

Cu(ATSM) Increases P-Glycoprotein Expression and Function at the Blood-Brain Barrier in C57BL6/J Mice

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
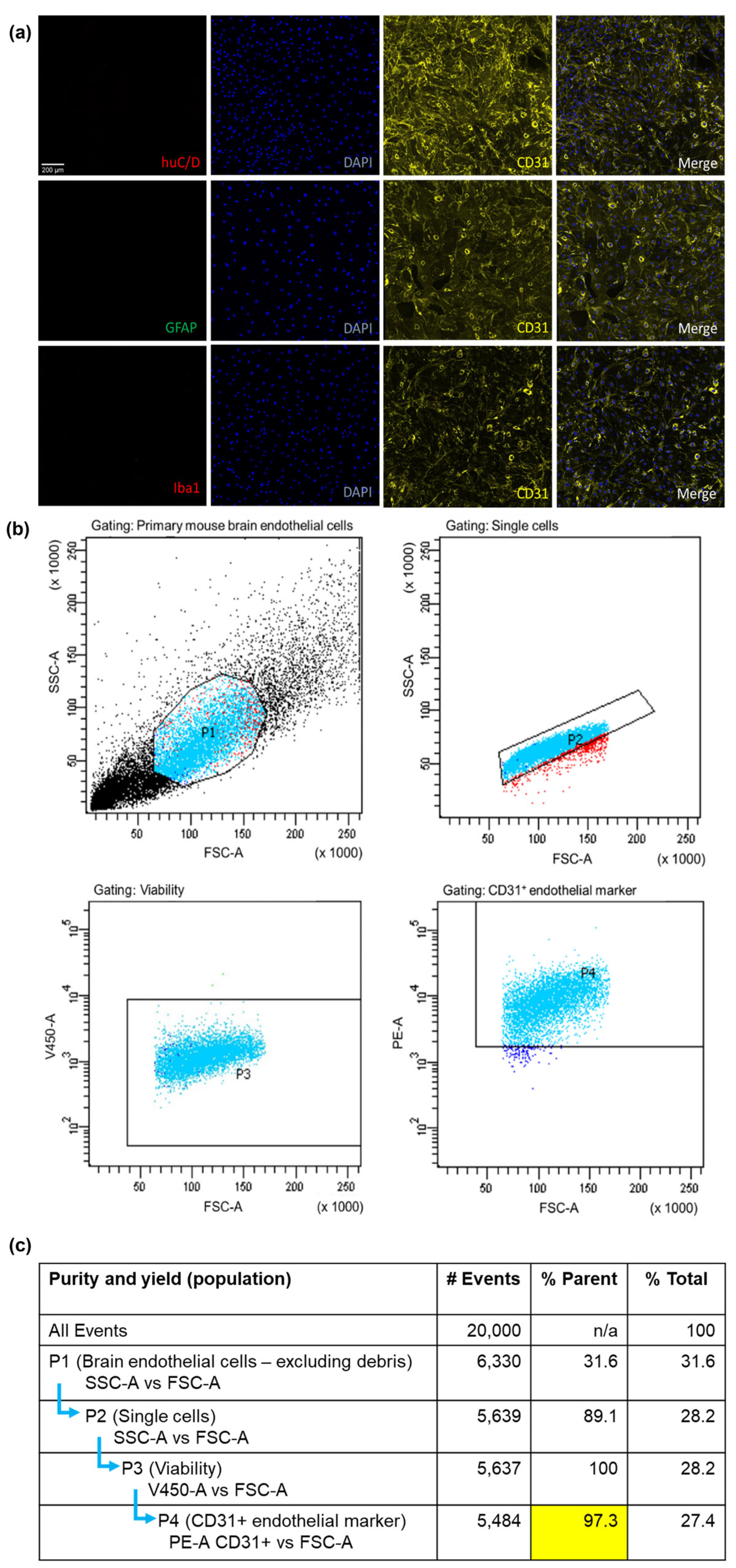
2.2. Isolation and Characterisation of mBECs
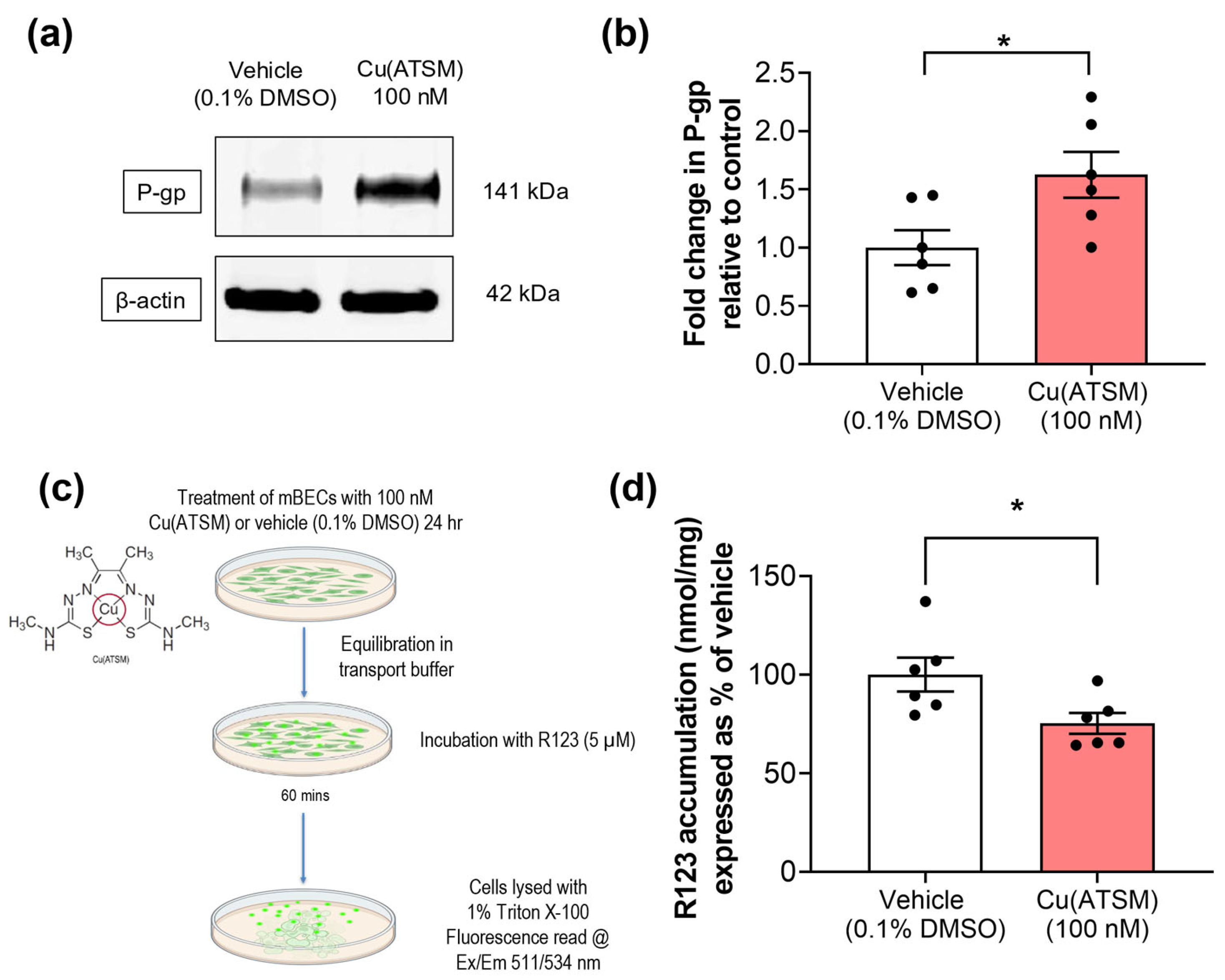
2.3. Treatment of mBECs with Cu(ATSM) to Assess P-gp Expression and Function
2.4. Dosing of Cu(ATSM) to C57BL/6J Mice
2.5. Brain Microvessel Membrane Fraction Isolation and Organ Harvesting
2.6. ICP-MS of Brain MEFs and Organ Homogenates
2.7. WB of Brain MEFs and Organ Homogenates
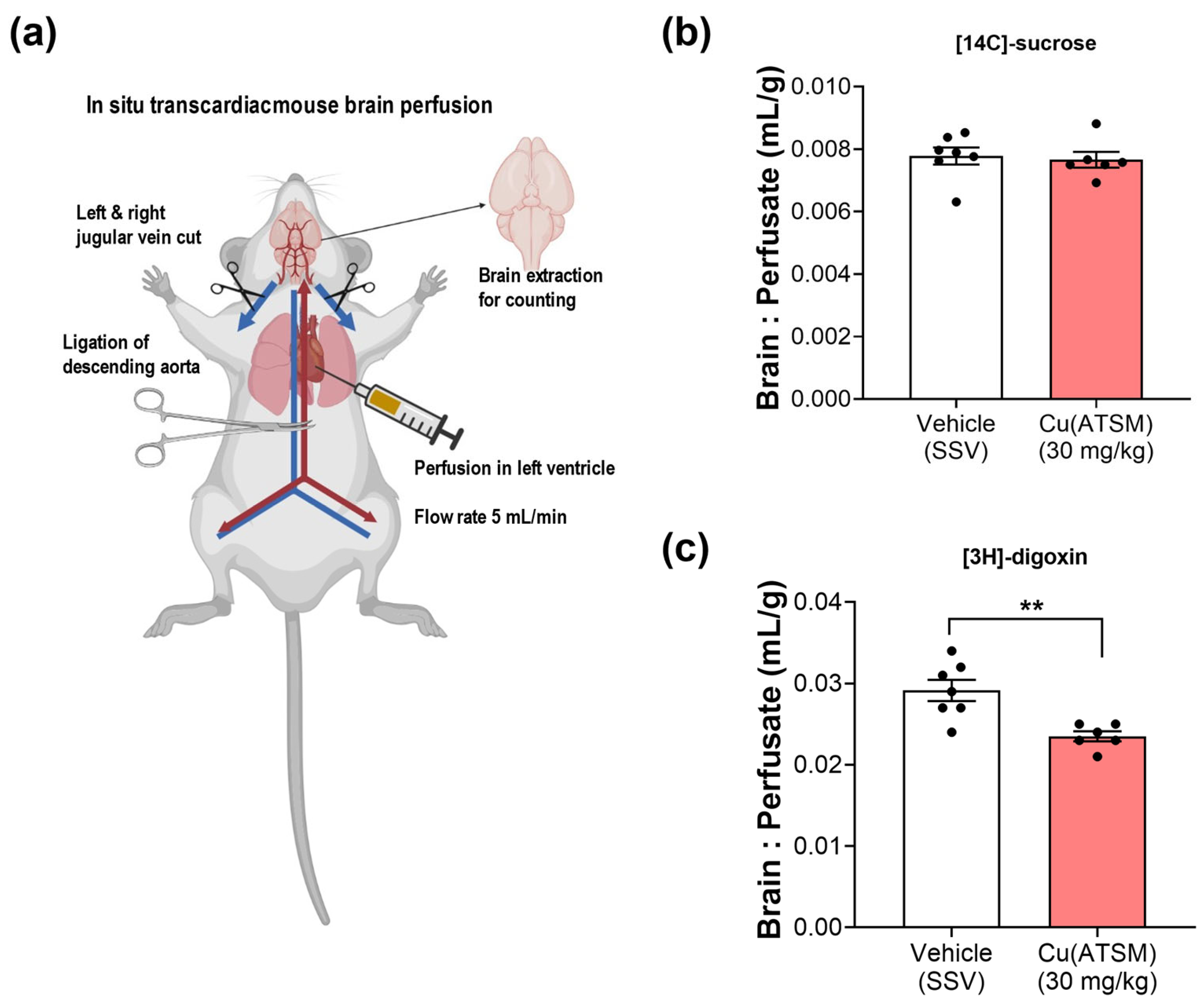
2.8. In situ Transcardiac Brain Perfusion to Assess BBB Transport
2.9. Statistical Analyses
3. Results
3.1. Characterisation and Purity of Isolated Primary mBECs
3.2. Cu(ATSM) Increases P-gp Expression and Function in Isolated Primary mBECs
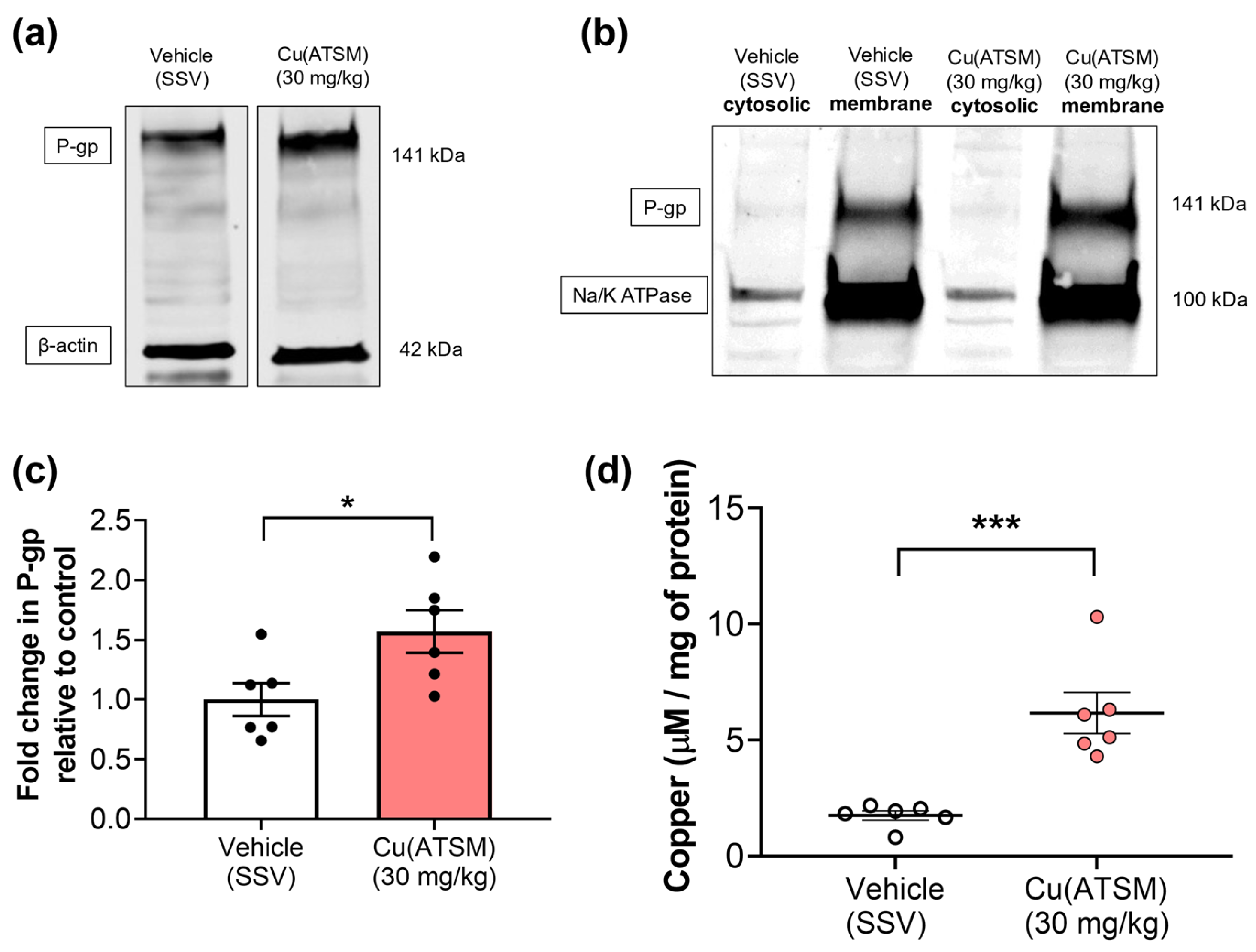
3.3. Mice Dosed with Cu(ATSM) Show Increased P-gp Abundance and Levels of Cu in Isolated Brain MEFs
3.4. Mice Dosed with Cu(ATSM) Show Increased BBB Function of P-gp
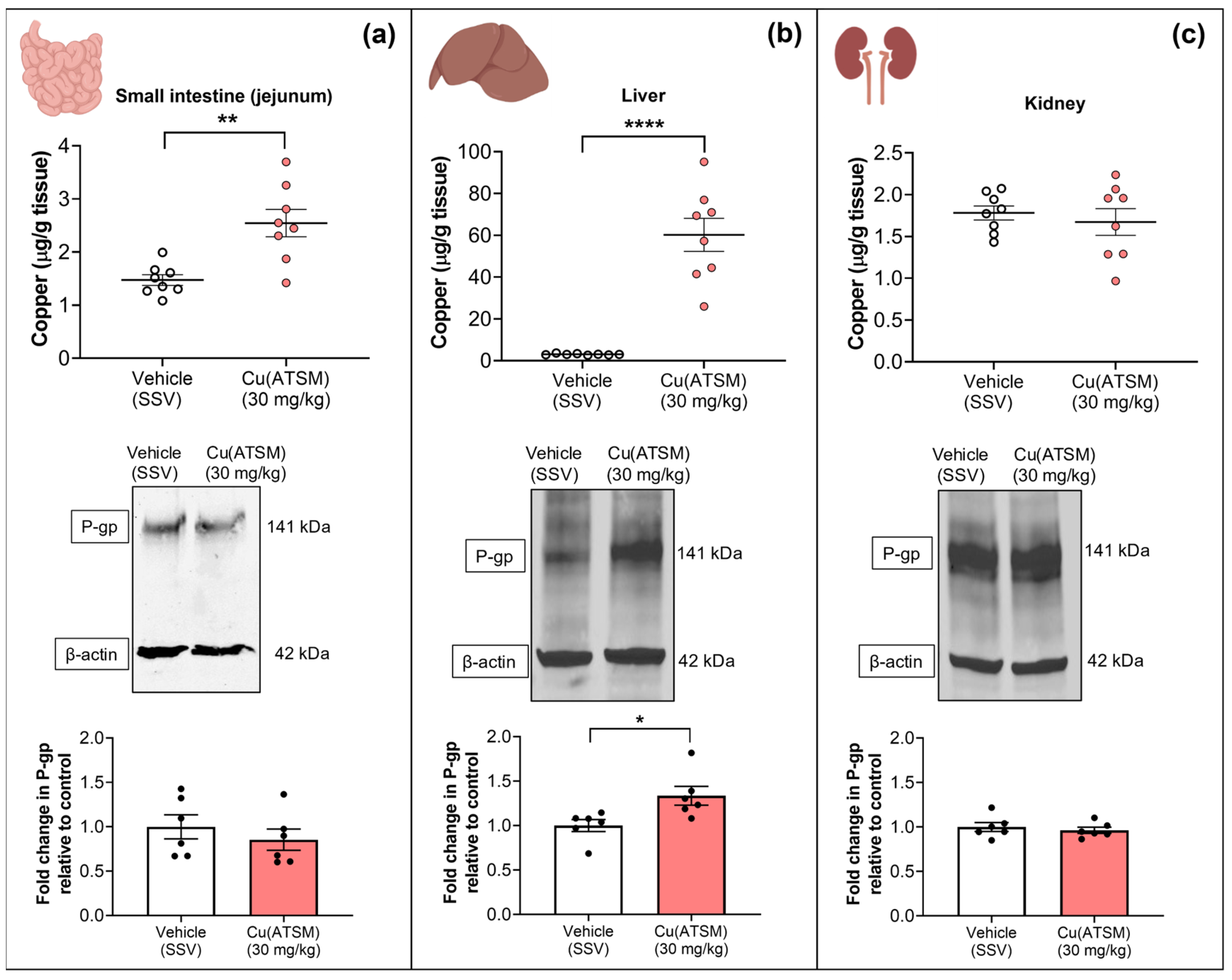
3.5. Cu(ATSM) Treatment Increases Copper and P-gp Expression in a Peripheral Organ-Specific Manner
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a gatekeeper in the blood–brain barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Clark, D.P.; Chen, C.J.; Roninson, I.B.; Gottesman, M.M.; Pastan, I. The human multidrug resistance (mdr1) gene. cDNA cloning and transcription initiation. J. Biol. Chem. 1987, 262, 505–508. [Google Scholar] [CrossRef]
- Borst, P.; Schinkel, A.H. P-glycoprotein ABCB1: A major player in drug handling by mammals. J. Clin. Investig. 2013, 123, 4131–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinkel, A.H.; Wagenaar, E.; van Deemter, L.; A Mol, C.; Borst, P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 1995, 96, 1698–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinkel, A.; Smit, J.; van Tellingen, O.; Beijnen, J.; Wagenaar, E.; van Deemter, L.; Mol, C.; van der Valk, M.; Robanus-Maandag, E.; Riele, H.T.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Wagenaar, E.; Mol, C.A.; Van Deemter, L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Investig. 1996, 97, 2517–2524. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the multidrug resistance P-glycoprotein: Time for a change of strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Atadja, P.; Watanabe, T.; Xu, H.; Cohen, D. PSC-833, a frontier in modulation of P-glycoprotein mediated multidrug resistance. Cancer Metastasis Rev. 1998, 17, 163–168. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, E.-H.; Han, M.; An, S.-H.; Park, J. Diminished expression of P-glycoprotein using focused ultrasound is associated with JNK-dependent signaling pathway in cerebral blood vessels. Front. Neurosci. 2019, 13, 1350. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.; Monteiro, A.; Silva, R.; Moreira, J.; Lobo, J.S.; Silva, A. Lipid nanoparticles strategies to modify pharmacokinetics of central nervous system targeting drugs: Crossing or circumventing the blood-brain barrier (BBB) to manage neurological disorders. Adv. Drug Deliv. Rev. 2022, 189, 114485. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, Y.; Gosselet, F.; Dehouck, M.-P.; Cecchelli, R. Blood–brain barrier proteomics: Towards the understanding of neurodegenerative diseases. Arch. Med. Res. 2014, 45, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Vautier, S.; Fernandez, C. ABCB1: The role in Parkinson’s disease and pharmacokinetics of antiparkinsonian drugs. Expert Opin. Drug Metab. Toxicol. 2009, 5, 1349–1358. [Google Scholar] [CrossRef]
- Qosa, H.; Lichter, J.; Sarlo, M.; Markandaiah, S.S.; McAvoy, K.; Richard, J.-P.; Jablonski, M.R.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-glycoprotein) in endothelial cells of the blood–brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia 2016, 64, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- van Assema, D.M.; Lubberink, M.; Bauer, M.; van der Flier, W.M.; Schuit, R.C.; Windhorst, A.D.; Comans, E.F.; Hoetjes, N.J.; Tolboom, N.; Langer, O.; et al. Blood–brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2011, 135, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2018, 99, 21–78. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Iturria-Medina, Y.I.; Sotero, R.C.; Toussaint, P.J.; Mateos-Pérez, J.M.; Evans, A.C. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat. Commun. 2016, 7, 11934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. β-amyloid efflux mediated by P-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer’s amyloid-β peptides—Implications for the mechanisms of Aβ clearance at the blood–brain barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, A.B.; Hartz, A.M.S.; Gao, X.; Yang, A.; Callaghan, R.; Gelissen, I.C. New evidence for P-gp-mediated export of amyloid-β peptides in molecular, blood-brain barrier and neuronal models. Int. J. Mol. Sci. 2021, 22, 246. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-β deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [Green Version]
- Vogelgesang, S.; Cascorbi, I.; Schroeder, E.; Pahnke, J.; Kroemer, H.K.; Siegmund, W.; Kunert-Keil, C.; Walker, L.C.; Warzok, R.W. Deposition of Alzheimer’s β-amyloid is inversely correlated with P-glycoprotein expression in the brains of elderly non-demented humans. Pharm. Genom. 2002, 12, 535–541. [Google Scholar] [CrossRef]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’sullivan, F.; et al. Activity of P-glycoprotein, a β-amyloid transporter at the blood–brain barrier, is compromised in patients with mild Alzheimer disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef] [Green Version]
- Jeynes, B.; Provias, J. An investigation into the role of P-glycoprotein in Alzheimer’s disease lesion pathogenesis. Neurosci. Lett. 2011, 487, 389–393. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Bush, A.I. Metallostasis in Alzheimer’s disease. Free Radic. Biol. Med. 2013, 62, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Ayton, S.; Agrawal, S.; Dhana, K.; Bennett, D.A.; Barnes, L.L.; Leurgans, S.E.; Bush, A.I.; Schneider, J.A. Brain copper may protect from cognitive decline and Alzheimer’s disease pathology: A community-based study. Mol. Psychiatry 2022, 27, 4307–4313. [Google Scholar] [CrossRef]
- McInerney, M.P.; Volitakis, I.; Bush, A.I.; Banks, W.A.; Short, J.L.; Nicolazzo, J.A. Ionophore and biometal modulation of P-glycoprotein expression and function in human brain microvascular endothelial cells. Pharm. Res. 2018, 35, 83. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Caragounis, A.; Du, T.; Laughton, K.M.; Volitakis, I.; Cherny, R.A.; Sharples, R.A.; Hill, A.F.; Li, Q.-X.; Masters, C.L.; et al. Selective intracellular release of copper and zinc ions from bis (thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-β peptide. J. Biol. Chem. 2008, 283, 4568–4577. [Google Scholar] [CrossRef] [Green Version]
- Fodero-Tavoletti, M.T.; Villemagne, V.L.; Paterson, B.M.; White, A.R.; Li, Q.X.; Camakaris, J.; O’keefe, G.; Cappai, R.; Barnham, K.J.; Donnelly, P.S. Bis (thiosemicarbazonato) Cu-64 complexes for positron emission tomography imaging of Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, 49–55. [Google Scholar] [CrossRef]
- Paterson, B.M.; Cullinane, C.; Crouch, P.; White, A.R.; Barnham, K.J.; Roselt, P.D.; Noonan, W.; Binns, D.; Hicks, R.J.; Donnelly, P.S. Modification of biodistribution and brain uptake of copper bis(thiosemicarbazonato) complexes by the incorporation of amine and polyamine functional groups. Inorg. Chem. 2019, 58, 4540–4552. [Google Scholar] [CrossRef]
- Pyun, J.; McInnes, L.E.; Donnelly, P.S.; Mawal, C.; Bush, A.I.; Short, J.L.; Nicolazzo, J.A. Copper bis (thiosemicarbazone) complexes modulate P-glycoprotein expression and function in human brain microvascular endothelial cells. J. Neurochem. 2022, 162, 226–244. [Google Scholar] [CrossRef]
- Syvänen, S.; Lindhe, O.; Palner, M.; Kornum, B.R.; Rahman, O.; Långström, B.; Knudsen, G.M.; Hammarlund-Udenaes, M. Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab. Dispos. 2009, 37, 635–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, A.K.; Theil, F.-P.; Nicolas, J.-M. Confounding parameters in preclinical assessment of blood–brain barrier permeation: An overview with emphasis on species differences and effect of disease states. Mol. Pharm. 2013, 10, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Blower, P.J.; Castle, T.C.; Cowley, A.R.; Dilworth, J.R.; Donnelly, P.S.; Labisbal, E.; Sowrey, F.E.; Teat, S.J.; Went, M.J. Structural trends in copper (II) bis (thiosemicarbazone) radiopharmaceuticals. Dalton Trans. 2003, 23, 4416–4425. [Google Scholar] [CrossRef]
- Gingras, B.; Suprunchuk, T.; Bayley, C.H. The preparation of some thiosemicarbazones and their copper complexes: Part III. Can. J. Chem. 1962, 40, 1053–1059. [Google Scholar] [CrossRef]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII (atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lim, N.K.; McAllum, E.J.; Donnelly, P.S.; Hare, D.J.; Doble, P.A.; Turner, B.J.; Price, K.A.; Lim, S.C.; Paterson, B.M.; et al. Oral treatment with CuII (atsm) increases mutant SOD1 in vivo but protects motor neurons and improves the phenotype of a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2014, 34, 8021–8031. [Google Scholar] [CrossRef] [Green Version]
- Soon, C.P.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.L.; Lim, N.K.H.; Lam, L.; Bica, L.; et al. Diacetylbis (N(4)-methylthiosemicarbazonato) copper (II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Short, J.L.; Newman, S.A.; Choy, K.H.; Tiwari, D.; Yap, C.; Senyschyn, D.; Banks, W.A.; Nicolazzo, J.A. Cognitive benefits of lithium chloride in APP/PS1 mice are associated with enhanced brain clearance of β-amyloid. Brain Behav. Immun. 2018, 70, 36–47. [Google Scholar] [CrossRef]
- Pan, Y.; Scanlon, M.J.; Owada, Y.; Yamamoto, Y.; Porter, C.J.H.; Nicolazzo, J.A. Fatty acid-binding protein 5 facilitates the blood–brain barrier transport of docosahexaenoic acid. Mol. Pharm. 2015, 12, 4375–4385. [Google Scholar] [CrossRef]
- McInerney, M.P.; Pan, Y.; Volitakis, I.; Bush, A.I.; Short, J.L.; Nicolazzo, J.A. The effects of clioquinol on P-glycoprotein expression and biometal distribution in the mouse brain microvasculature. J. Pharm. Sci. 2019, 108, 2247–2255. [Google Scholar] [CrossRef]
- Yap, C.; Short, J.L.; Nicolazzo, J.A. A combination of clioquinol, zinc and copper increases the abundance and function of breast cancer resistance protein in human brain microvascular endothelial cells. J. Pharm. Sci. 2020, 110, 338–346. [Google Scholar] [CrossRef]
- Xiao, Z.; Donnelly, P.S.; Zimmermann, M.; Wedd, A.G. Transfer of copper between bis (thiosemicarbazone) ligands and intracellular copper-binding proteins. Insights into mechanisms of copper uptake and hypoxia selectivity. Inorg. Chem. 2008, 47, 4338–4347. [Google Scholar] [CrossRef]
- Dearling, J.L.; Lewis, J.S.; Mullen, G.E.; Welch, M.J.; Blower, P.J. Copper bis (thiosemicarbazone) complexes as hypoxia imaging agents: Structure-activity relationships. J. Biol. Inorg. Chem. 2002, 7, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Hilton, J.B.; Kysenius, K.; Liddell, J.R.; Rautengarten, C.; Mercer, S.W.; Paul, B.; Beckman, J.S.; McLean, C.A.; White, A.R.; Donnelly, P.S.; et al. Disrupted copper availability in sporadic ALS: Implications for CuII (atsm) as a treatment option. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Hilton, J.B.; Mercer, S.W.; Lim, N.K.H.; Faux, N.G.; Buncic, G.; Beckman, J.S.; Roberts, B.R.; Donnelly, P.S.; White, A.R.; Crouch, P.J. Cu II (atsm) improves the neurological phenotype and survival of SOD1 G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci. Rep. 2017, 7, 42292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, L.E.; Noor, A.; Kysenius, K.; Cullinane, C.; Roselt, P.; McLean, C.A.; Chiu, F.C.K.; Powell, A.K.; Crouch, P.; White, J.M.; et al. Potential diagnostic imaging of Alzheimer’s disease with copper-64 complexes that bind to amyloid-β plaques. Inorg. Chem. 2019, 58, 3382–3395. [Google Scholar] [CrossRef]
- Price, K.A.; Crouch, P.J.; Lim, S.; Paterson, B.M.; Liddell, J.R.; Donnelly, P.S.; White, A.R. Subcellular localization of a fluorescent derivative of Cu II (atsm) offers insight into the neuroprotective action of Cu II (atsm). Metallomics 2011, 3, 1280–1290. [Google Scholar] [CrossRef]
- Bernard-Patrzynski, F.; Lécuyer, M.-A.; Puscas, I.; Boukhatem, I.; Charabati, M.; Bourbonnière, L.; Ramassamy, C.; Leclair, G.; Prat, A.; Roullin, V.G. Isolation of endothelial cells, pericytes and astrocytes from mouse brain. PLoS ONE 2019, 14, e0226302. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, H.C.; Wang, X.; Kuo, P.-C.; Furnas, D.; Scofield, B.A.; Chang, F.-L.; Yen, J.-H.; Yu, I.-C. Isolation of mouse cerebral microvasculature for molecular and single-cell analysis. Front. Cell. Neurosci. 2020, 14, 84. [Google Scholar] [CrossRef]
- Navone, S.E.; Marfia, G.; Invernici, G.; Cristini, S.; Nava, S.; Balbi, S.; Sangiorgi, S.; Ciusani, E.; Bosutti, A.; Alessandri, G.; et al. Isolation and expansion of human and mouse brain microvascular endothelial cells. Nat. Protoc. 2013, 8, 1680–1693. [Google Scholar] [CrossRef]
- Lee, G.; Schlichter, L.; Bendayan, M.; Bendayan, R. Functional expression of P-glycoprotein in rat brain microglia. J. Pharmacol. Exp. Ther. 2001, 299, 204–212. [Google Scholar] [PubMed]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.N.Y.; Hoque, T.; Cummins, C.L.; Bendayan, R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J. Neurochem. 2011, 118, 163–175. [Google Scholar] [CrossRef]
- Ding, Y.; Zhong, Y.; Baldeshwiler, A.; Abner, E.L.; Bauer, B.; Hartz, A.M.S. Protecting P-glycoprotein at the blood–brain barrier from degradation in an Alzheimer’s disease mouse model. Fluids Barriers CNS 2021, 18, 10. [Google Scholar] [CrossRef]
- Hartz, A.M.S.; Zhong, Y.; Shen, A.N.; Abner, E.L.; Bauer, B. Preventing P-gp ubiquitination lowers Aβ brain levels in an Alzheimer’s disease mouse model. Front. Aging Neurosci. 2018, 10, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nwaozuzu, O.M.; Sellers, L.A.; Barrand, M.A. Signalling pathways influencing basal and H2O2-induced P-glycoprotein expression in endothelial cells derived from the blood–brain barrier. J. Neurochem. 2003, 87, 1043–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, J.; Li, P.; Xie, Q.; Sun, B.; Li, Y.; Chen, Y.; Zhao, K.; Yang, T.; Zhu, L.; et al. Increase in P-glycoprotein levels in the blood-brain barrier of partial portal vein ligation/chronic hyperammonemia rats is medicated by ammonia/reactive oxygen species/ERK1/2 activation: In vitro and in vivo studies. Eur. J. Pharmacol. 2019, 846, 119–127. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, C.; Hong, Z.; Chen, Y. Inhibition of p38 mitogen-activated protein kinase signaling reduces multidrug transporter activity and anti-epileptic drug resistance in refractory epileptic rats. J. Neurochem. 2016, 136, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Blower, P.J.; Aubdool, A.A.; Hider, R.C.; Mann, G.E.; Siow, R.C. Cardioprotective effects of Cu (II) ATSM in human vascular smooth muscle cells and cardiomyocytes mediated by Nrf2 and DJ-1. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, K.M.; Hayne, D.J.; McInnes, L.E.; Noor, A.; Duncan, C.; Moujalled, D.; Volitakis, I.; Rigopoulos, A.; Barnham, K.J.; Villemagne, V.L.; et al. Effect of structural modifications to glyoxal-bis (thiosemicarbazonato) copper (II) complexes on cellular copper uptake, copper-mediated ATP7A trafficking, and P-glycoprotein mediated efflux. J. Med. Chem. 2018, 61, 711–723. [Google Scholar] [CrossRef]
- Crouch, P.J.; Hung, L.W.; Adlard, P.A.; Cortes, M.; Lal, V.; Filiz, G.; Perez, K.A.; Nurjono, M.; Caragounis, A.; Du, T.; et al. Increasing Cu bioavailability inhibits Aβ oligomers and tau phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Hsieh, H.L.; Shih, R.H.; Chi, P.L.; Cheng, S.E.; Yang, C.M. Up-regulation of COX-2/PGE 2 by endothelin-1 via MAPK-dependent NF-κB pathway in mouse brain microvascular endothelial cells. Cell Commun. Signal. 2013, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.-H.; Huang, W.; Mo, X.-A.; Chen, Y.-L.; Wu, X.-H. LPS induces occludin dysregulation in cerebral microvascular endothelial cells via MAPK signaling and augmenting MMP-2 levels. Oxidative Med. Cell. Longev. 2015, 2015, 120641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood–brain and blood–spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [Green Version]
- Chai, A.B.; Callaghan, R.; Gelissen, I.C. Regulation of P-Glycoprotein in the brain. Int. J. Mol. Sci. 2022, 23, 14667. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood–brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Kagawa, Y.; Sun, J.; Turner, B.J.; Huang, C.; Shah, A.D.; Schittenhelm, R.B.; Nicolazzo, J.A. Altered blood-brain barrier dynamics in the C9orf72 hexanucleotide repeat expansion mouse model of amyotrophic lateral sclerosis. Pharmaceutics 2022, 14, 2803. [Google Scholar] [CrossRef]
- Dagenais, C.; Rousselle, C.; Pollack, G.M.; Scherrmann, J.M. Development of an in situ mouse brain perfusion model and its application to mdr1a P-glycoprotein-deficient mice. J. Cereb. Blood Flow Metab. 2000, 20, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Cherny, R.A.; Atwood, C.S.; E Xilinas, M.; Gray, D.N.; Jones, W.D.; A McLean, C.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.-S.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: Review of clinical drug–drug interaction studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamoulis, I.; Kouraklis, G.; Theocharis, S. Zinc and the liver: An active interaction. Dig. Dis. Sci. 2007, 52, 1595–1612. [Google Scholar] [CrossRef] [PubMed]
- Hatano, R.; Ebara, M.; Fukuda, H.; Yoshikawa, M.; Sugiura, N.; Kondo, F.; Yukawa, M.; Saisho, H. Accumulation of copper in the liver and hepatic injury in chronic hepatitis C. J. Gastroenterol. Hepatol. 2000, 15, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Nikseresht, S.; Hilton, J.B.; Liddell, J.R.; Kysenius, K.; Bush, A.I.; Ayton, S.; Koay, H.; Donnelly, P.S.; Crouch, P.J. Transdermal application of soluble CuII (atsm) increases brain and spinal cord uptake compared to gavage with an insoluble suspension. Neuroscience 2023, 509, 125–131. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Secondary Alexa Fluor® Antibody | |
|---|---|---|
| mBEC marker | Rat anti-mouse CD31 (1:400) | Anti-rat Alexa 555 (1:500) |
| Neuronal marker | Mouse anti-human HuC/D (1:1000) | Anti-mouse Alexa 488 (1:500) |
| Astrocyte marker | Chicken anti-mouse GFAP (1:2000) | Anti-chicken Alexa 647 (1:500) |
| Microglial marker | Rabbit anti-mouse Iba1 (1:2000) | Anti-rabbit Alexa 488 (1:500) |
| Tissue | Element | Concentration in Vehicle-Treated Mice (n = 6–8) | Concentration in Cu(ATSM)-Treated Mice (n = 6–8) | p Value | Fold Change to Control (If Significant) |
|---|---|---|---|---|---|
| Microvessel Enriched Fraction (μg/mg protein) | Sodium | 237,338 ± 38,901 | 230,820 ± 37,462 | 0.9063 | |
| Magnesium | 280.3 ± 43.39 | 232.3 ± 25.60 | 0.3634 | ||
| Phosphorus | 20521 ± 2428 | 19953 ± 2564 | 0.8754 | ||
| Potassium | 9762 ± 1065 | 9590 ± 1095 | 0.9122 | ||
| Calcium | 1922 ± 542.1 | 1579 ± 273.2 | 0.5838 | ||
| Iron | 183.8 ± 8.56 | 180.6 ± 10.35 | 0.8166 | ||
| Copper | 1.74 ± 0.20 | 6.16 ± 0.88 | 0.0006 | 3.5 | |
| Zinc | 13.08 ± 2.07 | 13.82 ± 2.01 | 0.8038 | ||
| Small Intestines (μg/g tissue) | Sodium | 463.6 ± 34.09 | 401.6 ± 17.80 | 0.1295 | |
| Magnesium | 86.47 ± 4.91 | 92.14 ± 6.67 | 0.5046 | ||
| Phosphorus | 1222 ± 59.09 | 1082 ± 78.75 | 0.1746 | ||
| Potassium | 1394 ± 73.55 | 1163 ± 84.03 | 0.0578 | ||
| Calcium | 8226 ± 775.9 | 8970 ± 1133 | 0.5966 | ||
| Iron | 7.558 ± 0.57 | 7.558 ± 0.80 | 0.9997 | ||
| Copper | 1.473 ± 0.10 | 2.545 ± 0.26 | 0.0016 | 1.7 | |
| Zinc | 8.417 ±0.40 | 7.796 ± 0.56 | 0.3817 | ||
| Liver (μg/g tissue) | Sodium | 405.7 ± 13.95 | 398.1 ± 17.36 | 0.7376 | |
| Magnesium | 87.21 ± 2.48 | 77.25 ± 0.70 | 0.0017 | 0.9 | |
| Phosphorus | 1310 ± 36.79 | 1181 ± 19.62 | 0.0081 | 0.9 | |
| Potassium | 1547 ± 43.34 | 1355 ± 20.16 | 0.0012 | 0.9 | |
| Calcium | 1128 ± 97.08 | 1363 ± 125.6 | 0.1619 | ||
| Iron | 56.47 ± 2.99 | 46.99 ± 3.00 | 0.0421 | 0.8 | |
| Copper | 3.03 ± 0.12 | 60.19 ± 7.89 | <0.0001 | 19.9 | |
| Zinc | 10.94 ± 0.31 | 13.77 ± 0.24 | <0.0001 | 1.3 | |
| Kidneys (μg/g tissue) | Sodium | 428.2 ± 15.71 | 382.6 ± 30.94 | 0.2103 | |
| Magnesium | 49.20 ± 2.26 | 41.89 ± 3.60 | 0.1076 | ||
| Phosphorus | 816.1 ± 36.45 | 702.3 ± 64.41 | 0.1463 | ||
| Potassium | 868.3 ± 27.34 | 754.0 ± 58.65 | 0.0991 | ||
| Calcium | 2675 ± 203.8 | 2306 ± 178.0 | 0.1946 | ||
| Iron | 22.83 ± 1.23 | 20.38 ± 2.48 | 0.3910 | ||
| Copper | 1.781 ± 0.08 | 1.673 ± 0.16 | 0.5590 | ||
| Zinc | 4.380 ± 0.18 | 3.777 ± 0.32 | 0.1238 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyun, J.; Koay, H.; Runwal, P.; Mawal, C.; Bush, A.I.; Pan, Y.; Donnelly, P.S.; Short, J.L.; Nicolazzo, J.A. Cu(ATSM) Increases P-Glycoprotein Expression and Function at the Blood-Brain Barrier in C57BL6/J Mice. Pharmaceutics 2023, 15, 2084. https://doi.org/10.3390/pharmaceutics15082084
Pyun J, Koay H, Runwal P, Mawal C, Bush AI, Pan Y, Donnelly PS, Short JL, Nicolazzo JA. Cu(ATSM) Increases P-Glycoprotein Expression and Function at the Blood-Brain Barrier in C57BL6/J Mice. Pharmaceutics. 2023; 15(8):2084. https://doi.org/10.3390/pharmaceutics15082084
Chicago/Turabian StylePyun, Jae, HuiJing Koay, Pranav Runwal, Celeste Mawal, Ashley I. Bush, Yijun Pan, Paul S. Donnelly, Jennifer L. Short, and Joseph A. Nicolazzo. 2023. "Cu(ATSM) Increases P-Glycoprotein Expression and Function at the Blood-Brain Barrier in C57BL6/J Mice" Pharmaceutics 15, no. 8: 2084. https://doi.org/10.3390/pharmaceutics15082084
APA StylePyun, J., Koay, H., Runwal, P., Mawal, C., Bush, A. I., Pan, Y., Donnelly, P. S., Short, J. L., & Nicolazzo, J. A. (2023). Cu(ATSM) Increases P-Glycoprotein Expression and Function at the Blood-Brain Barrier in C57BL6/J Mice. Pharmaceutics, 15(8), 2084. https://doi.org/10.3390/pharmaceutics15082084