
Non-Invasive Vaccines: Challenges in Formulation and Vaccine Adjuvants



Abstract
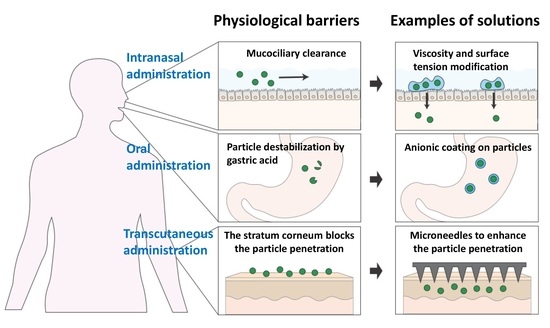
:
1. Introduction
2. Types of Vaccines and Their Formulation
2.1. Live Attenuated Vaccines
2.2. Inactivated Vaccines
2.3. Replicating and Non-Replicating Viral Vector Vaccines
2.4. DNA Vaccines
2.5. mRNA Vaccine
2.6. Subunit Vaccine
2.7. Adjuvants
2.8. Future Prospective
3. Administration of Non-Invasive Vaccines
3.1. Oral Administration
3.1.1. pH Sensitivity
3.1.2. Mucoadhesive Interaction
3.1.3. Intestinal Permeability
3.2. Intranasal Administration
3.2.1. Liquid Formulation
3.2.2. Powder Formulation
3.3. Transcutaneous Administration
3.3.1. Physical Delivery Systems
3.3.2. Chemical Enhancers
3.4. Adjuvants
3.5. Future Perspectives
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lloyd, J.; Lydon, P.; Ouhichi, R.; Zaffran, M. Reducing the loss of vaccines from accidental freezing in the cold chain: The experience of continuous temperature monitoring in Tunisia. Vaccine 2015, 33, 902–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criscuolo, E.; Caputo, V.; Diotti, R.A.; Sautto, G.A.; Kirchenbaum, G.A.; Clementi, N. Alternative methods of vaccine delivery: An overview of edible and intradermal vaccines. J. Immunol. Res. 2019, 2019, 8303648. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. COVID-19 Vaccination: Supply and Logistics Guidance. Available online: https://www.who.int/publications/i/item/who-2019-ncov-vaccine-deployment-logistics-2021-1 (accessed on 13 January 2023).
- Crommelin, D.J.A.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the cold reality of mRNA vaccine stability. J. Pharm. Sci. 2021, 110, 997–1001. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Vaccine Administration. Available online: https://www.cdc.gov/vaccines/hcp/acip-recs/general-recs/administration.html (accessed on 5 February 2023).
- Deisenhammer, S.; Radon, K.; Nowak, D.; Reichert, J. Needlestick injuries during medical training. J. Hosp. Infect. 2006, 63, 263–267. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, H.K. Delivery routes for COVID-19 vaccines. Vaccines 2021, 9, 524. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Martinez-Paniagua, M.; Rezvan, A.; Sefat, S.R.; Fathi, M.; Singh, S.; Biswas, S.; Pourpak, M.; Yee, C.; Liu, X. Single-dose intranasal vaccination elicits systemic and mucosal immunity against SARS-CoV-2. iScience 2021, 24, 103037. [Google Scholar] [CrossRef]
- Ma, Y.; Tao, W.; Krebs, S.J.; Sutton, W.F.; Haigwood, N.L.; Gill, H.S. Vaccine delivery to the oral cavity using coated microneedles induces systemic and mucosal immunity. Pharm. Res. 2014, 31, 2393–2403. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.A. Louis Pasteur, the father of immunology? Front. Immunol. 2012, 3, 68. [Google Scholar] [CrossRef] [Green Version]
- Trovato, M.; Sartorius, R.; D’Apice, L.; Manco, R.; De Berardinis, P. Viral emerging diseases: Challenges in developing vaccination strategies. Front. Immunol. 2020, 11, 2130. [Google Scholar] [CrossRef]
- Bellamkonda, N.; Lambe, U.P.; Sawant, S.; Nandi, S.S.; Chakraborty, C.; Shukla, D. Immune Response to SARS-CoV-2 Vaccines. Biomedicines 2022, 10, 1464. [Google Scholar] [CrossRef]
- Kutzler, M.A.; Weiner, D.B. DNA vaccines: Ready for prime time? Nat. Rev. Genet. 2008, 9, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA vaccines for infectious diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [Green Version]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Robert-Guroff, M. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 2007, 18, 546–556. [Google Scholar] [CrossRef]
- Liu, M.A. DNA vaccines: A review. J. Intern. Med. 2003, 253, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Berger, A. Th1 and Th2 responses: What are they? BMJ 2000, 321, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagawa, Y.; Iwabuchi, K.; Onoé, K. Co-operative action of interleukin-10 and interferon-γ to regulate dendritic cell functions. Immunology 2009, 127, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Shirouzu, T.; Nakata, K.; Yoshimura, N.; Ushigome, H. The role of major histocompatibility complex in organ transplantation-donor specific anti-major histocompatibility complex antibodies analysis goes to the next stage. Int. J. Mol. Sci. 2019, 20, 4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritz, S.A. The immune system in health and disease. In Clinical Immunology; Elsevier: Amsterdam, The Netherlands, 2004; Volume 6, pp. 393–416. [Google Scholar]
- Ebert, D. Experimental evolution of parasites. Science 1998, 282, 1432–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajra, D.; Datey, A.; Chakravortty, D. Attenuation Methods for Live Vaccines. Methods Mol. Biol. 2021, 2183, 331–356. [Google Scholar]
- Sutherland, I.A.; Shiels, B.R.; Jackson, L.; Brown, D.J.; Brown, C.G.D.; Preston, P.M. Theileria annulata: Altered Gene Expression and Clonal Selection during Continuousin VitroCulture. Exp. Parasitol. 1996, 83, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Pavan, O.H.; Boucias, D.G.; Pendland, J.C. The effects of serial passage of a nucleopolyhedrosis virus through an alternate host system. Entomophaga 1981, 26, 99–108. [Google Scholar] [CrossRef]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, B.; Koldijk, M.; Schuitemaker, H. Inactivated viral vaccines. In Vaccine Analysis: Strategies, Principles, and Control; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Lauring, A.S.; Jones, J.O.; Andino, R. Rationalizing the development of live attenuated virus vaccines. Nat. Biotechnol. 2010, 28, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.C.; Shaw, J.; Campagnoli, R.; Jorba, J.; Vincent, A.; Quay, J.; Kew, O. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 2006, 80, 3259–3272. [Google Scholar] [CrossRef] [Green Version]
- Delrue, I.; Verzele, D.; Madder, A.; Nauwynck, H.J. Inactivated virus vaccines from chemistry to prophylaxis: Merits, risks and challenges. Expert Rev. Vaccines 2012, 11, 695–719. [Google Scholar] [CrossRef] [Green Version]
- Food and Drug Administration. Emergency Use Authorization. Available online: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization (accessed on 25 April 2023).
- Sharma, R.; Tiwari, S.; Dixit, A. Covaxin: An overview of its immunogenicity and safety trials in India. Bioinformation 2021, 17, 840. [Google Scholar]
- Tanriover, M.D.; Doğanay, H.L.; Akova, M.; Güner, H.R.; Azap, A.; Akhan, S.; Köse, Ş.; Erdinç, F.Ş.; Akalın, E.H.; Tabak, Ö.F. Efficacy and safety of an inactivated whole-virion SARS-CoV-2 vaccine (CoronaVac): Interim results of a double-blind, randomised, placebo-controlled, phase 3 trial in Turkey. Lancet 2021, 398, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Tregoning, J.S.; Brown, E.S.; Cheeseman, H.M.; Flight, K.E.; Higham, S.L.; Lemm, N.M.; Pierce, B.F.; Stirling, D.C.; Wang, Z.; Pollock, K.M. Vaccines for COVID-19. Clin. Exp. Immunol. 2020, 202, 162–192. [Google Scholar] [CrossRef]
- Sánchez-Ramón, S.; Conejero, L.; Netea, M.G.; Sancho, D.; Palomares, Ó.; Subiza, J.L. Trained immunity-based vaccines: A new paradigm for the development of broad-spectrum anti-infectious formulations. Front. Immunol. 2018, 9, 2936. [Google Scholar] [CrossRef] [PubMed]
- Travieso, T.; Li, J.; Mahesh, S.; Mello, J.D.F.R.E.; Blasi, M. The use of viral vectors in vaccine development. NPJ Vaccines 2022, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Bezbaruah, R.; Athalye, M.; Parikh, P.K.; Chhipa, A.S.; Patel, S.; Apostolopoulos, V. Replicating viral vector-based vaccines for COVID-19: Potential avenue in vaccination arena. Viruses 2022, 14, 759. [Google Scholar] [CrossRef]
- Sm Wold, W.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Bailey, M.; Hensley, L.; Asiedu, C.; Geisbert, J.; Stanley, D.; Honko, A.; Johnson, J.; Mulangu, S.; Pau, M.G. Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J. Virol. 2011, 85, 4222–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, D.R.M.; Michelini, Z.; Bona, R.; Blasi, M.; Filati, P.; Leone, P.; Rossi, A.; Franco, M.; Cara, A. Integrase-defective lentiviral-vector-based vaccine: A new vector for induction of T cell immunity. Expert Opin. Biol. Ther. 2011, 11, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Schakowski, F.; Gorschlüter, M.; Junghans, C.; Schroff, M.; Buttgereit, P.; Ziske, C.; Schöttker, B.; König-Merediz, S.A.; Sauerbruch, T.; Wittig, B. A novel minimal-size vector (MIDGE) improves transgene expression in colon carcinoma cells and avoids transfection of undesired DNA. Mol. Ther. 2001, 3, 793–800. [Google Scholar] [CrossRef]
- Lundstrom, K. Self-replicating RNA viruses for vaccine development against infectious diseases and cancer. Vaccines 2021, 9, 1187. [Google Scholar] [CrossRef] [PubMed]
- Shafaati, M.; Saidijam, M.; Soleimani, M.; Hazrati, F.; Mirzaei, R.; Amirheidari, B.; Tanzadehpanah, H.; Karampoor, S.; Kazemi, S.; Yavari, B. A brief review on DNA vaccines in the era of COVID-19. Future Virol. 2022, 17, 49–66. [Google Scholar] [CrossRef] [PubMed]
- Carnes, A.E.; Hodgson, C.P.; Williams, J.A. Inducible Escherichia coli fermentation for increased plasmid DNA production. Biotechnol. Appl. Biochem. 2006, 45, 155–166. [Google Scholar] [PubMed]
- Tubulekas, I.; Berglund, P.; Fleeton, M.; Liljeström, P. Alphavirus expression vectors and their use as recombinant vaccines: A minireview. Gene 1997, 190, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Ying, H.; Restifo, N.P. DNA and RNA-based vaccines: Principles, progress and prospects. Vaccine 1999, 18, 765–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, C.O.; Fanslau, L.; Bistrovic Popov, A.; Tyagi, P.; Fruk, L. Biopolymer-based carriers for DNA vaccine design. Angew. Chem. Int. Ed. 2021, 60, 13225–13243. [Google Scholar] [CrossRef]
- Sasaki, S.; Takeshita, F.; Xin, K.-Q.; Ishii, N.; Okuda, K. Adjuvant formulations and delivery systems for DNA vaccines. Methods 2003, 31, 243–254. [Google Scholar] [CrossRef]
- Almeida, A.M.; Queiroz, J.A.; Sousa, F.; Sousa, Â. Minicircle DNA: The future for DNA-based vectors? Trends Biotechnol. 2020, 38, 1047–1051. [Google Scholar] [CrossRef]
- Eusébio, D.; Almeida, A.M.; Alves, J.M.; Maia, C.J.; Queiroz, J.A.; Sousa, F.; Sousa, Â. The performance of minicircle DNA versus parental plasmid in p53 gene delivery into HPV-18-Infected cervical cancer cells. Nucleic Acid Ther. 2021, 31, 82–91. [Google Scholar] [CrossRef]
- Arévalo-Soliz, L.M.; Hardee, C.L.; Fogg, J.M.; Corman, N.R.; Noorbakhsh, C.; Zechiedrich, L. Improving therapeutic potential of non-viral minimized DNA vectors. Cell Gene Ther. Insights 2020, 6, 1489. [Google Scholar] [CrossRef] [PubMed]
- Hardee, C.L.; Arévalo-Soliz, L.M.; Hornstein, B.D.; Zechiedrich, L. Advances in non-viral DNA vectors for gene therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef] [Green Version]
- Schakowski, F.; Gorschlüter, M.; Buttgereit, P.; Märten, A.; Lilienfeld-Toal, M.V.; Junghans, C.; Schroff, M.; König-Merediz, S.A.; Ziske, C.; Strehl, J.; et al. Minimal Size MIDGE Vectors Improve Transgene Expression In Vivo. In Vivo 2007, 21, 17. [Google Scholar]
- Wang, H.-S.; Chen, Z.-J.; Zhang, G.; Ou, X.-L.; Yang, X.-L.; Wong, C.K.C.; Giesy, J.P.; Du, J.; Chen, S.-Y. A Novel Micro-Linear Vector for In Vitro and In Vivo Gene Delivery and Its Application for EBV Positive Tumors. PLoS ONE 2012, 7, e47159. [Google Scholar] [CrossRef] [Green Version]
- Karda, R.; Counsell, J.R.; Karbowniczek, K.; Caproni, L.J.; Tite, J.P.; Waddington, S.N. Production of lentiviral vectors using novel, enzymatically produced, linear DNA. Gene Ther. 2019, 26, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Scott, V.L.; Patel, A.; Villarreal, D.O.; Hensley, S.E.; Ragwan, E.; Yan, J.; Sardesai, N.Y.; Rothwell, P.J.; Extance, J.P.; Caproni, L.J. Novel synthetic plasmid and Doggybone™ DNA vaccines induce neutralizing antibodies and provide protection from lethal influenza challenge in mice. Hum. Vaccines Immunother. 2015, 11, 1972–1982. [Google Scholar] [CrossRef]
- Grunwald, T.; Ulbert, S. Improvement of DNA vaccination by adjuvants and sophisticated delivery devices: Vaccine-platforms for the battle against infectious diseases. Clin. Exp. Vaccine Res. 2015, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chellasamy, G.; Kiriyanthan, R.M.; Govindaraju, S.; Radha, A.; Yun, K. Recent trends in the development of vaccine technologies to combat pandemic outbreaks and challenges. In Pandemic Outbreaks in the 21st Century; Elsevier: Amsterdam, The Netherlands, 2021; pp. 235–243. [Google Scholar]
- Tews, B.A.; Meyers, G. Self-Replicating RNA vaccines. Methods Mol. Biol. 2017, 1499, 15–35. [Google Scholar] [PubMed]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef]
- Kim, S.C.; Sekhon, S.S.; Shin, W.-R.; Ahn, G.; Cho, B.-K.; Ahn, J.-Y.; Kim, Y.-H. Modifications of mRNA vaccine structural elements for improving mRNA stability and translation efficiency. Mol. Cell. Toxicol. 2022, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Perri, S.; Greer, C.E.; Thudium, K.; Doe, B.; Legg, H.; Liu, H.; Romero, R.E.; Tang, Z.; Bin, Q.; Dubensky, T.W., Jr.; et al. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J. Virol. 2003, 77, 10394–10403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Caillaud, M.-C. Anionic lipids: A pipeline connecting key players of plant cell division. Front. Plant Sci. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Zhang, H.; Butowska, K.; Swingle, K.L.; Alameh, M.-G.; Weissman, D.; Mitchell, M.J. An ionizable lipid toolbox for RNA delivery. Nat. Commun. 2021, 12, 7233. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ma, Y.; Zhang, J.; Kuo, J.C.-T.; Zhang, Z.; Xie, H.; Zhu, J.; Liu, T. Modification of lipid-based nanoparticles: An efficient delivery system for nucleic acid-based immunotherapy. Molecules 2022, 27, 1943. [Google Scholar] [CrossRef]
- Lynn, D.M.; Langer, R. Degradable poly (β-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar] [CrossRef]
- Ko, Y.T.; Kale, A.; Hartner, W.C.; Papahadjopoulos-Sternberg, B.; Torchilin, V.P. Self-assembling micelle-like nanoparticles based on phospholipid–polyethyleneimine conjugates for systemic gene delivery. J. Control. Release 2009, 133, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Dey, B. Chitosan microspheres in novel drug delivery systems. Indian J. Pharm. Sci. 2011, 73, 355. [Google Scholar]
- Ramirez, J.E.V.; Sharpe, L.A.; Peppas, N.A. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev. 2017, 114, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Cecil, R.L.F.; Goldman, L.; Schafer, A.I. Goldman’s Cecil Medicine, Expert Consult Premium Edition—Enhanced Online Features and Print, Single Volume, 24: Goldman’s Cecil Medicine; Elsevier Health Sciences: Philadelphia, PA, USA, 2012; Volume 1. [Google Scholar]
- Rich, R.R.; Fleisher, T.A.; Shearer, W.T.; Schroeder, H.W., Jr.; Frew, A.J.; Weyand, C.M. Clinical Immunology E-Book: Principles and Practice; Elsevier Health Sciences: Philadelphia, PA, USA, 2012. [Google Scholar]
- Golos, M.; Eliakim-Raz, N.; Stern, A.; Leibovici, L.; Paul, M. Conjugated pneumococcal vaccine versus polysaccharide pneumococcal vaccine for prevention of pneumonia and invasive pneumococcal disease in immunocompetent and immunocompromised adults and children. Cochrane Database Syst. Rev. 2019, 2019, CD012306. [Google Scholar] [CrossRef]
- Pichichero, M.E. Protein carriers of conjugate vaccines: Characteristics, development, and clinical trials. Hum. Vaccines Immunother. 2013, 9, 2505–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, G.; Rappuoli, R.; Ratti, G. The amino-acid sequence of two non-toxic mutants of diphtheria toxin: CRM45 and CRM197. Nucleic Acids Res. 1984, 12, 4063–4069. [Google Scholar] [CrossRef] [PubMed]
- Sahu, U.; Khare, P. Chapter 2—Evolution and development of vaccines against major human infections. In System Vaccinology; Prajapati, V., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 17–30. [Google Scholar]
- Harrison, L.H.; Mohan, N.; Kirkpatrick, P. Meningococcal group A, C, Y and W-135 conjugate vaccine. Nat. Rev. Drug Discov. 2010, 9, 429. [Google Scholar] [CrossRef]
- Ruan, M.R.; Akkoyunlu, M.; Grubb, A.; Forsgren, A. Protein D of Haemophilus influenzae. A novel bacterial surface protein with affinity for human IgD. J. Immunol. 1990, 145, 3379–3384. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Vrba, S.M.; Kirk, N.; Liang, Y.; Ly, H. Emerging concepts and technologies in vaccine development. Front. Immunol. 2020, 11, 583077. [Google Scholar] [CrossRef]
- Donaldson, B.; Al-Barwani, F.; Young, V.; Scullion, S.; Ward, V.; Young, S. Virus-like particles, a versatile subunit vaccine platform. In Subunit Vaccine Delivery; Springer: New York, NY, USA, 2015; pp. 159–180. [Google Scholar]
- Yadav, D.; Yadav, N.; Khurana, S.M. Vaccines: Present Status and Application; Elsevier: Amsterdam, The Netherlands, 2013; pp. 491–508. [Google Scholar]
- Jones, R.G.A.; Liu, Y.; Rigsby, P.; Sesardic, D. An improved method for development of toxoid vaccines and antitoxins. J. Immunol. Methods 2008, 337, 42–48. [Google Scholar] [CrossRef]
- Huisman, W.; Martina, B.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef]
- Yang, S.; Li, Y.; Dai, L.; Wang, J.; He, P.; Li, C.; Fang, X.; Wang, C.; Zhao, X.; Huang, E.; et al. Safety and immunogenicity of a recombinant tandem-repeat dimeric RBD-based protein subunit vaccine (ZF2001) against COVID-19 in adults: Two randomised, double-blind, placebo-controlled, phase 1 and 2 trials. Lancet Infect. Dis. 2021, 21, 1107–1119. [Google Scholar] [CrossRef]
- He, Y.; Li, J.; Heck, S.; Lustigman, S.; Jiang, S. Antigenic and immunogenic characterization of recombinant baculovirus-expressed severe acute respiratory syndrome coronavirus spike protein: Implication for vaccine design. J. Virol. 2006, 80, 5757–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ulitzky, L.; Silberstein, E.; Taylor, D.R.; Viscidi, R. Immunogenicity and protection efficacy of monomeric and trimeric recombinant SARS coronavirus spike protein subunit vaccine candidates. Viral Immunol. 2013, 26, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.-S.; Webby Richard, J. Traditional and new influenza vaccines. Clin. Microbiol. Rev. 2013, 26, 476–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Zheng, B.-J.; Lu, L.; Zhou, Y.; Jiang, S.; Du, L. Advancements in the development of subunit influenza vaccines. Microbes Infect. 2015, 17, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobusawa, E. Structure and function of the hemagglutinin of influenza viruses. Nihon Rinsho Jpn. J. Clin. Med. 1997, 55, 2562–2569. [Google Scholar]
- Lee, Y.; Lee, Y.S.; Cho, S.Y.; Kwon, H.-J. Chapter four—perspective of peptide vaccine composed of epitope peptide, CpG-DNA, and liposome complex without carriers. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 99, pp. 75–97. [Google Scholar]
- Van der Weken, H.; Cox, E.; Devriendt, B. Advances in oral subunit vaccine design. Vaccines 2021, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Cai, W.; Jin, F. A novel oil-in-water emulsion as a potential adjuvant for influenza vaccine: Development, characterization, stability and in vivo evaluation. Int. J. Pharm. 2014, 468, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Sharma, T.; Kumar, G.S.; Chon, B.H.; Sangwai, J.S. Thermal stability of oil-in-water Pickering emulsion in the presence of nanoparticle, surfactant, and polymer. J. Ind. Eng. Chem. 2015, 22, 324–334. [Google Scholar] [CrossRef]
- Siva, S.P.; Kow, K.-W.; Chan, C.-H.; Tang, S.Y.; Ho, Y.K. Prediction of droplet sizes for oil-in-water emulsion systems assisted by ultrasound cavitation: Transient scaling law based on dynamic breakup potential. Ultrason. Sonochem. 2019, 55, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, W.; Lenzo, J.; Holden, J.; McCullough, M.; O’Connor, A.; O’Brien-Simpson, N. The potential of calcium phosphate nanoparticles as adjuvants and vaccine delivery vehicles. Front. Mater. 2021, 8, 788373. [Google Scholar] [CrossRef]
- Ferrando, R.M.; Lay, L.; Polito, L. Gold nanoparticle-based platforms for vaccine development. Drug Discov. Today Technol. 2020, 38, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ge, J.; Miao, Q.; Zhu, R.; Wen, L.; Zeng, J.; Gao, M. Biodegradable inorganic nanoparticles for cancer theranostics: Insights into the degradation behavior. Bioconjug Chem. 2020, 31, 315–331. [Google Scholar] [CrossRef]
- Duewell, P.; Kisser, U.; Heckelsmiller, K.; Hoves, S.; Stoitzner, P.; Koernig, S.; Morelli, A.B.; Clausen, B.E.; Dauer, M.; Eigler, A. ISCOMATRIX adjuvant combines immune activation with antigen delivery to dendritic cells in vivo leading to effective cross-priming of CD8+ T cells. J. Immunol. 2011, 187, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Lövgren Bengtsson, K.; Morein, B.; Osterhaus, A.D.M.E. ISCOM technology-based Matrix M™ adjuvant: Success in future vaccines relies on formulation. Expert Rev. Vaccines 2011, 10, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Levast, B.; Awate, S.; Babiuk, L.; Mutwiri, G.; Gerdts, V.; van Drunen Littel-van den Hurk, S. Vaccine potentiation by combination adjuvants. Vaccines 2014, 2, 297–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, J.M.; Tetley, L.; Richmond, J.; Liew, F.Y.; Alexander, J. Lipid vesicle size determines the Th1 or Th2 response to entrapped antigen. J. Immunol. 1998, 161, 4000–4007. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.A.; Essa, E.A.; Elebyary, T.T.; Faheem, A.M.; Elkordy, A.A. Brief on recent application of liposomal vaccines for lower respiratory tract viral infections: From influenza to COVID-19 vaccines. Pharmaceuticals 2021, 14, 1173. [Google Scholar] [CrossRef]
- Gregoriadis, G. Liposomes and mRNA: Two technologies together create a COVID-19 vaccine. Med. Drug Discov. 2021, 12, 100104. [Google Scholar] [CrossRef]
- Mann, J.F.S.; Shakir, E.; Carter, K.C.; Mullen, A.B.; Alexander, J.; Ferro, V.A. Lipid vesicle size of an oral influenza vaccine delivery vehicle influences the Th1/Th2 bias in the immune response and protection against infection. Vaccine 2009, 27, 3643–3649. [Google Scholar] [CrossRef]
- Corthésy, B.; Bioley, G. Lipid-based particles: Versatile delivery systems for mucosal vaccination against infection. Front. Immunol. 2018, 9, 431. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Alruwaili, N.K.; Imam, S.S.; Alotaibi, N.H.; Alharbi, K.S.; Afzal, M.; Ali, R.; Alshehri, S.; Alzarea, S.I.; Elmowafy, M. Bioactive Apigenin loaded oral nano bilosomes: Formulation optimization to preclinical assessment. Saudi Pharm. J. 2021, 29, 269–279. [Google Scholar] [CrossRef]
- Adamiak, N.; Krawczyk, K.T.; Locht, C.; Kowalewicz-Kulbat, M. Archaeosomes and gas vesicles as tools for vaccine development. Front. Immunol. 2021, 12, 746235. [Google Scholar] [CrossRef]
- Krishnan, L.; Dicaire, C.J.; Patel, G.B.; Sprott, G.D. Archaeosome vaccine adjuvants induce strong humoral, cell-mediated, and memory responses: Comparison to conventional liposomes and alum. Infect. Immun. 2000, 68, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCluskie, M.J.; Deschatelets, L.; Krishnan, L. Sulfated archaeal glycolipid archaeosomes as a safe and effective vaccine adjuvant for induction of cell-mediated immunity. Hum. Vaccines Immunother. 2017, 13, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Wilschut, J. Influenza vaccines: The virosome concept. Immunol. Lett. 2009, 122, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tu, Z.; Feng, F.; Shi, H.; Chen, K.; Xu, X. Virosome, a hybrid vehicle for efficient and safe drug delivery and its emerging application in cancer treatment. Acta Pharm. 2015, 65, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patravale, V.; Dandekar, P.; Jain, R. Nanoparticulate Drug Delivery: Perspectives on the Transition from Laboratory to Market; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Asadi, K.; Gholami, A. Virosome-based nanovaccines; a promising bioinspiration and biomimetic approach for preventing viral diseases: A review. Int. J. Biol. Macromol. 2021, 182, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Bourquin, C.; Anz, D.; Zwiorek, K.; Lanz, A.-L.; Fuchs, S.; Weigel, S.; Wurzenberger, C.; von der Borch, P.; Golic, M.; Moder, S. Targeting CpG oligonucleotides to the lymph node by nanoparticles elicits efficient antitumoral immunity. J. Immunol. 2008, 181, 2990–2998. [Google Scholar] [CrossRef] [Green Version]
- Sudheesh, M.S.; Vyas, S.P.; Kohli, D.V. Nanoparticle-based immunopotentiation via tetanus toxoid-loaded gelatin and aminated gelatin nanoparticles. Drug Deliv. 2011, 18, 320–330. [Google Scholar] [CrossRef]
- Hong, S.; Choi, D.W.; Kim, H.N.; Park, C.G.; Lee, W.; Park, H.H. Protein-based nanoparticles as drug delivery systems. Pharmaceutics 2020, 12, 604. [Google Scholar] [CrossRef]
- Sahoo, N.; Sahoo, R.K.; Biswas, N.; Guha, A.; Kuotsu, K. Recent advancement of gelatin nanoparticles in drug and vaccine delivery. Int. J. Biol. Macromol. 2015, 81, 317–331. [Google Scholar] [CrossRef]
- Zhu, G.; Lynn, G.M.; Jacobson, O.; Chen, K.; Liu, Y.; Zhang, H.; Ma, Y.; Zhang, F.; Tian, R.; Ni, Q. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat. Commun. 2017, 8, 1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Xue, J.; Chen, W.; Bai, S.; Zheng, T.; He, C.; Guo, Z.; Jiang, M.; Du, G.; Sun, X. Albumin-biomineralized nanoparticles to synergize phototherapy and immunotherapy against melanoma. J. Control. Release 2020, 322, 300–311. [Google Scholar] [CrossRef]
- Hassanin, I.; Elzoghby, A. Albumin-based nanoparticles: A promising strategy to overcome cancer drug resistance. Cancer Drug Resist. 2020, 3, 930. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, Y.; Liu, S.; Pan, X.; Liu, Y.; Zhao, H.; Yin, X.; Yu, C.; Kong, W.; Zhang, Y. The effect of size, dose, and administration route on zein nanoparticle immunogenicity in BALB/c mice. Int. J. Nanomed. 2019, 14, 9917–9928. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yu, M. Development of a nanoparticle delivery system based on zein/polysaccharide complexes. J. Food Sci. 2020, 85, 4108–4117. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.-J.; Kang, S.-M. Immunology and efficacy of MF59-adjuvanted vaccines. Hum. Vaccines Immunother. 2018, 14, 3041–3045. [Google Scholar] [CrossRef] [Green Version]
- Grigoryan, L.; Lee, A.; Walls, A.C.; Lai, L.; Franco, B.; Arunachalam, P.S.; Feng, Y.; Luo, W.; Vanderheiden, A.; Floyd, K. Adjuvanting a subunit SARS-CoV-2 vaccine with clinically relevant adjuvants induces durable protection in mice. NPJ Vaccines 2022, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Lodaya, R.; Gregory, S.; Amiji, M.; O’Hagan, D. Overview of Vaccine Adjuvants. In Practical Aspects of Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2021; pp. 9–25. [Google Scholar]
- De Rosa, S.C.; Cohen, K.W.; Bonaparte, M.; Fu, B.; Garg, S.; Gerard, C.; Goepfert, P.A.; Huang, Y.; Larocque, D.; McElrath, M.J. Whole-blood cytokine secretion assay as a high-throughput alternative for assessing the cell-mediated immunity profile after two doses of an adjuvanted SARS-CoV-2 recombinant protein vaccine candidate. Clin. Transl. Immunol. 2022, 11, e1360. [Google Scholar] [CrossRef]
- Klucker, M.F.; Dalençon, F.; Probeck, P.; Haensler, J. AF03, an alternative squalene emulsion-based vaccine adjuvant prepared by a phase inversion temperature method. J. Pharm. Sci. 2012, 101, 4490–4500. [Google Scholar] [CrossRef]
- Gao, L.; Li, J.; Song, T. Poly lactic-co-glycolic acid-based nanoparticles as delivery systems for enhanced cancer immunotherapy. Front. Chem. 2022, 10, 973666. [Google Scholar] [CrossRef]
- Bennewitz, N.L.; Babensee, J.E. The effect of the physical form of poly (lactic-co-glycolic acid) carriers on the humoral immune response to co-delivered antigen. Biomaterials 2005, 26, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Broaders, K.E.; Cohen, J.A.; Beaudette, T.T.; Bachelder, E.M.; Fréchet, J.M.J. Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy. Proc. Natl. Acad. Sci. USA 2009, 106, 5497–5502. [Google Scholar] [CrossRef]
- Chen, N.; Collier, M.A.; Gallovic, M.D.; Collins, G.C.; Sanchez, C.C.; Fernandes, E.Q.; Bachelder, E.M.; Ainslie, K.M. Degradation of acetalated dextran can be broadly tuned based on cyclic acetal coverage and molecular weight. Int. J. Pharm. 2016, 512, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, C.; Matos, A.I.; Conniot, J.; Sainz, V.; Zupančič, E.; Silva, J.M.; Graca, L.; Gaspar, R.S.; Preat, V.; Florindo, H.F. Poly (lactic acid)-based particulate systems are promising tools for immune modulation. Acta Biomater. 2017, 48, 41–57. [Google Scholar] [CrossRef]
- Baek, S.-W.; Kim, J.H.; Song, D.H.; Kim, D.-S.; Park, C.G.; Han, D.K. Enhanced Mechanical Properties and Anti–Inflammation of Poly (L–Lactic Acid) by Stereocomplexes of PLLA/PDLA and Surface–Modified Magnesium Hydroxide Nanoparticles. Polymers 2022, 14, 3790. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.-S.; Xu, Y.-L.; Zou, X.-T.; Xu, Z.-R. Chitosan nanoparticles act as an adjuvant to promote both Th1 and Th2 immune responses induced by ovalbumin in mice. Mar. Drugs 2011, 9, 1038–1055. [Google Scholar] [CrossRef] [Green Version]
- Lemke, C.D.; Graham, J.B.; Geary, S.M.; Zamba, G.; Lubaroff, D.M.; Salem, A.K. Chitosan is a surprising negative modulator of cytotoxic CD8+ T cell responses elicited by adenovirus cancer vaccines. Mol. Pharm. 2011, 8, 1652–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Wang, X.; Huang, X.; Zhang, J.; Xia, N.; Zhao, Q. Calcium phosphate nanoparticles as a new generation vaccine adjuvant. Expert Rev. Vaccines 2017, 16, 895–906. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Yoshiyuki, K.; Watanabe, Y.; Sogo, Y.; Ohno, T.; Tsuji, N.M.; Ito, A. Comprehensive mechanism analysis of mesoporous-silica-nanoparticle-induced cancer immunotherapy. Adv. Healthc. Mater. 2016, 5, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Bharti, C.; Nagaich, U.; Pal, A.K.; Gulati, N. Mesoporous silica nanoparticles in target drug delivery system: A review. Int. J. Pharm. Investig. 2015, 5, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Y.; Du, J.; Li, Y.; Zhou, Y.; Fu, Q.; Zhang, J.; Wang, X.; Zhan, L. Different-sized gold nanoparticle activator/antigen increases dendritic cells accumulation in liver-draining lymph nodes and CD8+ T cell responses. ACS Nano 2016, 10, 2678–2692. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.K.; Gonon, A.; Pécheur, E.-I.; Pezet, M.; Villiers, C.; Marche, P.N. Impact of gold nanoparticles on the functions of macrophages and dendritic cells. Cells 2021, 10, 96. [Google Scholar] [CrossRef]
- Balfourier, A.; Luciani, N.; Wang, G.; Lelong, G.; Ersen, O.; Khelfa, A.; Alloyeau, D.; Gazeau, F.; Carn, F. Unexpected intracellular biodegradation and recrystallization of gold nanoparticles. Proc. Natl. Acad. Sci. USA 2020, 117, 103–113. [Google Scholar] [CrossRef]
- Nguyen, B.; Tolia, N.H. Protein-based antigen presentation platforms for nanoparticle vaccines. NPJ Vaccines 2021, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Kianfar, E. Protein nanoparticles in drug delivery: Animal protein, plant proteins and protein cages, albumin nanoparticles. J. Nanobiotechnol. 2021, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Shen, L.; Xu, L.; Yang, Y. Controlled delivery of hollow corn protein nanoparticles via non-toxic crosslinking: In vivo and drug loading study. Biomed. Microdevices 2015, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Gulati, N.; Kaul, S.; Mukherjee, S.; Nagaich, U. Protein based nanostructures for drug delivery. J. Pharm. 2018, 2018, 9285854. [Google Scholar] [CrossRef] [Green Version]
- Marasini, N.; Ghaffar, K.A.; Skwarczynski, M.; Toth, I. Chapter twelve—Liposomes as a vaccine delivery system. In Micro and Nanotechnology in Vaccine Development; Skwarczynski, M., Toth, I., Eds.; William Andrew Elsevier: Amsterdam, The Netherlands, 2017; pp. 221–239. [Google Scholar]
- Kisakova, L.A.; Apartsin, E.K.; Nizolenko, L.F.; Karpenko, L.I. Dendrimer-mediated delivery of DNA and RNA vaccines. Pharmaceutics 2023, 15, 1106. [Google Scholar] [CrossRef] [PubMed]
- Gutjahr, A.; Phelip, C.; Coolen, A.L.; Monge, C.; Boisgard, A.S.; Paul, S.; Verrier, B. Biodegradable polymeric nanoparticles-based vaccine adjuvants for lymph nodes targeting. Vaccines 2016, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Vaid, R.; Yildirim, E.; Pasquinelli, M.A.; King, M.W. Hydrolytic degradation of polylactic acid fibers as a function of pH and exposure time. Molecules 2021, 26, 7554. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.; Patel, A.A. Organic and inorganic nanoparticle vaccines for prevention of infectious diseases. Nano Express 2020, 1, 012001. [Google Scholar] [CrossRef]
- Lang, S.; Huang, X. Carbohydrate conjugates in vaccine developments. Front. Chem. 2020, 8, 284. [Google Scholar] [CrossRef]
- de Carvalho Lima, E.N.; Diaz, R.S.; Justo, J.F.; Castilho Piqueira, J.R. Advances and perspectives in the use of carbon nanotubes in vaccine development. Int. J. Nanomed. 2021, 16, 5411–5435. [Google Scholar] [CrossRef]
- Bamrungsap, S.; Zhao, Z.; Chen, T.; Wang, L.; Li, C.; Fu, T.; Tan, W. Nanotechnology in therapeutics: A focus on nanoparticles as a drug delivery system. Nanomedicine 2012, 7, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, X.; Xiang, X.; Pang, X.; Chen, S.; Zhang, Y.; Ren, E.; Zhang, L.; Liu, X.; Lv, P.; et al. A nanovaccine for antigen self-presentation and immunosuppression reversal as a personalized cancer immunotherapy strategy. Nat. Nanotechnol. 2022, 17, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Ali, N. Nanovaccine: An emerging strategy. Expert Rev. Vaccines 2021, 20, 1273–1290. [Google Scholar] [CrossRef]
- Maina, T.W.; Grego, E.A.; Boggiatto, P.M.; Sacco, R.E.; Narasimhan, B.; McGill, J.L. Applications of Nanovaccines for Disease Prevention in Cattle. Front. Bioeng. Biotechnol. 2020, 8, 608050. [Google Scholar] [CrossRef]
- Azharuddin, M.; Zhu, G.H.; Sengupta, A.; Hinkula, J.; Slater, N.K.H.; Patra, H.K. Nano toolbox in immune modulation and nanovaccines. Trends Biotechnol. 2022, 40, 1195–1212. [Google Scholar] [CrossRef]
- Kim, L.; Martinez, C.J.; Hodgson, K.A.; Trager, G.R.; Brandl, J.R.; Sandefer, E.P.; Doll, W.J.; Liebowitz, D.; Tucker, S.N. Systemic and mucosal immune responses following oral adenoviral delivery of influenza vaccine to the human intestine by radio controlled capsule. Sci. Rep. 2016, 6, 37295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudie, D.M.; Amidon, G.L.; Amidon, G.E. Physiological parameters for oral delivery and in vitro testing. Mol. Pharm. 2010, 7, 1388–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Talton, J.; Zhang, G.; Cunningham, T.; Wang, Z.; Waters, R.C.; Kirk, J.; Eppler, B.; Klinman, D.M.; Sui, Y.; et al. Large intestine–targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat. Med. 2012, 18, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Le, T.; Aguilar, B.; Mangal, J.L.; Acharya, A.P. Oral drug delivery for immunoengineering. Bioeng. Transl. Med. 2022, 7, e10243. [Google Scholar] [CrossRef] [PubMed]
- Ahadian, S.; Finbloom, J.A.; Mofidfar, M.; Diltemiz, S.E.; Nasrollahi, F.; Davoodi, E.; Hosseini, V.; Mylonaki, I.; Sangabathuni, S.; Montazerian, H.; et al. Micro and nanoscale technologies in oral drug delivery. Adv. Drug Deliv. Rev. 2020, 157, 37–62. [Google Scholar] [CrossRef]
- Coffey, J.W.; Gaiha, G.D.; Traverso, G. Oral biologic delivery: Advances toward oral subunit, DNA, and mRNA vaccines and the potential for mass vaccination during pandemics. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 517–540. [Google Scholar] [CrossRef]
- de Santa Barbara, P.; van den Brink, G.R.; Roberts, D.J. Development and differentiation of the intestinal epithelium. Cell. Mol. Life Sci. CMLS 2003, 60, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M. Role of goblet cells in intestinal barrier and mucosal imunity. J. Inflamm. Res. 2021, 14, 3171–3183. [Google Scholar] [CrossRef]
- Jung, C.; Hugot, J.-P.; Barreau, F. Peyer’s patches: The immune sensors of the intestine. Int. J. Inflamm. 2010, 2010, 823710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homayun, B.; Lin, X.; Choi, H.-J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, R.; Allais, L.; Cuvelier, C.A. Recent advances in oral vaccine development: Yeast-derived β-glucan particles. Hum. Vaccines Immunother. 2014, 10, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, J.; Mastropietro, K.P.; Omidian, H. Polymers in Oral Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2017; pp. 430–444. [Google Scholar]
- dos Santos, J.; da Silva, G.S.; Velho, M.C.; Beck, R.C. Eudragit®: A versatile family of polymers for hot melt extrusion and 3D printing processes in pharmaceutics. Pharmaceutics 2021, 13, 1424. [Google Scholar] [CrossRef] [PubMed]
- Franco, P.; De Marco, I. Eudragit: A Novel Carrier for Controlled Drug Delivery in Supercritical Antisolvent Coprecipitation. Polymers 2020, 12, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snejdrova Eva, D.M. Pharmaceutical applications of plasticized polymers. Recent Adv. Plast. 2012, 21, 23–34. [Google Scholar]
- Vidal-Romero, G.; Rocha-Pérez, V.; Zambrano-Zaragoza, M.L.; Del Real, A.; Martínez-Acevedo, L.; Galindo-Pérez, M.J.; Quintanar-Guerrero, D. Development and characterization of pH-dependent cellulose acetate phthalate nanofibers by electrospinning technique. Nanomaterials 2021, 11, 3202. [Google Scholar] [CrossRef]
- Obara, S.; Tanno, F.K.; Sarode, A. Properties and applications of hypromellose acetate succinate (HPMCAS) for solubility enhancement using melt extrusion. In Melt Extrusion: Materials, Technology and Drug Product Design; Repka, M.A., Langley, N., DiNunzio, J., Eds.; Springer: New York, NY, USA, 2013; pp. 107–121. [Google Scholar]
- Bagre, A.P.; Jain, K.; Jain, N.K. Alginate coated chitosan core shell nanoparticles for oral delivery of enoxaparin: In vitro and in vivo assessment. Int. J. Pharm. 2013, 456, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, D.A.; Langer, R.; Traverso, G. Mucus interaction to improve gastrointestinal retention and pharmacokinetics of orally administered nano-drug delivery systems. J. Nanobiotechnol. 2022, 20, 362. [Google Scholar] [CrossRef] [PubMed]
- Bernkop-Schnürch, A. Mucoadhesive systems in oral drug delivery. Drug Discov. Today Technol. 2005, 2, 83–87. [Google Scholar] [CrossRef]
- Boddupalli, B.M.; Mohammed, Z.N.; Nath, R.A.; Banji, D. Mucoadhesive drug delivery system: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.-S.; Hwang, S.-K.; Gu, M.-J.; Kim, C.-G.; Kim, S.-K.; Ju, D.-B.; Yun, C.-H.; Kim, H.-J. Mucosal vaccine delivery using mucoadhesive polymer particulate systems. Tissue Eng. Regen. Med. 2021, 18, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.; Singh, T.R.R.; Garland, M.J.; Woolfson, A.D.; Donnelly, R.F. Mucoadhesive drug delivery systems. J. Pharm. Bioallied Sci. 2011, 3, 89. [Google Scholar]
- Pérez-González, G.L.; Villarreal-Gómez, L.J.; Serrano-Medina, A.; Torres-Martínez, E.J.; Cornejo-Bravo, J.M. Mucoadhesive electrospun nanofibers for drug delivery systems: Applications of polymers and the parameters’ roles. Int. J. Nanomed. 2019, 14, 5271–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Zhang, H.; Chen, K.; Jin, M.; Vu, S.H.; Jung, S.; He, N.; Zheng, Z.; Lee, M.-S. Application of chitosan/alginate nanoparticle in oral drug delivery systems: Prospects and challenges. Drug Deliv. 2022, 29, 1142–1149. [Google Scholar] [CrossRef]
- Li, X.; Kong, X.; Shi, S.; Zheng, X.; Guo, G.; Wei, Y.; Qian, Z. Preparation of alginate coated chitosan microparticles for vaccine delivery. BMC Biotechnol. 2008, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Forier, K.; Steukers, L.; Van Vlierberghe, S.; Dubruel, P.; Braeckmans, K.; Glorieux, S.; Nauwynck, H.J. Immobilization of pseudorabies virus in porcine tracheal respiratory mucus revealed by single particle tacking. PLoS ONE 2012, 7, e51054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Oral controlled and sustained drug delivery systems: Concepts, advances, preclinical, and clinical status. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Elsevier: Amsterdam, The Netherlands, 2018; pp. 567–626. [Google Scholar]
- Cevher, E.; Taha, M.A.M.; Orlu, M.; Araman, A. Evaluation of mechanical and mucoadhesive properties of clomiphene citrate gel formulations containing carbomers and their thiolated derivatives. Drug Deliv. 2008, 15, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.K.; Wang, Y.-Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Snook, J.D.; Chesson, C.B.; Peniche, A.G.; Dann, S.M.; Paulucci, A.; Pinchuk, I.V.; Rudra, J.S. Peptide nanofiber–CaCO3 composite microparticles as adjuvant-free oral vaccine delivery vehicles. J. Mater. Chem. B 2016, 4, 1640–1649. [Google Scholar] [CrossRef]
- Dong, W.; Ye, J.; Zhou, J.; Wang, W.; Wang, H.; Zheng, X.; Yang, Y.; Xia, X.; Liu, Y. Comparative study of mucoadhesive and mucus-penetrative nanoparticles based on phospholipid complex to overcome the mucus barrier for inhaled delivery of baicalein. Acta Pharm. Sin. B 2020, 10, 1576–1585. [Google Scholar] [CrossRef]
- Lamson, N.G.; Berger, A.; Fein, K.C.; Whitehead, K.A. Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nat. Biomed. Eng. 2020, 4, 84–96. [Google Scholar] [CrossRef]
- Lai, S.K.; Wang, Y.-Y.; Cone, R.; Wirtz, D.; Hanes, J. Altering Mucus Rheology to “Solidify” Human Mucus at the Nanoscale. PLoS ONE 2009, 4, e4294. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Boylan, N.J.; Suk, J.S.; Wang, Y.-Y.; Nance, E.A.; Yang, J.-C.; McDonnell, P.J.; Cone, R.A.; Duh, E.J.; Hanes, J. Nanoparticle diffusion in, and microrheology of, the bovine vitreous ex vivo. J. Control. Release 2013, 167, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.Y.; Lai, S.K.; Suk, J.S.; Pace, A.; Cone, R.; Hanes, J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that "slip" through the human mucus barrier. Angew. Chem. Int. Ed. Engl. 2008, 47, 9726–9729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Zhang, J.; Shan, W.; Huang, Y. Developments of mucus penetrating nanoparticles. Asian J. Pharm. Sci. 2015, 10, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Ensign, L.M.; Lai, S.K.; Wang, Y.-Y.; Yang, M.; Mert, O.; Hanes, J.; Cone, R. Pretreatment of human cervicovaginal mucus with pluronic F127 enhances nanoparticle penetration without compromising mucus barrier properties to herpes simplex virus. Biomacromolecules 2014, 15, 4403–4409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dünnhaupt, S.; Kammona, O.; Waldner, C.; Kiparissides, C.; Bernkop-Schnürch, A. Nano-carrier systems: Strategies to overcome the mucus gel barrier. Eur. J. Pharm. Biopharm. 2015, 96, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 4117–4129. [Google Scholar] [CrossRef]
- Partidos, C.D. Intranasal vaccines: Forthcoming challenges. Pharm. Sci. Technol. Today 2000, 3, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cai, L.; Hufnagel, S.; Cui, Z. Intranasal vaccine: Factors to consider in research and development. Int. J. Pharm. 2021, 609, 121180. [Google Scholar] [CrossRef]
- Takaki, H.; Ichimiya, S.; Matsumoto, M.; Seya, T. Mucosal immune response in nasal-associated lymphoid tissue upon intranasal administration by adjuvants. J. Innate Immun. 2018, 10, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Scherließ, R. Nasal formulations for drug administration and characterization of nasal preparations in drug delivery. Ther. Deliv. 2020, 11, 183–191. [Google Scholar] [CrossRef]
- Ramvikas, M.; Arumugam, M.; Chakrabarti, S.R.; Jaganathan, K.S. Nasal vaccine delivery. In Micro and Nanotechnology in Vaccine Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 279–301. [Google Scholar]
- Jadhav, K.R.; Gambhire, M.N.; Shaikh, I.M.; Kadam, V.J.; Pisal, S.S. Nasal drug delivery system-factors affecting and applications. Curr. Drug Ther. 2007, 2, 27–38. [Google Scholar] [CrossRef]
- Tirucherai, G.S.; Pezron, I.; Mitra, A.K. Novel approaches to nasal delivery of peptides and proteins. STP Pharma Sci. 2002, 12, 3–12. [Google Scholar]
- Marx, D.; Williams, G.; Birkhoff, M. Intranasal drug administration—An attractive delivery route for some drugs. In Drug Discovery and Development; IntechOpen: Rijeka, Croatia, 2015; pp. 299–320. [Google Scholar]
- Upadhyay, S.; Parikh, A.; Joshi, P.; Upadhyay, U.M.; Chotai, N.P. Intranasal drug delivery system-A glimpse to become maestro. J. Appl. Pharm. Sci. 2011, 1, 34–44. [Google Scholar]
- Han, K.; Woghiren, O.E.; Priefer, R. Surface tension examination of various liquid oral, nasal, and ophthalmic dosage forms. Chem. Cent. J. 2016, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Shen, X.; Mao, S. Factors influencing drug deposition in the nasal cavity upon delivery via nasal sprays. J. Pharm. Investig. 2020, 50, 251–259. [Google Scholar] [CrossRef]
- Djupesland, P.G. Nasal drug delivery devices: Characteristics and performance in a clinical perspective—A review. Drug Deliv. Transl. Res. 2013, 3, 42–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Food and Drug Administration. FluMist Quadrivalent. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/flumist-quadrivalent (accessed on 24 April 2023).
- Cardoso, F.M.C.; Petrovajová, D.; Horňáková, T. Viral vaccine stabilizers: Status and trends. Acta Virol. 2017, 61, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavda, V.P.; Vora, L.K.; Pandya, A.K.; Patravale, V.B. Intranasal vaccines for SARS-CoV-2: From challenges to potential in COVID-19 management. Drug Discov. Today 2021, 26, 2619–2636. [Google Scholar] [CrossRef] [PubMed]
- Kraan, H.; van Herpen, P.; Kersten, G.; Amorij, J.-P. Development of thermostable lyophilized inactivated polio vaccine. Pharm. Res. 2014, 31, 2618–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herdis, H.; Surachman, M.; Darmawan, I.W.A.; Afifah, A. The role of sucrose as extracellular cryoprotectant in maintaining the Garut rams’ frozen semen quality. In AIP Conference Proceedings; AIP Publishing: New York, NY, USA, 2019. [Google Scholar]
- Henriques, P.; Fortuna, A.; Doktorovová, S. Spray dried powders for nasal delivery: Process and formulation considerations. Eur. J. Pharm. Biopharm. 2022, 176, 1–20. [Google Scholar] [CrossRef]
- Kanojia, G.; Have, R.t.; Soema, P.C.; Frijlink, H.; Amorij, J.-P.; Kersten, G. Developments in the formulation and delivery of spray dried vaccines. Hum. Vaccines Immunother. 2017, 13, 2364–2378. [Google Scholar] [CrossRef]
- Bahamondez-Canas, T.F.; Cui, Z. Intranasal immunization with dry powder vaccines. Eur. J. Pharm. Biopharm. 2018, 122, 167–175. [Google Scholar] [CrossRef]
- Nižić Nodilo, L.; Ugrina, I.; Špoljarić, D.; Amidžić Klarić, D.; Jakobušić Brala, C.; Perkušić, M.; Pepić, I.; Lovrić, J.; Saršon, V.; Safundžić Kučuk, M. A dry powder platform for nose-to-brain delivery of dexamethasone: Formulation development and nasal deposition studies. Pharmaceutics 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, L.-J.; Zhan, L.-T.; Zhao, M.; Wu, G.-H.; Si, J.-Y.; Chen, L.; Lin, X.; Sun, Y.-P.; Lin, M. The Optimal Concentration of Formaldehyde is Key to Stabilizing the Pre-Fusion Conformation of Respiratory Syncytial Virus Fusion Protein. Viruses 2019, 11, 628. [Google Scholar] [CrossRef] [Green Version]
- Gockel, C.M.; Bao, S.; Beagley, K.W. Transcutaneous immunization induces mucosal and systemic immunity: A potent method for targeting immunity to the female reproductive tract. Mol. Immunol. 2000, 37, 537–544. [Google Scholar] [CrossRef]
- Karande, P.; Mitragotri, S. Transcutaneous immunization: An overview of advantages, disease targets, vaccines, and delivery technologies. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 175–201. [Google Scholar] [CrossRef] [PubMed]
- Hettinga, J.; Carlisle, R. Vaccination into the dermal compartment: Techniques, challenges, and prospects. Vaccines 2020, 8, 534. [Google Scholar] [CrossRef]
- Hirobe, S.; Okada, N.; Nakagawa, S. Transcutaneous vaccines–current and emerging strategies. Expert Opin. Drug Deliv. 2013, 10, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Manikkath, J.; Hegde, A.R.; Parekh, H.S.; Mutalik, S. Peptide dendrimers in delivery of bioactive molecules to skin. In Nanoscience in Dermatology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 89–97. [Google Scholar]
- Tezel, A.; Paliwal, S.; Shen, Z.; Mitragotri, S. Low-frequency ultrasound as a transcutaneous immunization adjuvant. Vaccine 2005, 23, 3800–3807. [Google Scholar] [CrossRef]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoppink, J.; Rivas, D.F. Jet injectors: Perspectives for small volume delivery with lasers. Adv. Drug Deliv. Rev. 2022, 182, 114109. [Google Scholar] [CrossRef]
- Trimzi, M.A.; Ham, Y.-B. A needle-free jet injection system for controlled release and repeated biopharmaceutical delivery. Pharmaceutics 2021, 13, 1770. [Google Scholar] [CrossRef]
- Lin, C.-H.; Aljuffali, I.A.; Fang, J.-Y. Lasers as an approach for promoting drug delivery via skin. Expert Opin. Drug Deliv. 2014, 11, 599–614. [Google Scholar] [CrossRef]
- Kakar, P.; Li, Z.; Li, Y.; Cao, Y.; Chen, X. Laser facilitates week-long sustained transdermal drug delivery at high doses. J. Control. Release 2020, 319, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Menon, I.; Bagwe, P.; Gomes, K.B.; Bajaj, L.; Gala, R.; Uddin, M.N.; D’souza, M.J.; Zughaier, S.M. Microneedles: A new generation vaccine delivery system. Micromachines 2021, 12, 435. [Google Scholar] [CrossRef] [PubMed]
- Alimardani, V.; Abolmaali, S.S.; Yousefi, G.; Rahiminezhad, Z.; Abedi, M.; Tamaddon, A.; Ahadian, S. Microneedle arrays combined with nanomedicine approaches for transdermal delivery of therapeutics. J. Clin. Med. 2021, 10, 181. [Google Scholar] [CrossRef]
- Wang, F.-Y.; Chen, Y.; Huang, Y.-Y.; Cheng, C.-M. Transdermal drug delivery systems for fighting common viral infectious diseases. Drug Deliv. Transl. Res. 2021, 11, 1498–1508. [Google Scholar] [CrossRef] [PubMed]
- Cárcamo-Martínez, Á.; Mallon, B.; Domínguez-Robles, J.; Vora, L.K.; Anjani, Q.K.; Donnelly, R.F. Hollow microneedles: A perspective in biomedical applications. Int. J. Pharm. 2021, 599, 120455. [Google Scholar] [CrossRef] [PubMed]
- Prausnitz, M.R. Engineering microneedle patches for vaccination and drug delivery to skin. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 177–200. [Google Scholar] [CrossRef]
- Tariq, N.; Ashraf, M.W.; Tayyaba, S. A review on solid microneedles for biomedical applications. J. Pharm. Innov. 2022, 17, 1464–1483. [Google Scholar] [CrossRef]
- Jung, J.H.; Jin, S.G. Microneedle for transdermal drug delivery: Current trends and fabrication. J. Pharm. Investig. 2021, 51, 503–517. [Google Scholar] [CrossRef]
- Aldawood, F.K.; Andar, A.; Desai, S. A comprehensive review of microneedles: Types, materials, processes, characterizations and applications. Polymers 2021, 13, 2815. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, J.; Zhuang, J.; Xu, H.; Liu, Y.; Wu, D. Microneedle system for transdermal drug and vaccine delivery: Devices, safety, and prospects. Dose-Response 2019, 17, 1559325819878585. [Google Scholar] [CrossRef] [Green Version]
- Leone, M.; Mönkäre, J.; Bouwstra, J.A.; Kersten, G. Dissolving microneedle patches for dermal vaccination. Pharm. Res. 2017, 34, 2223–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartawi, Z.; Blackshields, C.; Faisal, W. Dissolving microneedles: Applications and growing therapeutic potential. J. Control. Release 2022, 348, 186–205. [Google Scholar] [CrossRef]
- Turner, J.G.; White, L.R.; Estrela, P.; Leese, H.S. Hydrogel-forming microneedles: Current advancements and future trends. Macromol. Biosci. 2021, 21, 2000307. [Google Scholar] [CrossRef]
- Dardano, P.; Caliò, A.; Di Palma, V.; Bevilacqua, M.F.; Di Matteo, A.; De Stefano, L. A photolithographic approach to polymeric microneedles array fabrication. Materials 2015, 8, 8661–8673. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Zhao, B.; Wan, W.; Li, Y.; Bai, X.; Hu, C.; Wang, J.; Li, Y.; Xin, W.; Kang, L. Application of microneedle-based vaccines in biosecurity. J. Biosaf. Biosecur. 2022, 4, 75–83. [Google Scholar] [CrossRef]
- Donnelly, R.F.; McCrudden, M.T.C.; Zaid Alkilani, A.; Larrañeta, E.; McAlister, E.; Courtenay, A.J.; Kearney, M.-C.; Singh, T.R.R.; McCarthy, H.O.; Kett, V.L. Hydrogel-forming microneedles prepared from “super swelling” polymers combined with lyophilised wafers for transdermal drug delivery. PLoS ONE 2014, 9, e111547. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, H.; Li, W. Microneedle-mediated transdermal delivery of drug-carrying nanoparticles. Front. Bioeng. Biotechnol. 2022, 10, 840395. [Google Scholar] [CrossRef] [PubMed]
- Habibi, N.; Mauser, A.; Ko, Y.; Lahann, J. Protein nanoparticles: Uniting the power of proteins with engineering design approaches. Adv. Sci. 2022, 9, 2104012. [Google Scholar] [CrossRef]
- Karande, P.; Mitragotri, S. Enhancement of transdermal drug delivery via synergistic action of chemicals. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 2362–2373. [Google Scholar] [CrossRef] [Green Version]
- Barry, B.W. Lipid-protein-partitioning theory of skin penetration enhancement. J. Control. Release 1991, 15, 237–248. [Google Scholar] [CrossRef]
- Tan, G.; Xu, P.; Lawson, L.B.; He, J.; Freytag, L.C.; Clements, J.D.; John, V.T. Hydration effects on skin microstructure as probed by high-resolution cryo-scanning electron microscopy and mechanistic implications to enhanced transcutaneous delivery of biomacromolecules. J. Pharm. Sci. 2010, 99, 730–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Meirelles, N.C.; Yushmanov, V.E.; Tabak, M. Water increases the fluidity of intercellular membranes of stratum corneum: Correlation with water permeability, elastic, and electrical resistance properties. J. Investig. Dermatol. 1996, 106, 1058–1063. [Google Scholar] [CrossRef] [Green Version]
- Notman, R.; den Otter, W.K.; Noro, M.G.; Briels, W.J.; Anwar, J. The permeability enhancing mechanism of DMSO in ceramide bilayers simulated by molecular dynamics. Biophys. J. 2007, 93, 2056–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.C.; Barry, B.W. Terpenes and the lipid–protein–partitioning theory of skin penetration enhancement. Pharm. Res. 1991, 8, 17–24. [Google Scholar] [CrossRef]
- Osborne, D.W.; Musakhanian, J. Skin penetration and permeation properties of Transcutol®—Neat or diluted mixtures. AAPS PharmSciTech 2018, 19, 3512–3533. [Google Scholar] [CrossRef]
- Food and Drug Administration. VAXCHORA. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaxchora (accessed on 24 April 2023).
- Food and Drug Administration. ROTARIX. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/rotarix (accessed on 24 April 2023).
- Food and Drug Administration. RotaTeq. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/rotateq (accessed on 23 April 2023).
- Food and Drug Administration. VIVOTIF. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vivotif (accessed on 23 April 2023).
- Food and Drug Administration. Adenovirus Type 4 and Type 7 Vaccine, Live, Oral. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/adenovirus-type-4-and-type-7-vaccine-live-oral (accessed on 23 April 2023).
- Kool, M.; Fierens, K.; Lambrecht, B.N. Alum adjuvant: Some of the tricks of the oldest adjuvant. J. Med. Microbiol. 2012, 61, 927–934. [Google Scholar] [CrossRef]
- HogenEsch, H.; O’Hagan, D.T.; Fox, C.B. Optimizing the utilization of aluminum adjuvants in vaccines: You might just get what you want. NPJ Vaccines 2018, 3, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Van Riet, E.; Romeijn, S.; Kersten, G.F.A.; Jiskoot, W.; Bouwstra, J.A. Immune modulation by adjuvants combined with diphtheria toxoid administered topically in BALB/c mice after microneedle array pretreatment. Pharm. Res. 2009, 26, 1635–1643. [Google Scholar] [CrossRef] [Green Version]
- Stark, F.C.; Akache, B.; Deschatelets, L.; Tran, A.; Stuible, M.; Durocher, Y.; McCluskie, M.J.; Agbayani, G.; Dudani, R.; Harrison, B.A. Intranasal immunization with a proteosome-adjuvanted SARS-CoV-2 spike protein-based vaccine is immunogenic and efficacious in mice and hamsters. Sci. Rep. 2022, 12, 9772. [Google Scholar] [CrossRef]
- He, P.; Zou, Y.; Hu, Z. Advances in aluminum hydroxide-based adjuvant research and its mechanism. Hum. Vaccines Immunother. 2015, 11, 477–488. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Sztein, M.B.; Wasserman, S.S.; Losonsky, G.A.; DiLorenzo, S.C.; Walker, R.I. Safety and immunogenicity of oral inactivated whole-cell Helicobacter pylori vaccine with adjuvant among volunteers with or without subclinical infection. Infect. Immun. 2001, 69, 3581–3590. [Google Scholar] [CrossRef] [Green Version]
- Harro, C.; Bourgeois, A.L.; Sack, D.; Walker, R.; DeNearing, B.; Brubaker, J.; Maier, N.; Fix, A.; Dally, L.; Chakraborty, S. Live attenuated enterotoxigenic Escherichia coli (ETEC) vaccine with dmLT adjuvant protects human volunteers against virulent experimental ETEC challenge. Vaccine 2019, 37, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Qadri, F.; Akhtar, M.; Bhuiyan, T.R.; Chowdhury, M.I.; Ahmed, T.; Rafique, T.A.; Khan, A.; Rahman, S.I.A.; Khanam, F.; Lundgren, A. Safety and immunogenicity of the oral, inactivated, enterotoxigenic Escherichia coli vaccine ETVAX in Bangladeshi children and infants: A double-blind, randomised, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2020, 20, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Norton, E.B.; Lawson, L.B.; Freytag, L.C.; Clements, J.D. Characterization of a mutant Escherichia coli heat-labile toxin, LT (R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 2011, 18, 546–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjökvist Ottsjö, L.; Flach, C.-F.; Clements, J.; Holmgren, J.; Raghavan, S. A double mutant heat-labile toxin from Escherichia coli, LT (R192G/L211A), is an effective mucosal adjuvant for vaccination against Helicobacter pylori infection. Infect. Immun. 2013, 81, 1532–1540. [Google Scholar] [CrossRef] [Green Version]
- Sjökvist Ottsjö, L.; Jeverstam, F.; Yrlid, L.; Wenzel, A.U.; Walduck, A.K.; Raghavan, S. Induction of mucosal immune responses against Helicobacter pylori infection after sublingual and intragastric route of immunization. Immunology 2017, 150, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldarelli, G.A.; Matz, H.; Gao, S.; Chen, K.; Hamza, T.; Yfantis, H.G.; Feng, H.; Donnenberg, M.S. Pilin vaccination stimulates weak antibody responses and provides no protection in a C57Bl/6 murine model of acute Clostridium difficile infection. J. Vaccines Vaccin. 2016, 7, 321. [Google Scholar] [CrossRef] [Green Version]
- Hayden, C.A.; Streatfield, S.J.; Lamphear, B.J.; Fake, G.M.; Keener, T.K.; Walker, J.H.; Clements, J.D.; Turner, D.D.; Tizard, I.R.; Howard, J.A. Bioencapsulation of the hepatitis B surface antigen and its use as an effective oral immunogen. Vaccine 2012, 30, 2937–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalsiamthara, J.; Lee, J.H. Immunization with Salmonella Enteritidis secreting mucosal adjuvant labile toxin confers protection against wild type challenge via augmentation of CD3+ CD4+ T-cell proliferation and enhancement of IFN-γ, IL-6 and IL-10 expressions in chicken. Vaccine 2017, 35, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, J.; Nordqvist, S.; Blomquist, M.; Jeverstam, F.; Lebens, M.; Raghavan, S. Preclinical immunogenicity and protective efficacy of an oral Helicobacter pylori inactivated whole cell vaccine and multiple mutant cholera toxin: A novel and non-toxic mucosal adjuvant. Vaccine 2018, 36, 6223–6230. [Google Scholar] [CrossRef]
- Lebens, M.; Terrinoni, M.; Karlsson, S.L.; Larena, M.; Gustafsson-Hedberg, T.; Källgård, S.; Nygren, E.; Holmgren, J. Construction and preclinical evaluation of mmCT, a novel mutant cholera toxin adjuvant that can be efficiently produced in genetically manipulated Vibrio cholerae. Vaccine 2016, 34, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, T. Cholera toxin subunit B as adjuvant––An accelerator in protective immunity and a break in autoimmunity. Vaccines 2015, 3, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wu, W.; Xu, X. Oral vaccination with liposome-encapsulated recombinant fusion peptide of urease B epitope and cholera toxin B subunit affords prophylactic and therapeutic effects against H. pylori infection in BALB/c mice. Vaccine 2007, 25, 7664–7673. [Google Scholar] [CrossRef]
- Schussek, S.; Bernasconi, V.; Mattsson, J.; Wenzel, U.A.; Strömberg, A.; Gribonika, I.; Schön, K.; Lycke, N.Y. The CTA1-DD adjuvant strongly potentiates follicular dendritic cell function and germinal center formation, which results in improved neonatal immunization. Mucosal Immunol. 2020, 13, 545–557. [Google Scholar] [CrossRef]
- Guo, T.; Li, X.; Lin, M.; Zhang, L. Mucosal adjuvant activity of chitosan encapsulated nanoparticles as helicobacter pylori epitope vaccine carrier. Nanosci. Nanotechnol. Lett. 2016, 8, 1106–1111. [Google Scholar] [CrossRef]
- Laue, C.; Stevens, Y.; van Erp, M.; Papazova, E.; Soeth, E.; Pannenbeckers, A.; Stolte, E.; Böhm, R.; Gall, S.L.; Falourd, X. Adjuvant effect of orally applied preparations containing non-digestible polysaccharides on influenza vaccination in healthy seniors: A double-blind, randomised, controlled pilot trial. Nutrients 2021, 13, 2683. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, H.; Winarsih, S.; Prawiro, S.R.; Barlianto, W.; Sanarto, S.; Djunaedi, D.; Endharti, A.T.; Sardjono, T.W.; Khotimah, H.; Nugraheni, R.W. AdhO36 liposomes from Salmonella Typhi in combination with β-Glucan Immuno-adjuvant From Candida albicans cell wall as oral vaccine against typhoid fever in mice model. Open Access Maced. J. Med. Sci. 2020, 8, 441–448. [Google Scholar] [CrossRef]
- Kournikakis, B.; Mandeville, R.; Brousseau, P.; Ostroff, G. Anthrax-protective effects of yeast beta 1, 3 glucans. MedGenMed 2003, 5, 1. [Google Scholar] [PubMed]
- Courtney, A.N.; Nehete, P.N.; Nehete, B.P.; Thapa, P.; Zhou, D.; Sastry, K.J. Alpha-galactosylceramide is an effective mucosal adjuvant for repeated intranasal or oral delivery of HIV peptide antigens. Vaccine 2009, 27, 3335–3341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davitt, C.J.H.; Longet, S.; Albutti, A.; Aversa, V.; Nordqvist, S.; Hackett, B.; McEntee, C.P.; Rosa, M.; Coulter, I.S.; Lebens, M. Alpha-galactosylceramide enhances mucosal immunity to oral whole-cell cholera vaccines. Mucosal Immunol. 2019, 12, 1055–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longet, S.; Abautret-Daly, A.; Davitt, C.J.H.; McEntee, C.P.; Aversa, V.; Rosa, M.; Coulter, I.S.; Holmgren, J.; Raghavan, S.; Lavelle, E.C. An oral alpha-galactosylceramide adjuvanted Helicobacter pylori vaccine induces protective IL-1R-and IL-17R-dependent Th1 responses. NPJ Vaccines 2019, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Clements, J.D.; Norton, E.B. The mucosal vaccine adjuvant LT (R192G/L211A) or dmLT. mSphere 2018, 3, e00215–e00218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elieh Ali Komi, D.; Sharma, L.; Dela Cruz, C.S. Chitin and its effects on inflammatory and immune responses. Clin. Rev. Allergy Immunol. 2018, 54, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Honda-Okubo, Y.; Saade, F.; Petrovsky, N. Advax™, a polysaccharide adjuvant derived from delta inulin, provides improved influenza vaccine protection through broad-based enhancement of adaptive immune responses. Vaccine 2012, 30, 5373–5381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, Y.; Wang, Y.; Ren, S.; Li, Y.; Wang, B. Polyactin A is a novel and potent immunological adjuvant for peptide-based cancer vaccine. Int. Immunopharmacol. 2018, 54, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, W.; Smith, C.J.; Musa, H. Cereal-derived arabinoxylans as biological response modifiers: Extraction, molecular features, and immune-stimulating properties. Crit. Rev. Food Sci. Nutr. 2015, 55, 1035–1052. [Google Scholar] [CrossRef]
- Ichinohe, T.; Watanabe, I.; Ito, S.; Fujii, H.; Moriyama, M.; Tamura, S.-i.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T. Synthetic double-stranded RNA poly (I: C) combined with mucosal vaccine protects against influenza virus infection. J. Virol. 2005, 79, 2910–2919. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Galluzzi, L. Nucleic Acid Sensing and Immunity-PART B; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Ohto, U.; Shimizu, T. Structural aspects of nucleic acid-sensing Toll-like receptors. Biophys. Rev. 2016, 8, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Morere, J.; Hognon, C.; Miclot, T.; Jiang, T.; Dumont, E.; Barone, G.; Monari, A.; Bignon, E. How Fragile We Are: Influence of Stimulator of Interferon Genes (STING) Variants on Pathogen Recognition and Immune Response Efficiency. J. Chem. Inf. Model. 2022, 62, 3096–3106. [Google Scholar] [CrossRef] [PubMed]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Tandrup Schmidt, S.; Foged, C.; Smith Korsholm, K.; Rades, T.; Christensen, D. Liposome-based adjuvants for subunit vaccines: Formulation strategies for subunit antigens and immunostimulators. Pharmaceutics 2016, 8, 7. [Google Scholar] [CrossRef]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.K.; Akira, S.; Underhill, D.M.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 2001, 410, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, S.-H.; Ahn, D.-G.; Cho, H.; Sung, M.-H.; Han, S.H.; Oh, J.-W. The antiviral activity of poly-γ-glutamic acid, a polypeptide secreted by Bacillus sp., through induction of CD14-dependent type I interferon responses. Biomaterials 2013, 34, 9700–9708. [Google Scholar] [CrossRef]
- Jones, T.; Cyr, S.; Allard, F.; Bellerose, N.; Lowell, G.H.; Burt, D.S. Protollin™: A novel adjuvant for intranasal vaccines. Vaccine 2004, 22, 3691–3697. [Google Scholar] [CrossRef]
- Díaz-Dinamarca, D.A.; Salazar, M.L.; Castillo, B.N.; Manubens, A.; Vasquez, A.E.; Salazar, F.; Becker, M.I. Protein-based adjuvants for vaccines as immunomodulators of the innate and adaptive immune response: Current knowledge, challenges, and future opportunities. Pharmaceutics 2022, 14, 1671. [Google Scholar] [CrossRef]
- Food and Drug Administration. Common Ingredients in U.S. Licensed Vaccines. Available online: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/common-ingredients-us-licensed-vaccines (accessed on 23 April 2023).
- Petrovsky, N. Comparative safety of vaccine adjuvants: A summary of current evidence and future needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- McLenon, J.; Rogers, M.A.M. The fear of needles: A systematic review and meta-analysis. J. Adv. Nurs. 2019, 75, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Diaz-Arévalo, D.; Guan, H.; Zeng, M. Noninvasive vaccination against infectious diseases. Hum. Vaccines Immunother. 2018, 14, 1717–1733. [Google Scholar] [CrossRef]
- Mutsch, M.; Zhou, W.; Rhodes, P.; Bopp, M.; Chen, R.T.; Linder, T.; Spyr, C.; Steffen, R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 2004, 350, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, I.; Zambon, M.C.; Rudin, A.; Colegate, A.; Podda, A.; Bugarini, R.; Del Giudice, G.; Minutello, A.; Bonnington, S.; Holmgren, J. Phase I evaluation of intranasal trivalent inactivated influenza vaccine with nontoxigenic Escherichia coli enterotoxin and novel biovector as mucosal adjuvants, using adult volunteers. J. Virol. 2006, 80, 4962–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valli, E.; Harriett, A.J.; Nowakowska, M.K.; Baudier, R.L.; Provosty, W.B.; McSween, Z.; Lawson, L.B.; Nakanishi, Y.; Norton, E.B. LTA1 is a safe, intranasal enterotoxin-based adjuvant that improves vaccine protection against influenza in young, old and B-cell-depleted (μMT) mice. Sci. Rep. 2019, 9, 15128. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.T.; Wang, Y.L.; Su, Q.D.; Feng, Q.I.U.; Yao, Y.I.; Jia, Z.Y.; Da Yan, W.; Kun, Q.I.N.; Zou, Y.N.; Bi, S.L. Intranasal immunization using CTA1-DD as a mucosal adjuvant for an inactivated influenza vaccine. Biomed. Environ. Sci. 2019, 32, 531–540. [Google Scholar]
- Li, H.; Ren, H.; Zhang, Y.; Cao, L.; Xu, W. Intranasal vaccination with a recombinant protein CTA1-DD-RBF protects mice against hRSV infection. Sci. Rep. 2021, 11, 18641. [Google Scholar] [CrossRef]
- Andersen, C.S.; Dietrich, J.; Agger, E.M.; Lycke, N.Y.; Lövgren, K.; Andersen, P. The combined CTA1-DD/ISCOMs vector is an effective intranasal adjuvant for boosting prior Mycobacterium bovis BCG immunity to Mycobacterium tuberculosis. Infect. Immun. 2007, 75, 408–416. [Google Scholar] [CrossRef] [Green Version]
- De Filette, M.; Ramne, A.; Birkett, A.; Lycke, N.; Löwenadler, B.; Jou, W.M.; Saelens, X.; Fiers, W. The universal influenza vaccine M2e-HBc administered intranasally in combination with the adjuvant CTA1-DD provides complete protection. Vaccine 2006, 24, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Asahi-Ozaki, Y.; Itamura, S.; Ichinohe, T.; Strong, P.; Tamura, S.-i.; Takahashi, H.; Sawa, H.; Moriyama, M.; Tashiro, M.; Sata, T. Intranasal administration of adjuvant-combined recombinant influenza virus HA vaccine protects mice from the lethal H5N1 virus infection. Microbes Infect. 2006, 8, 2706–2714. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Arévalo, M.T.; Chen, Y.; Posadas, O.; Smith, J.A.; Zeng, M. Intranasal immunization with influenza antigens conjugated with cholera toxin subunit B stimulates broad spectrum immunity against influenza viruses. Hum. Vaccines Immunother. 2014, 10, 1211–1220. [Google Scholar] [CrossRef]
- Li, M.; Jiang, Y.; Gong, T.; Zhang, Z.; Sun, X. Intranasal vaccination against HIV-1 with adenoviral vector-based nanocomplex using synthetic TLR-4 agonist peptide as adjuvant. Mol. Pharm. 2016, 13, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.-T.; Zhang, X.-M.; Sun, P.-F.; Sun, L.-J.; Guo, X.; Tian, T.; Zhang, J.; Guo, Q.-Y.; Li, X.; Guo, L.-J. Intranasal immunization using mannatide as a novel adjuvant for an inactivated influenza vaccine and its adjuvant effect compared with MF59. PLoS ONE 2017, 12, e0169501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luczo, J.M.; Bousse, T.; Johnson, S.K.; Jones, C.A.; Pearce, N.; Neiswanger, C.A.; Wang, M.-X.; Miller, E.A.; Petrovsky, N.; Wentworth, D.E. Intranasal powder live attenuated influenza vaccine is thermostable, immunogenic, and protective against homologous challenge in ferrets. NPJ Vaccines 2021, 6, 59. [Google Scholar] [CrossRef]
- Doavi, T.; Mousavi, S.L.; Kamali, M.; Amani, J.; Ramandi, M.F. Chitosan-based intranasal vaccine against Escherichia coli O157: H7. Iran. Biomed. J. 2016, 20, 97. [Google Scholar]
- Mann, A.J.; Noulin, N.; Catchpole, A.; Stittelaar, K.J.; De Waal, L.; Veldhuis Kroeze, E.J.B.; Hinchcliffe, M.; Smith, A.; Montomoli, E.; Piccirella, S. Intranasal H5N1 vaccines, adjuvanted with chitosan derivatives, protect ferrets against highly pathogenic influenza intranasal and intratracheal challenge. PLoS ONE 2014, 9, e93761. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Wei, W.; Zhou, M.; Wang, Y.; Wu, J.; Ma, G.; Su, Z. Thermal-sensitive hydrogel as adjuvant-free vaccine delivery system for H5N1 intranasal immunization. Biomaterials 2012, 33, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Svindland, S.C.; Jul-Larsen, Å.; Pathirana, R.; Andersen, S.; Madhun, A.; Montomoli, E.; Jabbal-Gill, I.; Cox, R.J. The mucosal and systemic immune responses elicited by a chitosan-adjuvanted intranasal influenza H5N1 vaccine. Influenza Other Respir. Viruses 2012, 6, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.-B.; Shan, P.; Li, S.-Z.; Zhou, Y.; Deng, X.; Li, J.-L.; Zhang, Y.; Gao, J.-S.; Xu, J. The mechanism of action of acid-soluble chitosan as an adjuvant in the formulation of nasally administered vaccine against HBV. RSC Adv. 2016, 6, 96785–96797. [Google Scholar] [CrossRef]
- Hasegawa, H.; Ichinohe, T.; Strong, P.; Watanabe, I.; Ito, S.; Tamura, S.i.; Takahashi, H.; Sawa, H.; Chiba, J.; Kurata, T. Protection against influenza virus infection by intranasal administration of hemagglutinin vaccine with chitin microparticles as an adjuvant. J. Med. Virol. 2005, 75, 130–136. [Google Scholar] [CrossRef]
- Okamoto, S.; Matsuoka, S.; Takenaka, N.; Haredy, A.M.; Tanimoto, T.; Gomi, Y.; Ishikawa, T.; Akagi, T.; Akashi, M.; Okuno, Y. Intranasal immunization with a formalin-inactivated human influenza A virus whole-virion vaccine alone and intranasal immunization with a split-virion vaccine with mucosal adjuvants show similar levels of cross-protection. Clin. Vaccine Immunol. 2012, 19, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, E.; Kuramitsu, M.; Momose, H.; Kobiyama, K.; Aoshi, T.; Yamada, H.; Ishii, K.J.; Mizukami, T.; Hamaguchi, I. A novel vaccinological evaluation of intranasal vaccine and adjuvant safety for preclinical tests. Vaccine 2017, 35, 821–830. [Google Scholar] [CrossRef]
- Lin, Y.-L.; Chow, Y.-H.; Huang, L.-M.; Hsieh, S.-M.; Cheng, P.-Y.; Hu, K.-C.; Chiang, B.-L. A CpG-adjuvanted intranasal enterovirus 71 vaccine elicits mucosal and systemic immune responses and protects human SCARB2-transgenic mice against lethal challenge. Sci. Rep. 2018, 8, 10713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.-M.; Huang, C.-Y.; Yang, C.-H.; Liu, S.-J.; Chen, H.-W.; Yu, G.-Y.; Chen, J.-K.; Chuang, T.-H.; Huang, M.-H. Formulation of SARS-CoV-2 spike protein with CpG oligodeoxynucleotides and squalene nanoparticles modulates immunological aspects following intranasal delivery. Pharmaceutics 2022, 14, 2539. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Hamaoka, S.; Kinoshita, M.; Kainuma, A.; Shimizu, M.; Katoh, H.; Moriyama, K.; Ishii, K.J.; Sawa, T. The protective effects of nasal PcrV-CpG oligonucleotide vaccination against Pseudomonas aeruginosa pneumonia. Microbiol. Immunol. 2018, 62, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Poggio, T.V.; La Torre, J.L.; Scodeller, E.A. Intranasal immunization with a recombinant truncated FimH adhesin adjuvanted with CpG oligodeoxynucleotides protects mice against uropathogenic Escherichia coli challenge. Can. J. Microbiol. 2006, 52, 1093–1102. [Google Scholar] [CrossRef]
- Jearanaiwitayakul, T.; Limthongkul, J.; Kaofai, C.; Apichirapokey, S.; Chawengkirttikul, R.; Sapsutthipas, S.; Sunintaboon, P.; Ubol, S. The STING ligand and delivery system synergistically enhance the immunogenicity of an intranasal spike SARS-CoV-2 vaccine candidate. Biomedicines 2022, 10, 1142. [Google Scholar] [CrossRef]
- Du, Y.; Xu, Y.; Feng, J.; Hu, L.; Zhang, Y.; Zhang, B.; Guo, W.; Mai, R.; Chen, L.; Fang, J. Intranasal administration of a recombinant RBD vaccine induced protective immunity against SARS-CoV-2 in mouse. Vaccine 2021, 39, 2280–2287. [Google Scholar] [CrossRef]
- Falkeborn, T.; Asahara, N.; Hayashi, M.; Arai, M.; Hinkula, J.; Maltais, A.-K. Comparison of the mucosal adjuvant Endocine™ with two well-known adjuvants: Cholera toxin and alum. Jacobs J. Vaccine Vaccin. 2015, 1, 6. [Google Scholar]
- Yang, J.; Liu, M.-Q.; Liu, L.; Li, X.; Xu, M.; Lin, H.; Liu, S.; Hu, Y.; Li, B.; Liu, B. A triple-RBD-based mucosal vaccine provides broad protection against SARS-CoV-2 variants of concern. Cell. Mol. Immunol. 2022, 19, 1279–1289. [Google Scholar] [CrossRef]
- Hong, S.H.; Byun, Y.-H.; Nguyen, C.T.; Kim, S.Y.; Seong, B.L.; Park, S.; Woo, G.-J.; Yoon, Y.; Koh, J.T.; Fujihashi, K. Intranasal administration of a flagellin-adjuvanted inactivated influenza vaccine enhances mucosal immune responses to protect mice against lethal infection. Vaccine 2012, 30, 466–474. [Google Scholar] [CrossRef]
- Treanor, J.; Nolan, C.; O’Brien, D.; Burt, D.; Lowell, G.; Linden, J.; Fries, L. Intranasal administration of a proteosome-influenza vaccine is well-tolerated and induces serum and nasal secretion influenza antibodies in healthy human subjects. Vaccine 2006, 24, 254–262. [Google Scholar] [CrossRef]
- Novotny, L.A.; Clements, J.D.; Bakaletz, L.O. Transcutaneous immunization as preventative and therapeutic regimens to protect against experimental otitis media due to nontypeable Haemophilus influenzae. Mucosal Immunol. 2011, 4, 456–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddle, M.S.; Maciel, M., Jr.; Porter, C.K.; Poole, S.T.; Gutierrez, R.L.; Gormley, R.; Laird, R.M.; Sebeny, P.J.; Dori, K.E.; Greenleaf, M.E. A first in human clinical trial assessing the safety and immunogenicity of transcutaneously delivered enterotoxigenic Escherichia coli fimbrial tip adhesin with heat-labile enterotoxin with mutation R192G. Vaccine 2020, 38, 7040–7048. [Google Scholar] [CrossRef]
- Stickings, P.; Peyre, M.; Coombes, L.; Muller, S.; Rappuoli, R.; Del Giudice, G.; Partidos, C.D.; Sesardic, D. Transcutaneous immunization with cross-reacting material CRM197 of diphtheria toxin boosts functional antibody levels in mice primed parenterally with adsorbed diphtheria toxoid vaccine. Infect. Immun. 2008, 76, 1766–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, R.; Beignon, A.-S.; Rappuoli, R.; Muller, S.; Sesardic, D.; Partidos, C.D. Transcutaneous immunization with tetanus toxoid and mutants of Escherichia coli heat-labile enterotoxin as adjuvants elicits strong protective antibody responses. J. Infect. Dis. 2003, 188, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Skountzou, I.; Quan, F.-S.; Jacob, J.; Compans, R.W.; Kang, S.-M. Transcutaneous immunization with inactivated influenza virus induces protective immune responses. Vaccine 2006, 24, 6110–6119. [Google Scholar] [CrossRef]
- Guo, L.; Qiu, Y.; Chen, J.; Zhang, S.; Xu, B.; Gao, Y. Effective transcutaneous immunization against hepatitis B virus by a combined approach of hydrogel patch formulation and microneedle arrays. Biomed. Microdevices 2013, 15, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Bal, S.M.; Ding, Z.; Kersten, G.F.A.; Jiskoot, W.; Bouwstra, J.A. Microneedle-based transcutaneous immunisation in mice with N-trimethyl chitosan adjuvanted diphtheria toxoid formulations. Pharm. Res. 2010, 27, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weldon, W.C.; Zarnitsyn, V.G.; Esser, E.S.; Taherbhai, M.T.; Koutsonanos, D.G.; Vassilieva, E.V.; Skountzou, I.; Prausnitz, M.R.; Compans, R.W. Effect of adjuvants on responses to skin immunization by microneedles coated with influenza subunit vaccine. PLoS ONE 2012, 7, e41501. [Google Scholar] [CrossRef]
- Liu, C.; Hashizume, T.; Kurita-Ochiai, T.; Yamamoto, M. CpG oligodeoxynucleotide is an effective adjuvant for transcutaneous immunization. Int. J. Oral-Med. Sci. 2007, 6, 91–96. [Google Scholar] [CrossRef]
- Berry, L.J.; Hickey, D.K.; Skelding, K.A.; Bao, S.; Rendina, A.M.; Hansbro, P.M.; Gockel, C.M.; Beagley, K.W. Transcutaneous immunization with combined cholera toxin and CpG adjuvant protects against Chlamydia muridarum genital tract infection. Infect. Immun. 2004, 72, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Belyakov, I.M.; Hammond, S.A.; Ahlers, J.D.; Glenn, G.M.; Berzofsky, J.A. Transcutaneous immunization induces mucosal CTLs and protective immunity by migration of primed skin dendritic cells. J. Clin. Investig. 2004, 113, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Beignon, A.-S.; Brown, F.; Eftekhari, P.; Kramer, E.; Briand, J.-P.; Muller, S.; Partidos, C.D. A peptide vaccine administered transcutaneously together with cholera toxin elicits potent neutralising anti-FMDV antibody responses. Vet. Immunol. Immunopathol. 2005, 104, 273–280. [Google Scholar] [CrossRef]






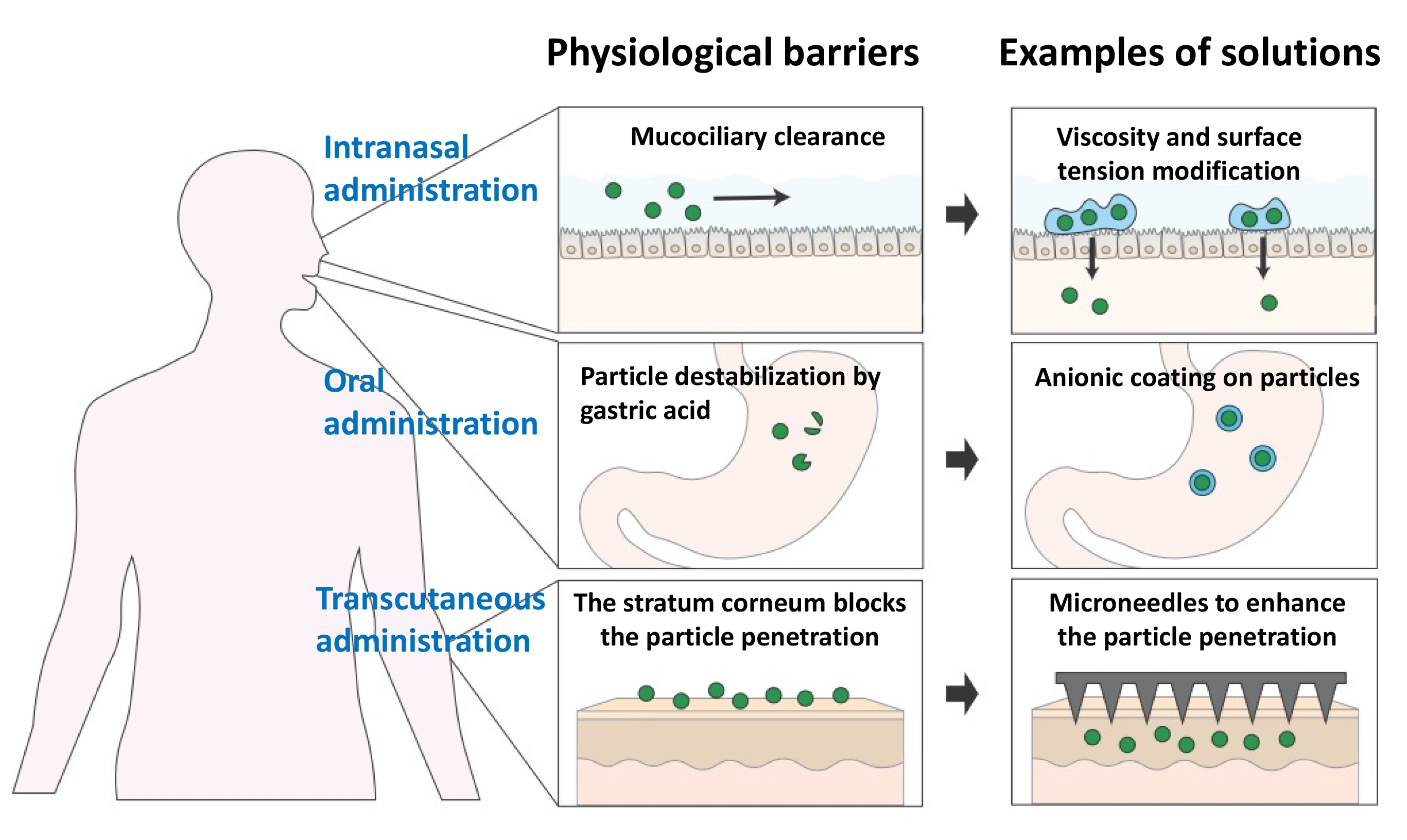{kind=link}
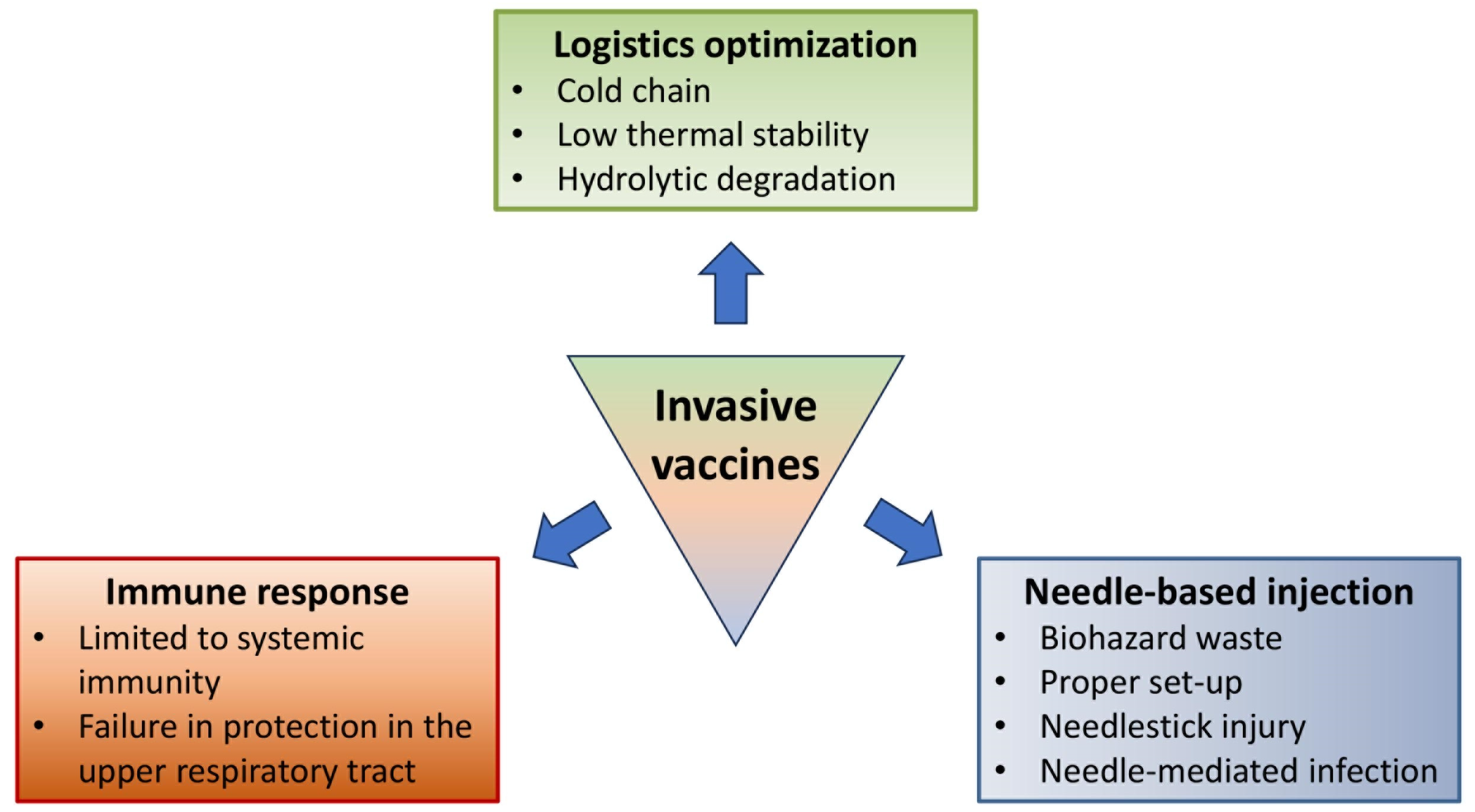{kind=link}
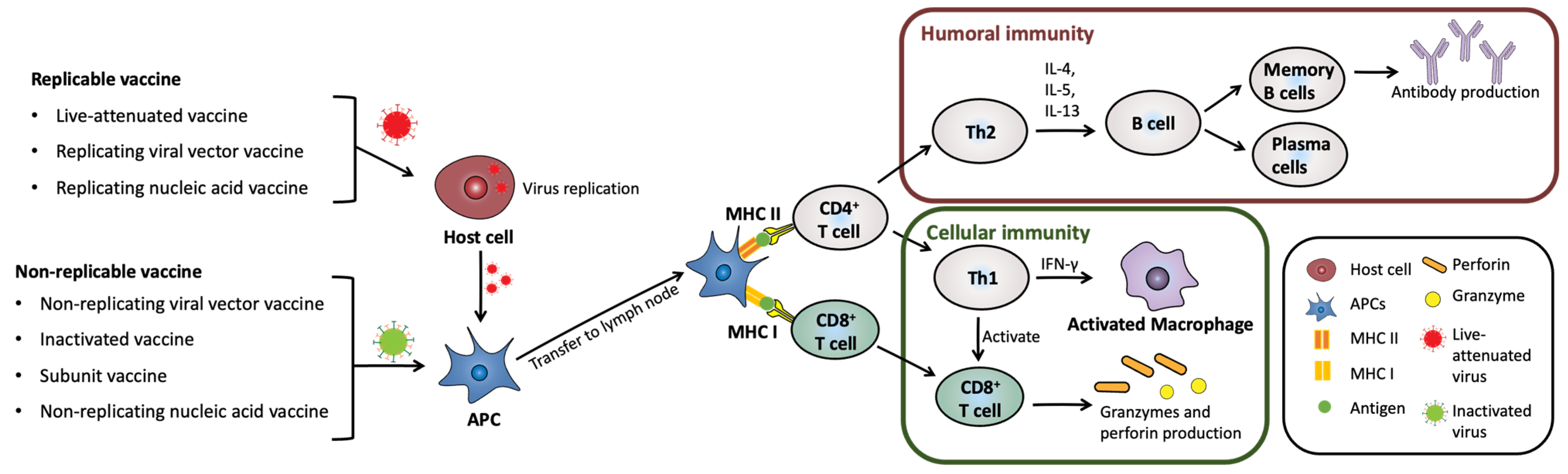{kind=link}
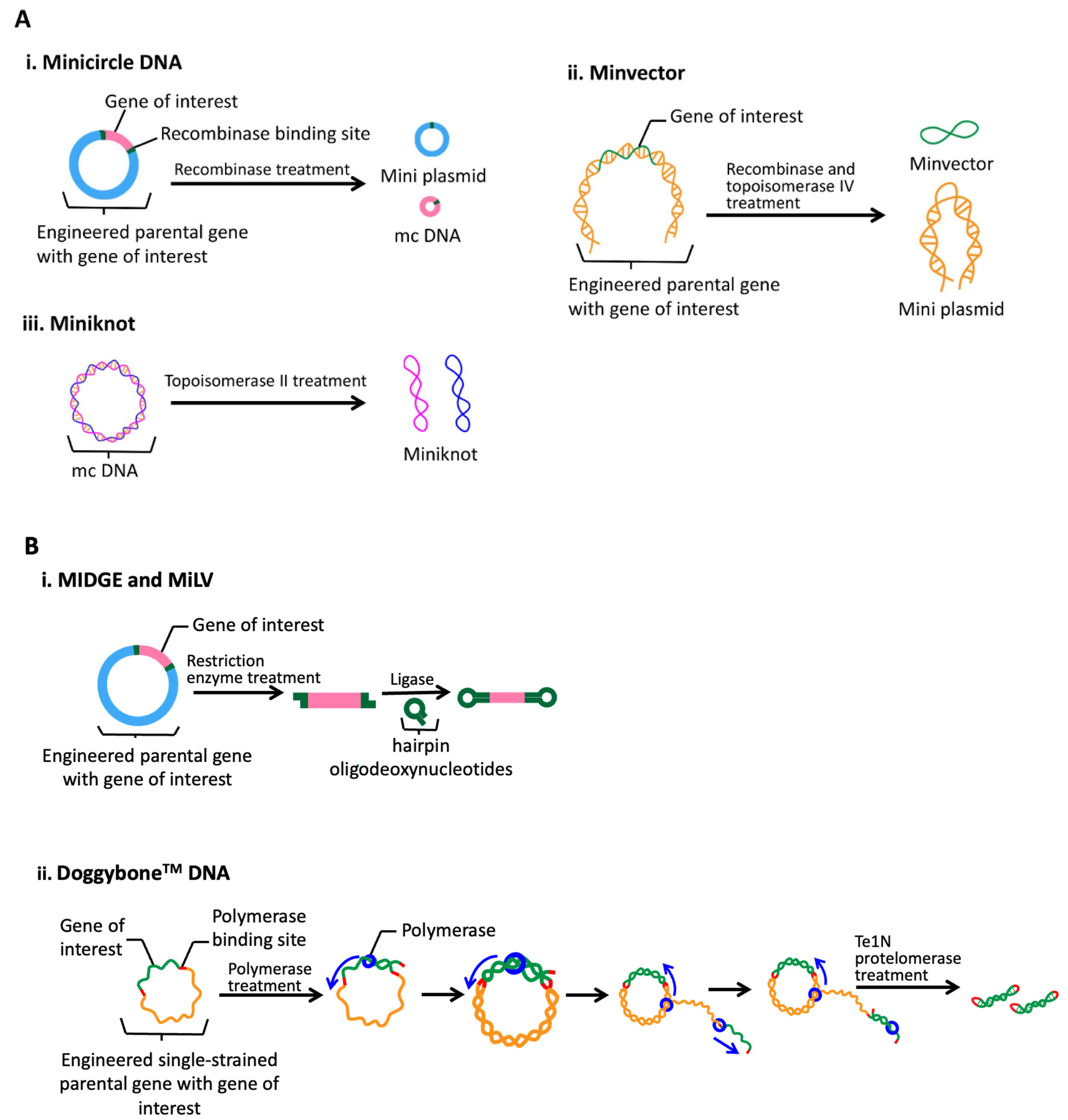{kind=link}
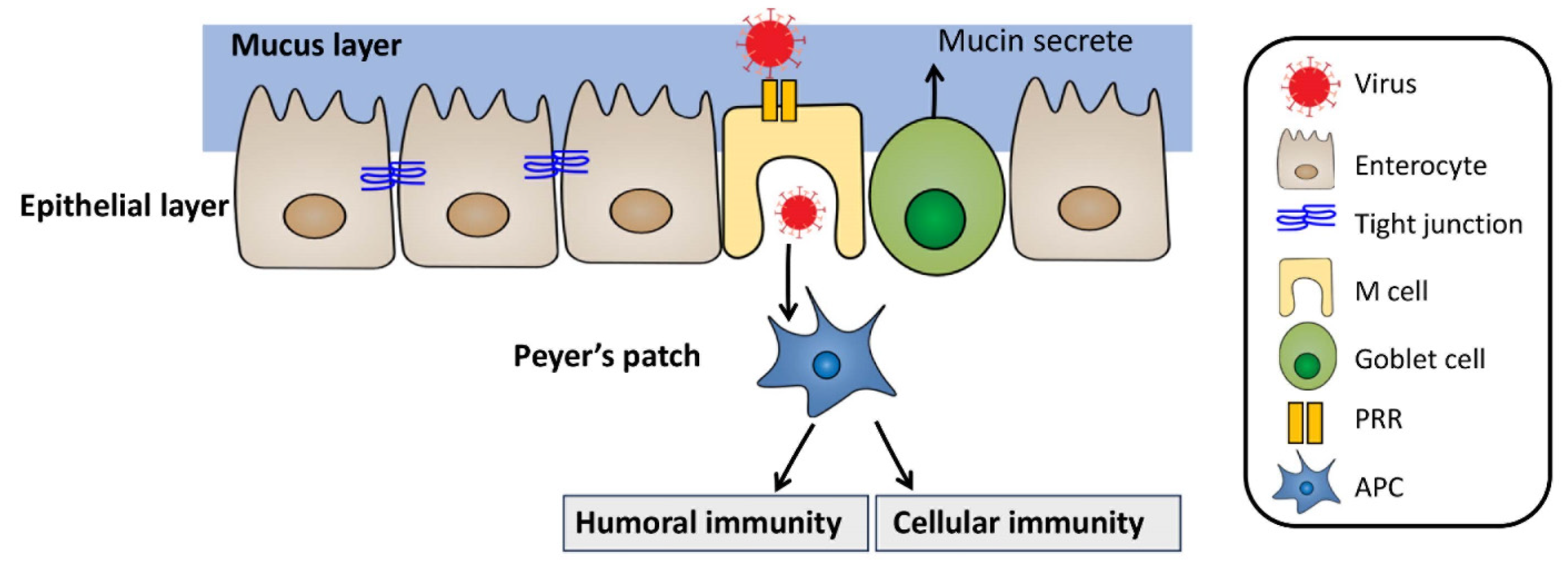{kind=link}
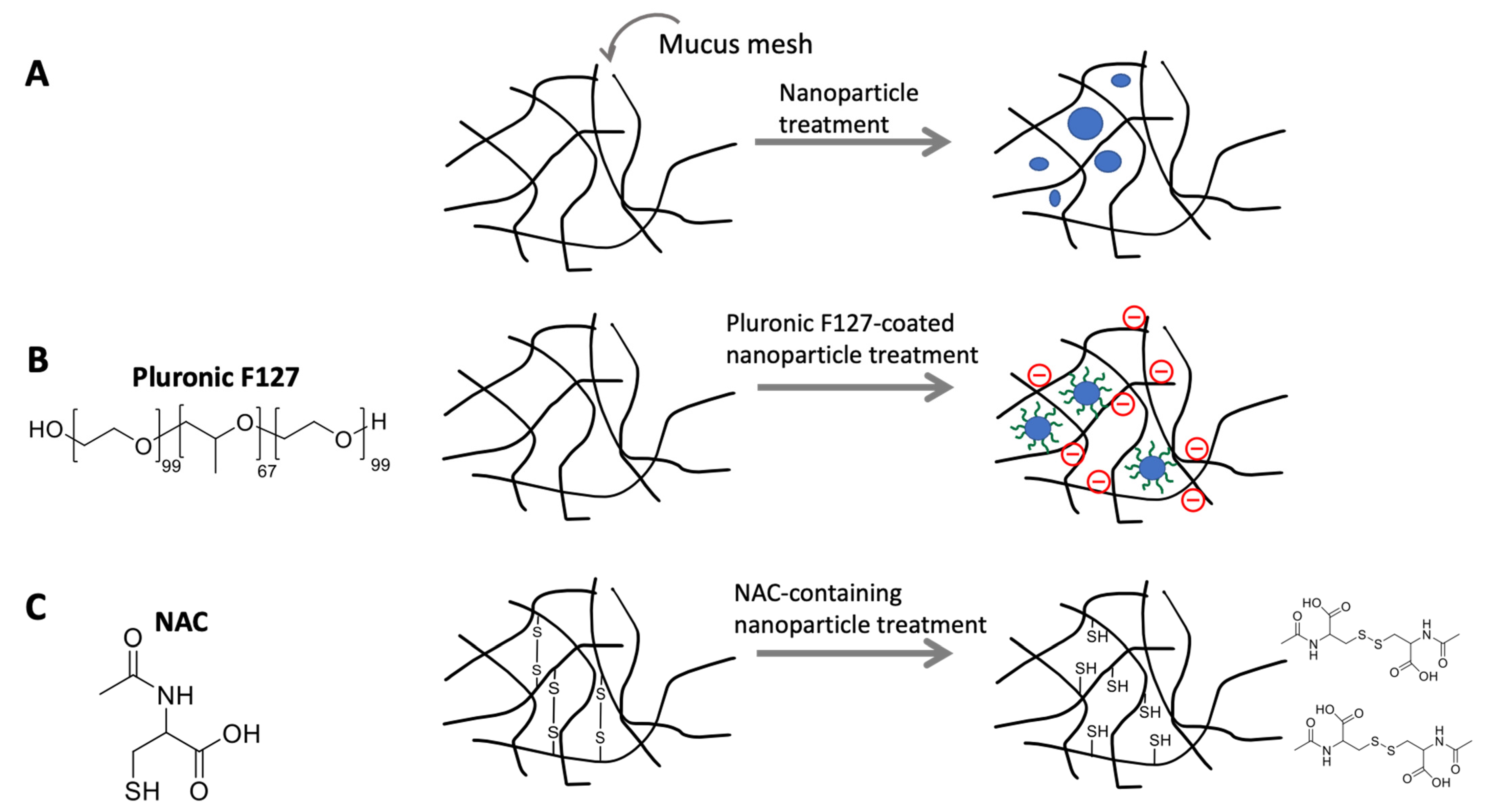{kind=link}
| Carriers/Adjuvants | Type | Immune Responses | Limitations | Status |
|---|---|---|---|---|
| Lipid-based carriers | Immune-stimulating complexes (ISCOM) | In clinical stage [107] | ||
| Liposome |
|
| In use in the vaccine industry; Pfizer/BioNTech and Moderna (SARS-CoV-2) [110] | |
| Bilosome |
|
| In preclinical stage [113] | |
| Archaeosomes |
| In preclinical stage [116] | ||
| Virosomes |
| In use in the vaccine industry; Epaxal® and Inflexal®V (influenza vaccine) [119,120] | ||
| Protein-based carriers | Gelatin |
| In clinical stage [124] | |
| Albumin |
| FDA-approved [123] | ||
| Zein |
|
| In preclinical stage [128] | |
| Emulsion | MF59 (oil-in-water emulsion) |
| In use in the vaccine industry; Fluad®, Focetria®, and Celtura® (influenza vaccine) [130] | |
| AS03 (oil-in-water emulsion) |
|
| In use in the vaccine industry; Pandemrix and Arepanrix (influenza vaccin) [132] | |
| AF03 (oil-in-water emulsion) |
|
| In use in the vaccine industry; Humenza™ (influenza vaccine) [134] | |
| Polymer | Poly(lactic-co-glycolic acid) | FDA-approved | ||
| Poly(lactic acid) |
|
| FDA-approved | |
| Chitosan |
|
| FDA-approved | |
| Dextran (acetalated) |
| FDA-approved | ||
| Inorganic | Calcium phosphate nanoparticles |
| FDA-approved | |
| Silica nanoparticles |
| In clinical stage [146] | ||
| Gold nanoparticles |
| FDA-approved |
| Adjuvant | Type | Vaccine Name | Vaccine Type | Target Disease | Status |
|---|---|---|---|---|---|
| Toxin-based adjuvant | Heat-labile enterotoxin (LT) | - |
| Helicobacter pylori | In clinical |
| Double-mutant heat-labile toxin (dmLT) | ETEC vaccine (ACE527) [276] |
| Enterotoxigenic Escherichia coli (ETEC) | In clinical | |
| ETVAX [277] |
| Enterotoxigenic Escherichia coli (ETEC) | In clinical | ||
| - |
| Helicobacter pylori | In preclinical | ||
| - |
| Clostridium difficile | In preclinical | ||
| - |
| Clostridium tetani | In preclinical | ||
| - |
| Hepatitis B virus | In preclinical | ||
| - |
| Salmonella enteritidis | In preclinical | ||
| Multiple-mutated cholera toxin (mmCT) | - |
| Helicobacter pylori | In preclinical | |
| - | Vibrio cholerae and influenza virus | In preclinical | |||
| Recombinant cholera toxin B subunit (rCTB) | Dukoral [286] |
| Vibrio cholerae | Prequalified by WHO | |
| - |
| Helicobacter pylori | In preclinical | ||
| Cholera-toxin-derived adjuvant (CTA1DD) | CTA1-3M2e-DD [288] |
| Influenza virus | In preclinical | |
| Polysaccharide-based adjuvant | Chitosan | - |
| Helicobacter pylori | In preclinical |
| β-glucans | - |
| Influenza virus | In clinical | |
| - |
| Salmonella Typhi | In preclinical | ||
| - |
| Bacillus anthracis | In preclinical | ||
| Arabinoxylan (AX) | - |
| Influenza virus | In clinical | |
| Bacterial exopolysaccharide (EPS) | - |
| Influenza virus | In clinical | |
| Lipid-based adjuvant | α-galactosyl ceramide | - |
| Human immunodeficiency virus | In preclinical |
| - |
| Vibrio cholerae | In preclinical | ||
| - |
| Helicobacter pylori | In preclinical |
| Adjuvant | Type | Vaccine Name | Vaccine Type | Target Disease | Status |
|---|---|---|---|---|---|
| Toxin-based adjuvant | Heat-labile enterotoxin (LT) | Nasalflu (Berna Biotech) [316] | Inactivated vaccine | Influenza virus | In clinical |
| - | Inactivated vaccine [317] | Influenza virus | In clinical | ||
| Enzymatic A1 domain of LT (LTA1) | - | Subunit vaccine [318] | Influenza virus | In preclinical | |
| Cholera-toxin-derived adjuvant (CTA1DD) | - | Inactivated vaccine [319] | Influenza virus | In clinical | |
| - | Inactivated vaccine [320] | Human respiratory syncytial virus (hRSV) | In preclinical | ||
| - | Subunit vaccine [321] | Mycobacterium tuberculosis | In preclinical | ||
| - | Subunit vaccine [322,323] | Influenza virus | In preclinical | ||
| Cholera toxin B subunit (CTB) | - | Live attenuated vaccine [323] | Influenza virus | In preclinical | |
| - | Subunit vaccine [324] | Influenza virus | In preclinical | ||
| Polysaccharide-based adjuvant | RS09 (Ala-Pro-Pro-His-Ala-Leu-Ser) | - | Viral vector vaccine [325] | Human immunodeficiency virus | In preclinical |
| Mannatide (polyactin A) | - | Inactivated vaccine [326] | Influenza virus | In preclinical | |
| Advax™ | - | Live attenuated [327] | Influenza virus | In preclinical | |
| Chitosan | - | Non-viral vector vaccine [328] | Enterohemorrhagic Escherichia coli (EHEC) O157:H7 | In preclinical | |
| - | Inactivated vaccine [329] | Influenza virus | In preclinical | ||
| - | Subunit vaccine [330,331] | Influenza virus | In preclinical | ||
| - | Subunit vaccine [332] | Hepatitis B virus | In preclinical | ||
| Chitin | - | Live attenuated [323] | Influenza virus | In preclinical | |
| - | Subunit vaccine [333] | Influenza virus | In preclinical | ||
| Nucleotide-based adjuvant, synthetic adjuvant | Synthetic double-stranded RNA polyriboinosinic acid-polyribocytidylic acid [poly (I:C)] | - | Live attenuated vaccine [323] | Influenza virus | In preclinical |
| - | Inactivated vaccine [334] | Influenza virus | In preclinical | ||
| Cytosine-phosphate-guanine oligodeoxynucleotide (CpG ODN) | - | Subunit vaccine [335] | Influenza virus | In preclinical | |
| - | Inactivated vaccine [336] | Enterovirus | In preclinical | ||
| - | Subunit vaccine [337] | SARS-CoV-2 | In preclinical | ||
| - | Non-viral vector vaccine [338] | Pseudomonas aeruginosa | In preclinical | ||
| - | Non-viral vector vaccine [339] | Escherichia coli | In preclinical | ||
| Cyclic guanosine monophosphate-adenosine monophosphate (2′3′-cGAMP or cGAMP) | - | Subunit vaccine [340] | SARS-CoV-2 | In preclinical | |
| Aluminum-based adjuvant | Alhydrogel® (aluminum oxyhydroxide gel) | - | Subunit vaccine [341] | SARS-CoV-2 | In preclinical |
| Lipid-based adjuvant | Endocine™ (lipids monoolein and oleic acid) | - | Subunit vaccine [342] | Influenza virus | In preclinical |
| Protein-based adjuvant | Flagellin | - | Inactivated vaccine [343] | SARS-CoV-2 | In preclinical |
| - | Inactivated vaccine [344] | Influenza virus | In preclinical | ||
| Poly(γ-glutamic acid) | - | Inactivated vaccine [334] | Influenza virus | In preclinical | |
| Proteosome | - | Subunit vaccine [273] | SARS-CoV-2 | In preclinical | |
| - | Inactivated vaccine [345] | Influenza virus | In preclinical | ||
| ProtollinTM | - | Subunit vaccine [310] | Influenza virus | In preclinical |
| Adjuvant | Type | Vaccine Name | Vaccine Type | Target Disease | Administration Method | Status |
|---|---|---|---|---|---|---|
| Toxin derivates | Double-mutant heat-labile toxin (dmLT) | - | Subunit vaccine [346] | Nontypeable Haemophilus influenzae (NTHI) | Hydration (sterile, pyrogen-free 0.9% sodium chloride) | In preclinical |
| Single-mutant heat-labile enterotoxin | - | Subunit vaccine [347] | Enterotoxigenic Escherichia coli (ETEC) | Hydration and abrasion (70% isopropyl alcohol, glycerol 10%, and 10 swipes of medical grade sandpaper) | In clinical | |
| - | Subunit vaccine [348] | Corynebacterium diphtheriae | Hydration (sterile PBS) | In preclinical | ||
| - | Subunit vaccine [349] | Clostridium tetani | Hydration | In preclinical | ||
| Cholera toxin (CT) | - | Subunit vaccine [272] | Corynebacterium diphtheriae | Hollow microneedle (pre-treatment purpose) | In preclinical | |
| - | Inactivated vaccine [350] | Influenza virus | Hydration (saline-soaked gauze) | In preclinical | ||
| Cholera toxin B subunit (CTB) | - | Subunit vaccine [351] | hepatitis B virus | Solid microneedle and hydrogel patch | In preclinical | |
| Polysaccharide-based adjuvant | Chitosan | - | Subunit vaccine [352] | Corynebacterium diphtheriae | Hollow microneedle (pre- or post-treatment purpose) | In preclinical |
| Nucleotide-based adjuvant, synthetic adjuvant | Synthetic double-stranded RNA polyriboinosinic acid-polyribocytidylic acid [poly (I:C)] | - | Subunit vaccine [353] | Influenza virus | Coated microneedle | In preclinical |
| Cytosine-phosphate-guanine oligodeoxynucleotide (CpG ODN) | - | Subunit vaccine [354] | Clostridium tetani | Hydration (PBS-drenched wound plaster) | In preclinical | |
| - | Subunit vaccine [272] | Corynebacterium diphtheriae | Hollow microneedle (pre-treatment purpose) | In preclinical | ||
| - | Non-viral vector vaccine [355] | Chlamydia muridarum | Hydration (sterile PBS) | In preclinical | ||
| - | Subunit vaccine [356] | Human immunodeficiency virus | Hydration (saline-drenched gauze) | In preclinical | ||
| - | Subunit vaccine [357] | Foot-and-mouth disease virus | Hydration | In preclinical | ||
| Immune-stimulating complexes (ISCOMs) | Quil A | - | Subunit vaccine [272] | Corynebacterium diphtheriae | Hollow microneedle (pre-treatment purpose) | In preclinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.; Lee, P.; Choi, H.-J. Non-Invasive Vaccines: Challenges in Formulation and Vaccine Adjuvants. Pharmaceutics 2023, 15, 2114. https://doi.org/10.3390/pharmaceutics15082114
Han S, Lee P, Choi H-J. Non-Invasive Vaccines: Challenges in Formulation and Vaccine Adjuvants. Pharmaceutics. 2023; 15(8):2114. https://doi.org/10.3390/pharmaceutics15082114
Chicago/Turabian StyleHan, Sumin, Panjae Lee, and Hyo-Jick Choi. 2023. "Non-Invasive Vaccines: Challenges in Formulation and Vaccine Adjuvants" Pharmaceutics 15, no. 8: 2114. https://doi.org/10.3390/pharmaceutics15082114
APA StyleHan, S., Lee, P., & Choi, H. -J. (2023). Non-Invasive Vaccines: Challenges in Formulation and Vaccine Adjuvants. Pharmaceutics, 15(8), 2114. https://doi.org/10.3390/pharmaceutics15082114