
Cavitation-Mediated Immunomodulation and Its Use with Checkpoint Inhibitors


Abstract
:1. Cancer Immunology
1.1. Cancer-Immunity Cycle
1.2. Evading Immune Recognition
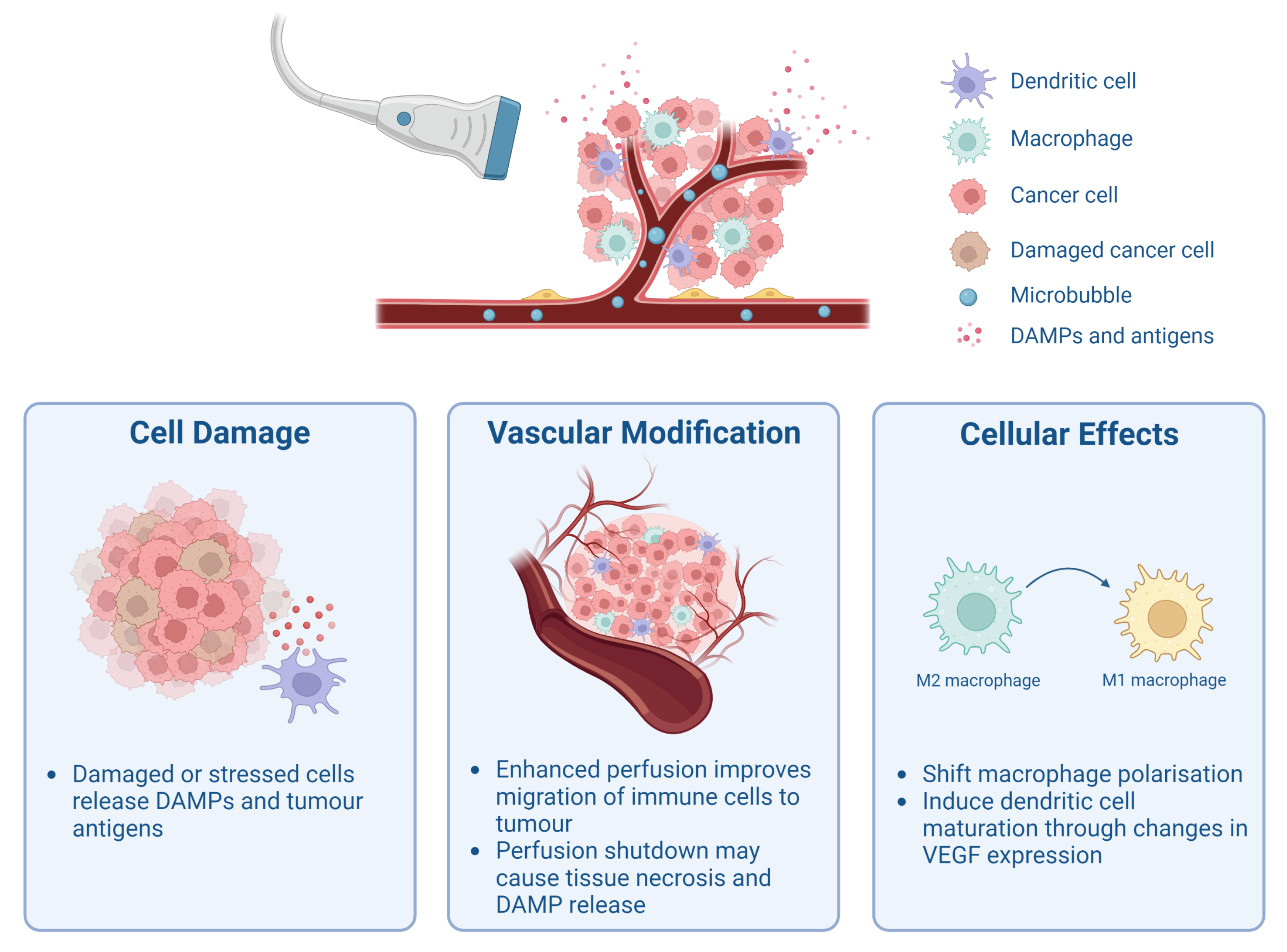
2. Cavitation-Mediated Immunomodulation
2.1. Release of Tumour-Associated Antigens and Damage-Associated Molecular Patterns
2.2. Modulation of Tumour Vasculature and Perfusion
2.3. Cellular Effects of Cavitation

{kind=link}
| Author | Treatment Protocol | US Parameters | Main Findings |
|---|---|---|---|
| Liu et al. [40] | CT26 tumours were divided into three treatment groups: (1) untreated, (2) US-only, and (3) US+CA. SonoVue MBs were used as CAs. Bolus of CAs was injected immediately before US exposure. Mice were sacrificed 1, 3, and 18 days after treatment. | F1 = , PRF 2 = 1 , PL 3 = 100 , and PNP 4 = and . Exposure time: 20 per spot, and 9–12 spots were sonicated to cover the entire tumour. Transducer details: element diameter of 64 and radius curvature of 55 . | Tumour growth was inhibited for both pressures, albeit it was greater for the higher pressure. The percentage of CD45+CD8+ T cells increased in tumours treated with PNP of compared to untreated tumours across all three time points. |
| Joiner et al. [41] | Mice with KPC tumours were divided into two groups: (1) untreated and (2) US+CA. Lipid MBs with in the core were used as CAs. CAs were infused for the entire duration of US treatment. Tumours were excised either 2 or 15 days after treatment. | F = 1 , PRF = 100 , PL = 1 , and PNP = . Exposure time: 7– 10 depending on tumour size. Transducer details: eight-element annular array, 80 focus, 1 × 1 focal spot. | Significant reduction in tumour growth after treatment. The number of CD4+ T cells, CD8+ T cells, and Ly6C-F4/80+CD11b+ macrophages in lymph nodes was significantly higher in treated tumours 2 days after treatment. However, the observed immune response was likely transient due to no significant difference in these immune cell populations being observed 15 days after treatment. A substantial increase in HMGB1 was measured in treated tumours. It was suggested that stable cavitation was the dominant bubble behaviour in this study. |
| Hu et al. [42] | RM1, MC38, and B19 tumours were divided into four groups: (1) untreated, (2) US+CA, (3) aPD-1, and (4) US+CA+aPD-1. The CA was a lipid nanobubble with as core gas. US was applied to tumours 5 after intravenously administering the CAs. aPD-1 was administered intraperitoneally into mice once every 3 days during the treatment period for a total of four doses. | F = 1 , PRF = 100 , PL = 6 , and I 5 = 1 /2. Exposure time: 30 . Transducer details: collimated beam and an effective probe radiation area of 2 2. | US+CA and US+CA+aPD-1 substantially inhibited tumour growth compared to untreated controls. These two groups had significantly more CD44+CD8+ cells compared to untreated tumours, but the difference was greater for the US+CA+aPD-1 group. There were significantly more granzyme B and IFN--secreting CD8+ T cells in the combination therapy group and the US+CA group compared to the untreated group. They also reported TAA release and DAMP release in vitro when RM-1 cells were treated with US+CA compared to the untreated, US-only, and CA-only groups. |
| Wu et al. [43] | For the first study, 4T1 tumours were divided into one of the following groups: (1) no treatment, (2) US, (3) CA, or (4) US+CA. Lipid MBs with as core gas were used as CAs. Tumours were exposed to US immediately after the injection of lipid MBs on days 0, 1, 2, 3, and 4. In another in vivo, they divided the tumour-bearing mice into these four groups: (1) control, (2) aPD-L1, (3) US+CA, and (4) US+CA+aPD-L1. The treatment strategy was similar to the first in vivo, but aPD-L1 was injected intravenously on days 1, 4, and 7. Mice were sacrificed on day 11 after treatment. | F = 1 , PRF = not specified, PL = not specified, I = /2, and duty cycle = 50%. Exposure time: 5 . Transducer details: diameter of 1 , focal length of , and focus area of 2. | In the first study, a substantial difference was reported in tumour volume after treating with US+CA compared to the untreated, US-only, and CA-only groups. The tumour blood perfusion was blocked even 24 after treatment. There was a significant increase in CD11c+CD80+CD86+ cells (matured DCs) and CD3+CD8+ T cells, as well as the level of IL-12 and TNF- cytokines. In the second study, it was observed that the tumour growth was even more inhibited by the US+CA+aPD-L1 and there was a remarkable increase in activated CD8+ T cell infiltration compared to untreated tumours and aPD-L1-treated tumours. |
| Huang et al. [44] | LL/2 and CT26 tumour cells and tumours were treated with either (1) CA or (2) US+CA. The CA was a lipid MB with in the core. CAs with or without US exposure were administered every 3 days, for a total of 6 treatments (18 days), and mice were observed for 28 days. US exposure occurred 1 after CA administration. | F = , PRF = 1 , PL = 10 , and PNP = . Exposure time: 10 . Transducer details: diameter of 20 and focal length of 50 . | Tumour cells treated with only CA or US+CA did not show induced translocation of calreticulin or Erp57, or release of HMGB1 or ATP in vitro. There was no substantial difference in CD80+CD86+ cells (mature DCs) or IFN--secreting cells in vivo between the two groups. |
| Bulner et al. [34] | Mice with CT26 tumours were randomised into these groups: (1) CA, (2) aPD-1, (3) US+CA, and (4) US+CA+aPD-1. The CAs were MBs consisting of lipids encapsulating gas. US exposure commenced immediately after intravenous injection of CAs. For the acute experiments, animals sacrificed at day 3 received US+CA or CA treatment at day 3 with or without aPD-1 at day 0. Animals sacrificed at day 7 received US+CA or CA treatment with or without aPD-1 at day 0, 3, and 6. For the longitudinal experiment, the treatment schedule was similar but aPD-1 was administered on day 9 and 12 as well, and mice were sacrificed at day 30. | F = 1 , PRF = 100 , PL = , and PNP = . Exposure time: 50 pulses were repeated at 20 interval for a duration of 2 . Transducer details: spherically focused, diameter, 15 focal length, and beam width. | The improved tumour growth inhibition was attributed to the shutdown of blood flow due to no evidence supporting a T cell-dependent mechanism (CD45+CD8+ cells and CD45+CD4+ cells). However, the re-challenge experiment suggested an engagement of adaptive memory response. Passive cavitation detection detected broadband noise. |
| Hunt et al. [35] | Animals with K1735 tumours were initially divided into three treatment groups: (1) 3 US exposure, (2) US+CA with 1 exposure, and (3) US+CA with 3 exposure. Definity MBs were used as CAs. Tumours were insonated immediately after intravenous injection of CAs. Mice were sacrificed 24 after treatment. | F = 3 , PRF = continuous, PL = continuous, PNP = , and exposure time: 1 or 3 . Transducer details: unfocused, power level 3, and spatial average intensity /2. | A significant shutdown of blood flow after both US+CA treatments compared to US-treated tumours was reported. There was a significant increase in the mean number of CD45+ cells and CD3+ cells after US+CA ( 3 ) compared to untreated tumours. |
| Li et al. [33] | MC38 tumour-bearing mice were divided into the following groups for the perfusion study: (1) US+CA at , (2) US+CA at , (3) US at , and (4) untreated control. For the second study, the groups were: (1) untreated, (2) US+CA at , (3) aPD-L1, and (4) US+CA+aPD-L1 at . Sonazoid MBs were used as CAs. The CAs were slowly injected through tail vein during US exposure. For the perfusion study, mice were sacrificed 24 after treatment. For the combination therapy, the mice were injected with aPD-L1 on days 4, 7, 10, and 13, US+CA treatment was performed 24 after each aPD-L1 administration, and mice were sacrificed 24 after the final treatment. | F = 4 , PRF = 1 , PL = , and PNP = and . Exposure time: 1 on and 1 off for 10 . Transducer details: phased focus. | The exposure enhanced perfusion substantially, whereas the reduced blood perfusion. There was a significant increase in CD8+ T cells for the exposure compared to the untreated tumours and US+CA treatment at . Moreover, US+CA+aPD-L1 had significantly better therapeutic effect and more CD8+ T cells than US+CA and aPD-L1 only. Additionally, the US+CA+aPD-L1 boosted IFN- and granzyme B secretion. |
| Lin et al. [36] | Mice with SW1990 tumours were randomised into (1) untreated and (2) US+CA groups. MBs from Bracco were used as CAs. Tumours were treated 5 days per week. The CAs were slowly injected via the tail vein. | F = 1 , PRF = 1 , PL = , and I = /2. Exposure time: 2 five times. Transducer details: not specified. | It was demonstrated that the induction of vessel normalisation by US+CA mainly relied on shifting TAM polarisation from M2-type to M1-type. |
| Zhang et al. [37] | For the VEGF expression experiment, RM-1 cells were divided into the following groups: (1) US, (2) CA, (3) US+CA, and (4) untreated. The CAs were SonoVue MBs. To detect DC and T lymphocyte phenotype, RM-1 cells either treated with US+CA or untreated were co-cultured with DCs and T lymphocytes. | F = 800 , PRF = 1 , PL = , and ISATA 6 = 360 /2. Exposure time: 30 . Transducer details: cylindrical probe with a diameter of 13 . | VEGF expression was significantly decreased after US+CA treatment compared to the other groups. There was a significant increase in CD11c+ DCs and CD8a+ T cells in the US+CA group compared to the untreated group. |
| Tan et al. [49] | Spleens of LLC tumour-bearing mice were in the first study divided into two groups: (1) US+CA and (2) untreated. The CAs were Sonazoid MBs. Diluted CA of was injected at the first 100 , per 100 for three times, and per 50 at the rest of the treatment. Tumours and spleens were excised 24 after treatment. In the second study, mice were divided into the following groups: (1) US+CA, (2) aPD-L1, (3) US+CA+aPD-L1, and (4) untreated. Spleens were treated once every 3 days for a total of 3 times with US+CA, and aPD-L1 was injected intraperitoneally on the following day. Spleens and tumours were excised on day 12. | F = 5 , PRF = 500 , PL = , PNP = , and . Exposure time: transmitting and intermittent time of , and total duration 600 . Transducer details: linear array probe. | In the first study, the results showed a significant reduction in splenic CECs and an increase in CD8+ T cells when treated with US+CA compared to untreated spleens. There was no substantial difference in CD11b+Gr1+ cells (MDSCs), CD11b+CD11c+ cells (DCs), CD11b+F4/80+ cells (macrophages), or B220+ cells (B cells). In the second study, tumour growth was only inhibited when US+CA was combined with aPD-L1. The US+CA+aPD-L1 treatment demonstrated a significant increase in number of IFN--producing CD8+ T cells and CD4+ T cells. |
3. Cavitation-Enhanced Checkpoint Inhibitor Therapy
3.1. Improved Delivery
3.2. Favourable Tumour Immune Microenvironment
3.3. Reduced Adverse Effects
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| APC | Antigen-presenting cell |
| aPD-L1 | Anti-programmed death ligand 1 |
| aPD-1 | Anti-programmed cell death protein 1 |
| CA | Cavitation agent |
| CEC | CD71+ erythroid progenitor cell |
| CTL | Cytotoxic T lymphocyte |
| CTLA-4 | Cytotoxic T lymphocyte-associated protein 4 |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cell |
| HSP | Heat shock protein |
| HMGB1 | High-mobility-group box 1 |
| HPF | High power field |
| ICI | Immune checkpoint inhibitor |
| irAE | Immune-related adverse effect |
| IFN- | Interferon gamma |
| IL | Interleukin |
| MHC | Major histocompatibility complex |
| MB | Microbubble |
| MDSC | Myeloid-derived suppressor cell |
| NK | Natural killer |
| OVA | Ovalbumin |
| PNP | Peak negativepressure |
| PD-L1 | Programmed death ligand 1 |
| PD-1 | Programmed cell death protein 1 |
| Treg | Regulatory T |
| TNF- | Transforming growth factor beta |
| TAA | Tumour-associated antigen |
| TAM | Tumour-associated macrophage |
| TME | Tumour microenvironment |
| TNF- | Tumour necrosis factor alpha |
| TSA | Tumour-specific antigen |
| US | Ultrasound |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Virchow, R. An address on the value of pathological experiments. Br. Med. J. 1881, 2, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, P. Ueber den Jetzigen Stand der Karzinomforschung. 1909, Volume 5, pp. 117–164. Available online: https://www.pei.de/SharedDocs/Downloads/DE/institut/veroeffentlichungen-von-paul-ehrlich/1906-1914/1909-karzinomforschung.pdf?__blob=publicationFile&v=2 (accessed on 10 June 2023).
- Zamarron, B.F.; Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 2011, 7, 651. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savina, A.; Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 2007, 219, 143–156. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Garrido, F.; Cabrera, T.; Concha, A.; Glew, S.; Ruiz-Cabello, F.; Stern, P.L. Natural history of HLA expression during tumour development. Immunol. Today 1993, 14, 491–499. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef]
- Lisiecka, U.; Kostro, K. Mechanisms of tumour escape from immune surveillance. J. Vet. Res. 2016, 60, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Weeks, A.; Yuzhalin, A.E. Cancer extracellular matrix proteins regulate tumour immunity. Cancers 2020, 12, 3331. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J. Clin. Oncol. 2017, 35, 1803. [Google Scholar] [CrossRef]
- Nemunaitis, J. GVAX (GMCSF gene modified tumor vaccine) in advanced stage non small cell lung cancer. J. Control. Release 2003, 91, 225–231. [Google Scholar] [CrossRef]
- Nemunaitis, J. Vaccines in cancer: GVAX®, a GM-CSF gene vaccine. Expert Rev. Vaccines 2005, 4, 259–274. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 blockade in mismatch repair–deficient, locally advanced rectal cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.I.; Ho, A.Y.; McArthur, H.L. Combined radiation therapy and immune checkpoint blockade therapy for breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Nagata, H.; Crosby, E.J.; Inoue, Y.; Kaneko, K.; Liu, C.X.; Yang, X.; Wang, T.; Acharya, C.R.; Agarwal, P.; et al. Combination of ultrasound-based mechanical disruption of tumor with immune checkpoint blockade modifies tumor microenvironment and augments systemic antitumor immunity. J. Immunother. Cancer 2022, 10, e003717. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wang, Z.B.; Lu, P.; Xu, Z.L.; Chen, W.Z.; Zhu, H.; Jin, C.B. Activated anti-tumor immunity in cancer patients after high intensity focused ultrasound ablation. Ultrasound Med. Biol. 2004, 30, 1217–1222. [Google Scholar] [CrossRef]
- Xia, J.Z.; Xie, F.L.; Ran, L.F.; Xie, X.P.; Fan, Y.M.; Wu, F. High-intensity focused ultrasound tumor ablation activates autologous tumor-specific cytotoxic T lymphocytes. Ultrasound Med. Biol. 2012, 38, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.M.; Li, H.; Yang, M.; Zha, H.; Sun, H.; Li, X.R.; Li, A.F.; Gu, Y.; Duan, L.; Luo, J.Y.; et al. High intensity focused ultrasound enhances anti-tumor immunity by inhibiting the negative regulatory effect of miR-134 on CD86 in a murine melanoma model. Oncotarget 2015, 6, 37626. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, X.Y.; Liu, Y.; Sankin, G.N.; Pua, E.C.; Morse, M.A.; Lyerly, H.K.; Clay, T.M.; Zhong, P. Investigation of HIFU-induced anti-tumor immunity in a murine tumor model. J. Transl. Med. 2007, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, M.; Eikelenboom, D.; den Brok, M.H.; Heerschap, A.; Fütterer, J.J.; Adema, G.J. Mechanical high-intensity focused ultrasound destruction of soft tissue: Working mechanisms and physiologic effects. Ultrasound Med. Biol. 2015, 41, 1500–1517. [Google Scholar] [CrossRef]
- Stride, E.; Coussios, C. Nucleation, mapping and control of cavitation for drug delivery. Nat. Rev. Phys. 2019, 1, 495–509. [Google Scholar] [CrossRef]
- Kooiman, K.; Roovers, S.; Langeveld, S.A.; Kleven, R.T.; Dewitte, H.; O’Reilly, M.A.; Escoffre, J.M.; Bouakaz, A.; Verweij, M.D.; Hynynen, K.; et al. Ultrasound-responsive cavitation nuclei for therapy and drug delivery. Ultrasound Med. Biol. 2020, 46, 1296–1325. [Google Scholar] [CrossRef]
- Li, N.; Tang, J.; Yang, J.; Zhu, B.; Wang, X.; Luo, Y.; Yang, H.; Jang, F.; Zou, J.; Liu, Z.; et al. Tumor perfusion enhancement by ultrasound stimulated microbubbles potentiates PD-L1 blockade of MC38 colon cancer in mice. Cancer Lett. 2021, 498, 121–129. [Google Scholar] [CrossRef]
- Bulner, S.; Prodeus, A.; Gariepy, J.; Hynynen, K.; Goertz, D.E. Enhancing checkpoint inhibitor therapy with ultrasound stimulated microbubbles. Ultrasound Med. Biol. 2019, 45, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.J.; Gade, T.; Soulen, M.C.; Pickup, S.; Sehgal, C.M. Antivascular ultrasound therapy: Magnetic resonance imaging validation and activation of the immune response in murine melanoma. J. Ultrasound Med. 2015, 34, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Du, Y.; Hao, J.; Wu, R.; Du, L. UTMD inhibits pancreatic cancer growth and metastasis by inducing macrophage polarization and vessel normalization. Biomed. Pharmacother. 2023, 160, 114322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shou, W.D.; Xu, Y.J.; Bai, W.K.; Hu, B. Low-frequency ultrasound-induced VEGF suppression and synergy with dendritic cell-mediated anti-tumor immunity in murine prostate cancer cells in vitro. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Gallucci, S.; Matzinger, P. Danger signals: SOS to the immune system. Curr. Opin. Immunol. 2001, 13, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Hsieh, H.Y.; Lu, L.A.; Kang, C.W.; Wu, M.F.; Lin, C.Y. Low-pressure pulsed focused ultrasound with microbubbles promotes an anticancer immunological response. J. Transl. Med. 2012, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Joiner, J.B.; Kren, N.P.; Durham, P.G.; McRee, A.J.; Dayton, P.A.; Pylayeva-Gupta, Y. Low-Intensity Focused Ultrasound Produces Immune Response in Pancreatic Cancer. Ultrasound Med. Biol. 2022, 48, 2344–2353. [Google Scholar] [CrossRef]
- Hu, J.; He, J.; Wang, Y.; Zhao, Y.; Fang, K.; Dong, Y.; Chen, Y.; Zhang, Y.; Zhang, C.; Wang, H.; et al. Ultrasound combined with nanobubbles promotes systemic anticancer immunity and augments anti-PD1 efficacy. J. Immunother. Cancer 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Cao, Y.; Liu, Y.; Zhou, Y.; He, H.; Tang, R.; Wan, L.; Wang, C.; Xiong, X.; Zhong, L.; et al. Low-intensity focused ultrasound targeted microbubble destruction reduces tumor blood supply and sensitizes anti-PD-L1 immunotherapy. Front. Bioeng. Biotechnol. 2023, 11, 1173381. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.Y.; Lei, J.; Sun, Y.; Yan, F.; Chen, B.; Zhang, L.; Lu, Z.; Cao, R.; Lin, Y.Y.; Wang, C.C.; et al. Induction of enhanced immunogenic cell death through ultrasound-controlled release of doxorubicin by liposome-microbubble complexes. Oncoimmunology 2018, 7, e1446720. [Google Scholar] [CrossRef]
- Bianchi, M.E.; Crippa, M.P.; Manfredi, A.A.; Mezzapelle, R.; Rovere Querini, P.; Venereau, E. High-mobility group box 1 protein orchestrates responses to tissue damage via inflammation, innate and adaptive immunity, and tissue repair. Immunol. Rev. 2017, 280, 74–82. [Google Scholar] [CrossRef] [Green Version]
- van der Woude, L.L.; Gorris, M.A.; Halilovic, A.; Figdor, C.G.; de Vries, I.J.M. Migrating into the tumor: A roadmap for T cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Tesniere, A.; Schlemmer, F.; Michaud, M.; Senovilla, L.; Zitvogel, L.; Kroemer, G. Immunogenic cell death modalities and their impact on cancer treatment. Apoptosis 2009, 14, 364–375. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, S.; Zhao, Y.; Li, P.; Liu, J.; Li, P.; Tan, K.; Xie, F. Disruption of tumor neovasculature by microbubble enhanced ultrasound: A potential new physical therapy of anti-angiogenesis. Ultrasound Med. Biol. 2012, 38, 253–261. [Google Scholar] [CrossRef]
- Tan, X.; Yi, C.; Zhang, Y.; Tang, N.; Xu, Y.; Liu, Z. Ultrasound-targeted microbubble destruction alleviates immunosuppression induced by CD71+ erythroid progenitor cells and promotes PDL-1 blockade immunotherapy in the lewis lung cancer model. Front. Oncol. 2021, 11, 768222. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Brekken, R.A. Direct and indirect regulation of the tumor immune microenvironment by VEGF. J. Leukoc. Biol. 2022, 111, 1269–1286. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qiao, Y.D.; Li, X.; Xu, J.L.; Ye, Q.J.; Jiang, N.; Zhang, H.; Wu, X.Y. Intratumoral CD45+ CD71+ erythroid cells induce immune tolerance and predict tumor recurrence in hepatocellular carcinoma. Cancer Lett. 2021, 499, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Justyniarska, M.; Nowis, D.; Golab, J. Tumor immune evasion induced by dysregulation of erythroid progenitor cells development. Cancers 2021, 13, 870. [Google Scholar] [CrossRef]
- Sano, Y.; Yoshida, T.; Choo, M.K.; Jiménez-Andrade, Y.; Hill, K.R.; Georgopoulos, K.; Park, J.M. Multiorgan signaling mobilizes tumor-associated erythroid cells expressing immune checkpoint molecules. Mol. Cancer Res. 2021, 19, 507–515. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Chowdhury, S.M.; Abou-Elkacem, L.; Lee, T.; Dahl, J.; Lutz, A.M. Ultrasound and microbubble mediated therapeutic delivery: Underlying mechanisms and future outlook. J. Control. Release 2020, 326, 75–90. [Google Scholar] [CrossRef]
- Grundy, M.; Bau, L.; Hill, C.; Paverd, C.; Mannaris, C.; Kwan, J.; Crake, C.; Coviello, C.; Coussios, C.; Carlisle, R. Improved therapeutic antibody delivery to xenograft tumors using cavitation nucleated by gas-entrapping nanoparticles. Nanomedicine 2020, 16, 37–50. [Google Scholar] [CrossRef]
- Kim, D.; Lee, S.S.; Moon, H.; Park, S.Y.; Lee, H.J. PD-L1 targeting immune-microbubble complex enhances therapeutic index in murine colon cancer models. Pharmaceuticals 2020, 14, 6. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K.; et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint BlockadeImmune Signatures of Response to Checkpoint Blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Macy, E. Immune-related adverse drug reactions and immunologically mediated drug hypersensitivity. Immunol. Allergy Clin. 2020, 40, 635–647. [Google Scholar] [CrossRef]
- Benoot, T.; Piccioni, E.; De Ridder, K.; Goyvaerts, C. TNFα and immune checkpoint inhibition: Friend or foe for lung cancer? Int. J. Mol. Sci. 2021, 22, 8691. [Google Scholar] [CrossRef] [PubMed]
- Mortezaee, K.; Majidpoor, J. Checkpoint inhibitor/interleukin-based combination therapy of cancer. Cancer Med. 2022, 11, 2934–2943. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Hwang, J.H.; Tu, J.; Brayman, A.A.; Matula, T.J.; Crum, L.A. Correlation between inertial cavitation dose and endothelial cell damage in vivo. Ultrasound Med. Biol. 2006, 32, 1611–1619. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Matsuura, N.; Koonar, E.; Zhu, S.; Leung, B.; Seo, M.; Sivapalan, N.; Goertz, D. Inducing antivascular effects in tumors with ultrasound stimulated micron-sized bubbles. In Proceedings of the 2015 IEEE International Ultrasonics Symposium (IUS), Taipei, Taiwan, 21–24 October 2015; pp. 1–4. [Google Scholar]
- Ho, Y.J.; Li, J.P.; Fan, C.H.; Liu, H.L.; Yeh, C.K. Ultrasound in tumor immunotherapy: Current status and future developments. J. Control. Release 2020, 323, 12–23. [Google Scholar] [CrossRef]
- Martinez, P.; Bottenus, N.; Borden, M. Cavitation characterization of size-isolated microbubbles in a vessel phantom using focused ultrasound. Pharmaceutics 2022, 14, 1925. [Google Scholar] [CrossRef]
- Gray, M.D.; Elbes, D.; Paverd, C.; Lyka, E.; Coviello, C.M.; Cleveland, R.O.; Coussios, C.C. Dual-array passive acoustic mapping for cavitation imaging with enhanced 2-D resolution. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2020, 68, 647–663. [Google Scholar] [CrossRef]
- Choi, J.J.; Carlisle, R.C.; Coviello, C.; Seymour, L.; Coussios, C.C. Non-invasive and real-time passive acoustic mapping of ultrasound-mediated drug delivery. Phys. Med. Biol. 2014, 59, 4861. [Google Scholar] [CrossRef]
- Graham, S.M.; Carlisle, R.; Choi, J.J.; Stevenson, M.; Shah, A.R.; Myers, R.S.; Fisher, K.; Peregrino, M.B.; Seymour, L.; Coussios, C.C. Inertial cavitation to non-invasively trigger and monitor intratumoral release of drug from intravenously delivered liposomes. J. Control. Release 2014, 178, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Yasui, K.; Tuziuti, T.; Lee, J.; Kozuka, T.; Towata, A.; Iida, Y. Numerical simulations of acoustic cavitation noise with the temporal fluctuation in the number of bubbles. Ultrason. Sonochem. 2010, 17, 460–472. [Google Scholar] [CrossRef]
- Hou, G.Y.; Marquet, F.; Wang, S.; Konofagou, E.E. Multi-parametric monitoring and assessment of high-intensity focused ultrasound (HIFU) boiling by harmonic motion imaging for focused ultrasound (HMIFU): An ex vivo feasibility study. Phys. Med. Biol. 2014, 59, 1121. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maardalen, M.; Carlisle, R.; Coussios, C. Cavitation-Mediated Immunomodulation and Its Use with Checkpoint Inhibitors. Pharmaceutics 2023, 15, 2110. https://doi.org/10.3390/pharmaceutics15082110
Maardalen M, Carlisle R, Coussios C. Cavitation-Mediated Immunomodulation and Its Use with Checkpoint Inhibitors. Pharmaceutics. 2023; 15(8):2110. https://doi.org/10.3390/pharmaceutics15082110
Chicago/Turabian StyleMaardalen, Matilde, Robert Carlisle, and Constantin Coussios. 2023. "Cavitation-Mediated Immunomodulation and Its Use with Checkpoint Inhibitors" Pharmaceutics 15, no. 8: 2110. https://doi.org/10.3390/pharmaceutics15082110
APA StyleMaardalen, M., Carlisle, R., & Coussios, C. (2023). Cavitation-Mediated Immunomodulation and Its Use with Checkpoint Inhibitors. Pharmaceutics, 15(8), 2110. https://doi.org/10.3390/pharmaceutics15082110