; Takai, M.; Kato, D.; Iinuma, K.; Koie, T.; Ito, M. Gene therapy of prostate cancer using liposomes containing perforin expression vector driven by the promoter of prostate-specific antigen gene. Sci. Rep. 2022, 12, 1442. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xie, H.; Liu, Y.; Xia, C.; Cun, X.; Long, Y.; Chen, X.; Deng, M.; Guo, R.; Zhang, Z.; et al. Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer. J. Control Release 2019, 304, 204–215. [Google Scholar] [CrossRef]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yao, R.; Ren, M.; Yuan, K.; Du, Y.; He, Y.; Kang, H.; Yuan, S.; Ju, W.; Qiao, J.; et al. Liposomes trigger bone marrow niche macrophage “foam” cell formation and affect hematopoiesis in mice. J. Lipid Res. 2022, 63, 100273. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Choi, E.-J.; Webster, T.J.; Kim, S.-H.; Khang, D. Effect of the protein corona on nanoparticles for modulating cytotoxicity and immunotoxicity. Int. J. Nanomed. 2015, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Lila, A.S.A.; Kiwada, H.; Ishida, T. The accelerated blood clearance (ABC) phenomenon: Clinical challenge and approaches to manage. J. Control Release 2013, 172, 38–47. [Google Scholar] [CrossRef]
- Kedmi, R.; Ben-Arie, N.; Peer, D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials 2010, 31, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Sellins, K.; Fradkin, L.; Liggitt, D.; Dow, S. Type I interferons potently suppress gene expression following gene delivery using liposome–DNA complexes. Mol. Ther. 2005, 12, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Al-Zahrani, S.S.; Bora, R.S.; Al-Garni, S.M. Antimicrobial activity of chitosan nanoparticles. Biotechnol. Biotechnol. Equip. 2021, 35, 1874–1880. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Z.; Chen, L.; Gu, W.; Li, Y. Chitosan N-betainates/DNA self-assembly nanoparticles for gene delivery: In vitro uptake and transfection efficiency. Int. J. Pharm. 2009, 371, 156–162. [Google Scholar] [CrossRef]
- Poor, E.M.; Eslaminejad, M.B.; Gheibi, N.; Bagheri, F.; Atyabi, F. Chitosan—pDNA nanoparticle characteristics determine the transfection efficacy of gene delivery to human mesenchymal stem cells. Artif. Cells Nanomed. Biotechnol. 2014, 42, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.M.; Köping-Höggård, M.; Artursson, P. Chitosan and the mucosal delivery of biotechnology drugs. Drug Discov. Today Technol. 2005, 2, 1–6. [Google Scholar] [CrossRef]
- Kumar, M.; Behera, A.K.; Lockey, R.F.; Zhang, J.; Bhullar, G.; De La Cruz, C.P.; Chen, L.-C.; Leong, K.W.; Huang, S.-K.; Mohapatra, S.S. Intranasal gene transfer by chitosan-DNA nanospheres protects BALB/c mice against acute respiratory syncytial virus infection. Hum. Gene Ther. 2002, 13, 1415–1425. [Google Scholar] [CrossRef]
- Iqbal, M.; Lin, W.; Jabbal-Gill, I.; Davis, S.; Steward, M.; Illum, L. Nasal delivery of chitosan-DNA plasmid expressing epitopes of respiratory syncytial virus (RSV) induces protective CTL responses in BALB/c mice. Vaccine 2003, 21, 1478–1485. [Google Scholar] [CrossRef]
- Sato, T.; Nakata, M.; Yang, Z.; Torizuka, Y.; Kishimoto, S.; Ishihara, M. In vitro and in vivo gene delivery using chitosan/hyaluronic acid nanoparticles: Influences of molecular mass of hyaluronic acid and lyophilization on transfection efficiency. J. Gene Med. 2017, 19, e2968. [Google Scholar] [CrossRef]
- Deng, R.-H.; Qiu, B.; Zhou, P.-H. Chitosan/hyaluronic acid/plasmid-DNA nanoparticles encoding interleukin-1 receptor antagonist attenuate inflammation in synoviocytes induced by interleukin-1 beta. J. Mater. Sci. Mater. Med. 2018, 29, 155. [Google Scholar] [CrossRef] [Green Version]
- Aldawsari, H.M.; Dhaliwal, H.K.; Aljaeid, B.M.; Alhakamy, N.A.; Banjar, Z.M.; Amiji, M.M. Optimization of the Conditions for Plasmid DNA Delivery and Transfection with Self-Assembled Hyaluronic Acid-Based Nanoparticles. Mol. Pharm. 2019, 16, 128–140. [Google Scholar] [CrossRef]
- Santos-Carballal, B.; Fernández, E.F.; Goycoolea, F.M. Chitosan in Non-Viral Gene Delivery: Role of Structure, Characterization Methods, and Insights in Cancer and Rare Diseases Therapies. Polymers 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, M.W.J.; Venkatraman, S. Recent Advances in Chitosan-Based Carriers for Gene Delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef]
- Qin, J.; Xu, Q. Functions and application of exosomes. Acta Pol. Pharm. 2014, 71, 537–543. [Google Scholar] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Luan, X.; Sansanaphongpricha, K.; Myers, I.; Chen, H.; Yuan, H.; Sun, D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol. Sin. 2017, 38, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Simeone, P.; Bologna, G.; Lanuti, P.; Pierdomenico, L.; Guagnano, M.T.; Pieragostino, D.; Del Boccio, P.; Vergara, D.; Marchisio, M.; Miscia, S. Extracellular vesicles as signaling mediators and disease biomarkers across biological barriers. Int. J. Mol. Sci. 2020, 21, 2514. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Ouyang, Y.; Wang, Z.; Zhang, R.; Huang, P.H.; Chen, C.; Li, H.; Li, P.; Quinn, D.; Dao, M.; et al. Isolation of exosomes from whole blood by integrating acoustics and microfluidics. Proc. Natl. Acad. Sci. USA 2017, 114, 10584–10589. [Google Scholar] [CrossRef] [Green Version]
- Vaswani, K.; Mitchell, M.D.; Holland, O.J.; Koh, Y.Q.; Hill, R.J.; Harb, T.; Davies, P.S.W.; Peiris, H. A Method for the Isolation of Exosomes from Human and Bovine Milk. J. Nutr. Metab. 2019, 2019, 5764740. [Google Scholar] [CrossRef] [PubMed]
- Comfort, N.; Bloomquist, T.R.; Shephard, A.P.; Petty, C.R.; Cunningham, A.; Hauptman, M.; Phipatanakul, W.; Baccarelli, A. Isolation and characterization of extracellular vesicles in saliva of children with asthma. Extracell. Vesicles Circ. Nucleic Acids 2021, 2, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yang, H.C.; Rhee, W.J. Development and comparative analysis of human urine exosome isolation strategies. Process Biochem. 2020, 88, 197–203. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Rawat, S.; Arora, V.; Kottarath, S.K.; Dinda, A.K.; Vaishnav, P.K.; Nayak, B.; Mohanty, S. An improvised one-step sucrose cushion ultracentrifugation method for exosome isolation from culture supernatants of mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Chopra, N.; Arya, B.D.; Jain, N.; Yadav, P.; Wajid, S.; Singh, S.P.; Choudhury, S. Biophysical Characterization and Drug Delivery Potential of Exosomes from Human Wharton’s Jelly-Derived Mesenchymal Stem Cells. ACS Omega 2019, 4, 13143–13152. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, M.M.; Bennit, H.R.F.; Gonda, A.; Osterman, C.J.D.; Hibma, A.; Khan, S.; Wall, N.R. Exosomes Secreted from Human Cancer Cell Lines Contain Inhibitors of Apoptosis (IAP). Cancer Microenviron. 2015, 8, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.m.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.F.; Wiegmans, A.P.; Leimgruber, A.; Möller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef]
- Xu, W.-M.; Li, A.; Chen, J.-J.; Sun, E.-J. Research Development on Exosome Separation Technology. J. Membr. Biol. 2023, 256, 25–34. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.T.; Wang, Y.K.; Tseng, Y.J. Exosomal Proteins and Lipids as Potential Biomarkers for Lung Cancer Diagnosis, Prognosis, and Treatment. Cancers 2022, 14, 732. [Google Scholar] [CrossRef] [PubMed]
- Kibria, G.; Ramos, E.K.; Lee, K.E.; Bedoyan, S.; Huang, S.; Samaeekia, R.; Athman, J.J.; Harding, C.V.; Lotvall, J.; Harris, L.; et al. A rapid, automated surface protein profiling of single circulating exosomes in human blood. Sci. Rep. 2016, 6, 36502. [Google Scholar] [CrossRef] [Green Version]
- Oldenborg, P.A. CD47: A Cell Surface Glycoprotein Which Regulates Multiple Functions of Hematopoietic Cells in Health and Disease. ISRN Hematol. 2013, 2013, 614619. [Google Scholar] [CrossRef] [Green Version]
- Wallen, M.; Aqil, F.; Spencer, W.; Gupta, R.C. Milk/colostrum exosomes: A nanoplatform advancing delivery of cancer therapeutics. Cancer Lett. 2023, 561, 216141. [Google Scholar] [CrossRef] [PubMed]
- Hammon, H.M.; Liermann, W.; Frieten, D.; Koch, C. Review: Importance of colostrum supply and milk feeding intensity on gastrointestinal and systemic development in calves. Animal 2020, 14, s133–s143. [Google Scholar] [CrossRef] [Green Version]
- Geiger, A.J. Colostrum: Back to basics with immunoglobulins. J. Anim. Sci. 2020, 98 (Suppl. 1), S126–S132. [Google Scholar] [CrossRef]
- Playford, R.J.; Weiser, M.J. Bovine Colostrum: Its Constituents and Uses. Nutrients 2021, 13, 265. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Kandimalla, R.; Wallen, M.; Tyagi, N.; Wilcher, S.; Yan, J.; Schultz, D.J.; Spencer, W.; et al. Exosome-mediated delivery of RNA and DNA for gene therapy. Cancer Lett. 2021, 505, 58–72. [Google Scholar] [CrossRef]
- Kandimalla, R.; Aqil, F.; Alhakeem, S.S.; Jeyabalan, J.; Tyagi, N.; Agrawal, A.; Yan, J.; Spencer, W.; Bondada, S.; Gupta, R.C. Targeted Oral Delivery of Paclitaxel Using Colostrum-Derived Exosomes. Cancers 2021, 13, 3700. [Google Scholar] [CrossRef]
- Munagala, R.; Aqil, F.; Jeyabalan, J.; Gupta, R.C. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016, 371, 48–61. [Google Scholar] [CrossRef] [Green Version]
- FDA. Immunotoxicity Testing Guidance. 1999. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunotoxicity-testing-guidance (accessed on 6 June 2023).
- Agrawal, A.K.; Aqil, F.; Jeyabalan, J.; Spencer, W.A.; Beck, J.; Gachuki, B.W.; Alhakeem, S.S.; Oben, K.; Munagala, R.; Bondada, S.; et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomedicine 2017, 13, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- FDA. Immunogenicity of Protein-Based Therapeutics. 2020. Available online: https://www.fda.gov/vaccines-blood-biologics/biologics-research-projects/immunogenicity-protein-based-therapeutics (accessed on 6 June 2023).
- Lopez-Castejon, G.; Brough, D. Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Kanneganti, T.D. Function and regulation of IL-1α in inflammatory diseases and cancer. Immunol. Rev. 2018, 281, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [Green Version]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Junttila, I.S. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef] [Green Version]
- Greenfeder, S.; Umland, S.P.; Cuss, F.M.; Chapman, R.W.; Egan, R.W. Th2 cytokines and asthma. The role of interleukin-5 in allergic eosinophilic disease. Respir. Res. 2001, 2, 71–79. [Google Scholar] [CrossRef]
- Somiya, M.; Yoshioka, Y.; Ochiya, T. Biocompatibility of highly purified bovine milk-derived extracellular vesicles. J. Extracell. Vesicles 2018, 7, 1440132. [Google Scholar] [CrossRef] [Green Version]
- Khanam, A.; Ngu, A.; Zempleni, J. Bioavailability of orally administered small extracellular vesicles from bovine milk in C57BL/6J mice. Int. J. Pharm. 2023, 639, 122974. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, P.S.; Kularatne, S.A. Folate-targeted therapeutic and imaging agents for cancer. Curr. Opin. Chem. Biol. 2009, 13, 256–262. [Google Scholar] [CrossRef]
- Sega, E.I.; Low, P.S. Tumor detection using folate receptor-targeted imaging agents. Cancer Metastasis Rev. 2008, 27, 655–664. [Google Scholar] [CrossRef]
- Li, D.; Yao, S.; Zhou, Z.; Shi, J.; Huang, Z.; Wu, Z. Hyaluronan decoration of milk exosomes directs tumor-specific delivery of doxorubicin. Carbohydr. Res. 2020, 493, 108032. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, T.N.; Raiker, R.S.; Jay, S.M. Exogenous DNA Loading into Extracellular Vesicles via Electroporation is Size-Dependent and Enables Limited Gene Delivery. Mol. Pharm. 2015, 12, 3650–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAndrews, K.M.; Xiao, F.; Chronopoulos, A.; LeBleu, V.S.; Kugeratski, F.G.; Kalluri, R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic KrasG12D in pancreatic cancer. Life Sci. Alliance 2021, 4, e202000875. [Google Scholar] [CrossRef]
- Kim, S.M.; Yang, Y.; Oh, S.J.; Hong, Y.; Seo, M.; Jang, M. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J. Control Release 2017, 266, 8–16. [Google Scholar] [CrossRef]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Christiansen, G.; Gurevich, L.; Moos, T.; Duroux, M. Evaluation of electroporation-induced adverse effects on adipose-derived stem cell exosomes. Cytotechnology 2016, 68, 2125–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, J.L.; Scott, M.J.; Wickline, S.A. Maximizing exosome colloidal stability following electroporation. Anal. Biochem. 2014, 448, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Nizamudeen, Z.A.; Xerri, R.; Parmenter, C.; Suain, K.; Markus, R.; Chakrabarti, L.; Sottile, V. Low-Power Sonication Can Alter Extracellular Vesicle Size and Properties. Cells 2021, 10, 2413. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Elsner, H.I.; Lindblad, E.B. Ultrasonic degradation of DNA. DNA 1989, 8, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wu, J.; Gu, W.; Huang, Y.; Tong, Z.; Huang, L.; Tan, J. Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Adv. Sci. 2018, 5, 1700611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Xu, X.; Xu, L.; Iqbal, Z.; Ouyang, K.; Zhang, H.; Wen, C.; Li, D.; Jiang, X. Chondrocyte-specific genomic editing enabled by hybrid exosomes for osteoarthritis treatment. Theranostics 2022, 12, 4866–4878. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Zhao, Y.; Harrison, E.B.; Mahajan, V.; Ahmed, S.; He, Z.; Suresh, P.; Hingtgen, S.D.; Klyachko, N.L.; Mosley, R.L.; et al. Specific Transfection of Inflamed Brain by Macrophages: A New Therapeutic Strategy for Neurodegenerative Diseases. PLoS ONE 2013, 8, e61852. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Q.; Zhang, X.; Huang, H.; Tang, S.; Chai, Y.; Xu, Z.; Li, M.; Chen, X.; Liu, J.; et al. Recent advances in exosome-mediated nucleic acid delivery for cancer therapy. J. Nanobiotechnol. 2022, 20, 279. [Google Scholar] [CrossRef]
- Duan, L.; Xu, L.; Xu, X.; Qin, Z.; Zhou, X.; Xiao, Y.; Liang, Y.; Xia, J. Exosome-mediated delivery of gene vectors for gene therapy. Nanoscale 2021, 13, 1387–1397. [Google Scholar] [CrossRef]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Wallen, M.; Aqil, F.; Kandimalla, R.; Jeyabalan, J.; Auwardt, S.; Tyagi, N.; Schultz, D.J.; Spencer, W.; Gupta, R.C. A model system for antiviral siRNA therapeutics using exosome-based delivery. Mol. Ther. Nucleic Acids 2022, 29, 691–704. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Delivered | Size of Plasmid (kb) | Therapeutic Application | In Vitro Application | In Vivo Application | Reference |
|---|---|---|---|---|---|
| emGFP | 6.2 | Proof-of-concept | ✓ | ✓ | [131] |
| p53 | 6.4 | Cancer treatment | ✓ | ✓ | [131] |
| SARS-CoV-2 Spike protein | 9 | Development of antiviral therapeutics including vaccines | ✓ | [167] | |
| SARS-CoV-2 Spike protein subunit 1 | 7.2 | ✓ | ✓ | [167] | |
| SARS-CoV-2 nucleocapsid | 6.5 | ✓ | ✓ | [167] | |
| SARS-CoV-2 replicase | 8 | ✓ | ✓ | [167] | |
| CRISPR/Cas9 and sgRNA ab | 9.3 | Genome editing | ✓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wallen, M.; Aqil, F.; Spencer, W.; Gupta, R.C. Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy. Pharmaceutics 2023, 15, 1832. https://doi.org/10.3390/pharmaceutics15071832
Wallen M, Aqil F, Spencer W, Gupta RC. Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy. Pharmaceutics. 2023; 15(7):1832. https://doi.org/10.3390/pharmaceutics15071832
Chicago/Turabian StyleWallen, Margaret, Farrukh Aqil, Wendy Spencer, and Ramesh C. Gupta. 2023. "Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy" Pharmaceutics 15, no. 7: 1832. https://doi.org/10.3390/pharmaceutics15071832
APA StyleWallen, M., Aqil, F., Spencer, W., & Gupta, R. C. (2023). Exosomes as an Emerging Plasmid Delivery Vehicle for Gene Therapy. Pharmaceutics, 15(7), 1832. https://doi.org/10.3390/pharmaceutics15071832