

Evaluation of Pharmacokinetic Feasibility of Febuxostat/L-pyroglutamic Acid Cocrystals in Rats and Mice



Abstract
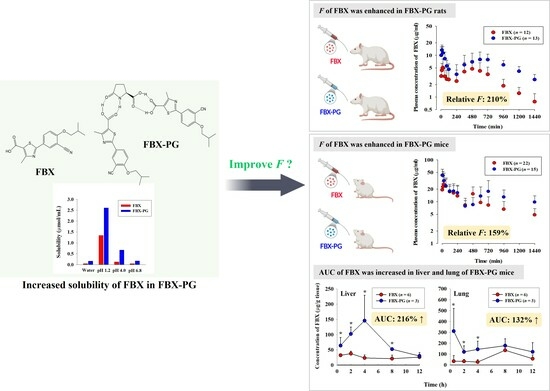
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animals
2.3. LC-MS/MS Analysis of FBX in the Biological Samples
2.4. Pharmacokinetics of FBX after Oral Administration of FBX or FBX-PG to Rats
2.5. Pharmacokinetics of FBX after Oral Administration of FBX or FBX-PG to Mice
2.6. Tissue Distribution of FBX after Oral Administration of FBX or FBX-PG to Mice
2.7. Plasma Protein Binding of FBX and FBX-PG in Rats and Mice
2.8. Pharmacokinetic Analysis
2.9. Statistical Analysis
3. Results
3.1. Pharmacokinetics of FBX after the Oral Administration of FBX or FBX-PG to Rats
3.2. Pharmacokinetics of FBX after the Oral Administration of FBX or FBX-PG to Mice
3.3. Tissue Distribution of FBX after the Oral Administration of FBX or FBX-PG to Mice
3.4. Plasma Protein Binding of FBX and FBX-PG in Rats and Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maddileti, D.; Jayabun, S.K.; Nangia, A. Soluble cocrystals of the xanthine oxidase inhibitor febuxostat. Cryst. Growth Des. 2013, 13, 3188–3196. [Google Scholar] [CrossRef]
- Samineni, R.; Chimakurthy, J.; Palei, N.N.; Yella, P.K.; Sabareesh, M.; Guru, P.M. Lamotrigine novel cocrystals: An attempt to enhance physicochemical parameters. J. Pharm. Negat. 2022, 13, 622–633. [Google Scholar] [CrossRef]
- Vishweshwar, P.; McMahon, J.A.; Bis, J.A.; Zaworotko, M.J. Pharmaceutical co-crystals. J. Pharm. Sci. 2006, 95, 499–516. [Google Scholar] [CrossRef]
- Guo, M.; Sun, X.; Chen, J.; Cai, T. Pharmaceutical cocrystals: A review of preparations, physicochemical properties and applications. Acta. Pharm. Sin. B. 2021, 11, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Maryam, K.-J.; Padrela, L.; Walker, G.M.; Croker, D.M. Creating cocrystals: A review of pharmaceutical cocrystal preparation routes and applications. Cryst. Growth Des. 2018, 18, 6370–6387. [Google Scholar]
- Ronik, D.F.V.; Hosni, A.P.; Brancalione, R.C.; Biscaia, I.F.B.; Bernardi, L.S.; Oliveira, P.R. Synthesis and characterization of ezetimibe pharmaceutical cocrystal: A reaction crystallization method to improve physicochemical properties and hypolipemic activity evaluation. J. Braz. Chem. Soc. 2023, e-20230114. [Google Scholar] [CrossRef]
- Shen, D.; Jin, T.; Xiao, Y.; Zhu, X.; Hua, Y. Preparation of pazopanib-fumarate disodium glycyrrhizinate nanocrystalline micelles by liquid-assisted ball milling. Eur. J. Pharm. Sci. 2023, 188, 106530. [Google Scholar] [CrossRef]
- Shubham; Wadhwa, R.; Usman, R.; Sharma, R.B.; Sakshi, T. Exploring cocrystals of imatinib: Synthesis, characterization, and in vitro evaluation. J. Surv. Fish. Sci. 2023, 10, 734–743. [Google Scholar]
- Chen, Y.; Li, L.; Yao, J.; Ma, Y.-Y.; Chen, J.-M.; Lu, T.-B. Improving the solubility and bioavailability of apixaban via apixaban–oxalic acid cocrystal. Cryst. Growth Des. 2016, 16, 2923–2930. [Google Scholar] [CrossRef]
- Jung, M.S.; Kim, J.S.; Kim, M.S.; Alhalaweh, A.; Cho, W.; Hwang, S.J.; Velaga, S.P. Bioavailability of indomethacin-saccharin cocrystals. J. Pharm. Pharmacol. 2010, 62, 1560–1568. [Google Scholar] [CrossRef]
- Zhang, L.; Kong, D.; Wang, H.; Jiao, L.; Zhao, X.; Song, J.; Yang, D.; Yang, H.; Yang, S.; Du, G.; et al. Cocrystal of apixaban–quercetin: Improving solubility and bioavailability of drug combination of two poorly soluble drugs. Molecules 2021, 26, 2677. [Google Scholar] [CrossRef] [PubMed]
- Khosravan, R.; Grabowski, B.; Wu, J.-T.; Joseph-Ridge, N.; Vernillet, L. Effect of food or antacid on pharmacokinetics and pharmacodynamics of febuxostat in healthy subjects. Br. J. Clin. Pharmacol. 2008, 65, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Roubenoff, R.; Klag, M.J.; Mead, L.A.; Liang, K.Y.; Seidler, A.J.; Hochberg, M.C. Incidence and risk factors for gout in white men. JAMA 1991, 266, 3004–3007. [Google Scholar] [CrossRef] [PubMed]
- Amin, O.M.; Ammar, A.; Eladawy, S.A. Febuxostat loaded β-cyclodextrin based nanosponge tablet: An in vitro and in vivo evaluation. J. Pharm. Investig. 2019, 50, 399–411. [Google Scholar] [CrossRef]
- White, W.B.; Saag, K.G.; Becker, M.A.; Borer, J.S.; Gorelick, P.B.; Whelton, A.; Hunt, B.; Castillo, M.; Gunawardhana, L.; CARES Investigators. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N. Engl. J. Med. 2018, 378, 1200–1210. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, J.; Yang, D.; Hu, Y.; Hu, X.; Jiang, B.; Ruan, Z.; Lou, H. Development of LC-MS/MS determination method and backpropagation artificial neural networks pharmacokinetic model of febuxostat in healthy subjects. J. Clin. Pharm. Ther. 2021, 46, 333–342. [Google Scholar] [CrossRef]
- Ahuja, B.K.; Jena, S.K.; Paidi, S.K.; Bagri, S.; Suresh, S. Formulation, optimization and in vitro-in vivo evaluation of febuxostat nanosuspension. Int. J. Pharm. 2015, 478, 540–552. [Google Scholar] [CrossRef]
- An, J.-H.; Lim, C.; Ryu, H.C.; Kim, J.S.; Kim, H.M.; Kiyonga, A.N.; Park, M.; Suh, Y.G.; Park, G.H.; Jung, K. Structural characterization of febuxostat/L-pyroglutamic acid cocrystal using solid-state 13C-NMR and investigational study of its water solubility. Crystals 2017, 7, 365. [Google Scholar] [CrossRef]
- Gurumukhi, V.C.; Sonawane, V.P.; Tapadiya, G.G.; Bari, S.B.; Surana, S.J.; Chalikwar, S.S. Quality-by-design based fabrication of febuxostat-loaded nanoemulsion: Statistical optimization, characterizations, permeability, and bioavailability studies. Heliyon 2023, 9, e15404. [Google Scholar] [CrossRef]
- Habib, B.A.; Abd El-Samiae, A.S.; El-Houssieny, B.M.; Tag, R. Formulation, characterization, optimization, and in-vivo performance of febuxostat self-nano-emulsifying system loaded sublingual films. Drug. Deliv. 2021, 28, 1321–1333. [Google Scholar] [CrossRef]
- Haq, N.; Alghaith, A.F.; Alshehri, S.; Shakeel, F. Solubility and thermodynamic data of febuxostat in various mono solvents at different temperatures. Molecules 2022, 27, 4043. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, X. Cocrystallization of febuxostat with pyridine coformers: Crystal structural and physicochemical properties analysis. J Chem. 2021, 2021, 3834368. [Google Scholar] [CrossRef]
- Han, X.; Qi, W.; Dong, W.; Guo, M.; Ma, P.; Wang, J. Preparation, optimization and in vitro–in vivo investigation for capsules of the choline salt of febuxostat. Asian J. Pharm. Sci. 2016, 11, 715–721. [Google Scholar] [CrossRef]
- Kang, Y.; Gu, J.; Hu, X. Syntheses, structure characterization and dissolution of two novel cocrystals of febuxostat. Mol. Struct. 2016, 15, 480–486. [Google Scholar] [CrossRef]
- Sohn, J.S.; Choi, J.S. Development and evaluation of febuxostat solid dispersion through screening method. Saudi Pharm. J. 2023, 31, 101724. [Google Scholar] [CrossRef]
- Banerjee, R.; Bhatt, P.M.; Ravindra, N.V.; Desiraju, G.R. Saccharin salts of active pharmaceutical ingredients, their crystal structures, and increased water solubilities. Cryst. Growth Des. 2005, 5, 2299–2309. [Google Scholar] [CrossRef]
- Basavoju, S.; Boström, D.; Velaga, S.P. Pharmaceutical cocrystal and salts of norfloxacin. Cryst. Growth Des. 2006, 6, 2699–2708. [Google Scholar] [CrossRef]
- Byrn, S.R.; Pfeiffer, R.R.; Stowell, J.G. Solid-State Chemistry of Drugs, 2nd ed.; SSCI: West Lafayette, IN, USA, 1999. [Google Scholar]
- Good, D.J.; Rodríguez-Hornedo, N. Solubility advantage of pharmaceutical cocrystals. Cryst. Growth Des. 2009, 9, 2252–2264. [Google Scholar] [CrossRef]
- McNamara, D.P.; Childs, S.L.; Giordano, J.; Iarriccio, A.; Cassidy, J.; Shet, M.S.; Mannion, R.; O’Donnell, E.; Park, A. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm. Res. 2006, 23, 1888–1897. [Google Scholar] [CrossRef]
- Remenar, J.F.; Morissette, S.L.; Peterson, M.L.; Moulton, B.; MacPhee, J.M.; Guzmán, H.R.; Almarsson, O. Crystal engineering of novel cocrystals of a triazole drug with 1,4-dicarboxylic acids. J. Am. Chem. Soc. 2003, 125, 8456–8457. [Google Scholar] [CrossRef]
- Sanphui, P.; Goud, N.R.; Khandavilli, U.B.; Nangia, A. Fast dissolving curcumin cocrystals. Cryst. Growth Des. 2011, 11, 4135–4145. [Google Scholar] [CrossRef]
- Sanphui, P.; Goud, N.R.; Khandavilli, U.B.; Bhanoth, S.; Nangia, A. New polymorphs of curcumin. Chem. Commun. 2011, 47, 5013–5015. [Google Scholar] [CrossRef]
- Jagia, M.; Kale, D.P.; Bansal, A.K.; Patel, S. Novel co-crystals and eutectics of febuxostat: Characterization, mechanism of formation, and improved dissolution. AAPS PharmSciTech. 2021, 23, 43. [Google Scholar] [CrossRef] [PubMed]
- Safety Data Sheet (L-pyroglutamic Acid) Spectrum Chemical Home Page. Available online: https://www.spectrumchemical.com/media/sds/P1835_AGHS.pdf (accessed on 3 May 2018).
- Chen, J.; Sarma, B.; Evans, J.M.B.; Myerson, A.S. Pharmaceutical Crystallization. Cryst. Growth Des. 2011, 11, 887–895. [Google Scholar] [CrossRef]
- Abuhelwa, A.Y.; Williams, D.B.; Upton, R.N.; Foster, D.J. Food, gastrointestinal pH, and models of oral drug absorption. Eur. J. Pharm. Biopharm. 2017, 112, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.C. Crystalline Febuxostat Pidolate Salt and Method for Preparing Thereof. K.R. Patent 10-1501253, 4 March 2015. [Google Scholar]
- Han, S.Y.; Chae, H.S.; You, B.H.; Chin, Y.-W.; Kim, H.J.; Choi, H.S.; Choi, Y.H. Lonicera japonica extract increases metformin distribution in the liver without change of systemic exposed metformin in rats. J. Ethnopharmacol. 2019, 238, 111892. [Google Scholar] [CrossRef] [PubMed]
- You, B.H.; Chin, Y.-W.; Kim, H.J.; Choi, H.S.; Choi, Y.H. Houttuynia cordata extract increased systemic exposure and liver concentrations of metformin through OCTs and MATEs in rats. Phytother. Res. 2018, 32, 1004–1013. [Google Scholar] [CrossRef]
- You, B.H.; Bae, M.; Han, S.Y.; Jung, J.; Jung, K.; Choi, Y.H. Pharmacokinetic feasibility of stability-enhanced solid-state (SESS) tenofovir disoproxil free base crystal. Pharmaceutics 2023, 15, 1392. [Google Scholar] [CrossRef]
- Diehl, K.H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.M.; van de Vorstenbosch, C. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [CrossRef]
- Golde, W.T.; Gollobin, P.; Rodriguez, L.L. A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab. Anim. 2005, 34, 39–43. [Google Scholar] [CrossRef]
- Lee, C.W.; You, B.H.; Yim, S.; Han, S.Y.; Chae, H.S.; Bae, M.; Kim, S.Y.; Yu, J.E.; Jung, J.; Nhoek, P.; et al. Change of metformin concentrations in the liver as a pharmacological target site of metformin after long-term combined treatment with ginseng berry extract. Front. Pharmacol. 2023, 14, 1148155. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; You, B.H.; Kim, Y.C.; Chin, Y.-W.; Choi, Y.H. Dose-independent ADME properties and tentative identification of metabolites of α-mangostin from Garcinia mangostana in mice by automated microsampling and UPLC-MS/MS methods. PLoS ONE 2015, 10, e0131587. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Han, S.Y.; Seo, J.S.; Chin, Y.-W.; Choi, Y.H. Pharmacokinetics, tissue distribution, and tentative metabolite identification of sauchinone in mice by microsampling and HPLC-MS/MS methods. Biol. Pharm. Bull. 2015, 38, 218–227. [Google Scholar] [CrossRef]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel-Dekker: Boca Raton, FL, USA, 1982. [Google Scholar]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet. Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, B.A.; Khosravan, R.; Vernillet, L.; Mulford, D.J. Metabolism and excretion of [14C] febuxostat, a novel nonpurine selective inhibitor of xanthine oxidase, in healthy male subjects. J. Clin. Pharmacol. 2011, 51, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Khosravan, R.; Grabowski, B.A.; Wu, J.-T.; Joseph-Ridge, N.; Vernillet, L. Pharmacokinetics, pharmacodynamics and safety of febuxostat, a non-purine selective inhibitor of xanthine oxidase, in a dose escalation study in healthy subjects. Clin. Pharmacokinet. 2006, 45, 821–841. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug. Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Lin, J.H. Tissue distribution and pharmacodynamics: A complicated relationship. Curr. Drug. Metab. 2006, 7, 39–65. [Google Scholar] [CrossRef]
- Bryda, E.C. The mighty mouse: The impact of rodents on advances in biomedical research. Mo. Med. 2013, 110, 207–211. [Google Scholar]
- Sivakrishnan, S.; Anbiah, S.V. Animals used in experimental pharmacology and 3 Rs. Pharmacophore 2021, 12, 1–7. [Google Scholar] [CrossRef]
- An, G.; Morris, M.E. A Physiologically based pharmacokinetic model of mitoxantrone in mice and scale-up to humans: A semi-mechanistic model incorporating DNA and protein binding. AAPS J. 2012, 14, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Han, S.Y.; Kim, Y.J.; Kim, Y.M.; Chin, Y.W. Absorption, tissue distribution, tissue metabolism and safety of α-mangostin in mangosteen extract using mouse models. Food Chem. Toxicol. 2014, 66, 140–146. [Google Scholar] [CrossRef]
- Wiberg, G.S.; Trenholm, H.L.; Coldwell, B.B. Increased ethanol toxicity in old rats: Changes in LD50, in vivo and in vitro metabolism, and liver alcohol dehydrogenase activity. Toxicol. Appl. Pharmacol. 1970, 16, 718–727. [Google Scholar] [CrossRef] [PubMed]
- The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) Home Page. Available online: https://database.ich.org/sites/default/files/ICH_Q3C-R8_Guideline_Step4_2021_0422_1.pdf (accessed on 21 August 2011).
- Tsibulsky, V.L.; Amit, Z. Tolerance to effects of high doses of ethanol: 1. Lethal effects in mice. Pharmacol. Biochem. Behav. 1993, 45, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Fitz, Y.; Li, Y.; Fernandez, M.; Cortes Puch, I.; Wang, D.; Pazniokas, S.; Bucher, B.; Cui, X.; Solomon, S.B. Catheterization of the carotid artery and jugular vein to perform hemodynamic measures, infusions and blood sampling in a conscious rat model. J. Vis. Exp. 2015, 95, 51881. [Google Scholar]
- Hoggatt, J.; Hoggatt, A.F.; Tate, T.A.; Fortman, J.; Pelus, L.M. Bleeding the laboratory mouse: Not all methods are equal. Exp. Hematol. 2016, 44, 132–137. [Google Scholar] [CrossRef]
- Parasuraman, S.; Raveendran, R.; Kesavan, R. Blood sample collection in small laboratory animals. J. Pharmacol. Pharmacother. 2010, 1, 87–93. [Google Scholar]
- Arunachalam, K.; Sasidharan, S.P. Bioassays in Experimental and Preclinical Pharmacology, 1st ed.; Springer: New York, NY, USA, 2021. [Google Scholar]
- Florida Atlantic University Institutional Animal Care and Use Committee (FAU IACUC). Guidelines for Rodent Survival Blood Collection; Florida Atlantic University: Boca Raton, FL, USA, 2021. [Google Scholar]
- Wolforth, J.B. Methods of blood collection in the mouse. Lab. Anim. 2000, 29, 47–53. [Google Scholar]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, F.; Zhao, X.; Wang, S.; Yang, Q.; Zhang, X. Crystal structure, solubility, and pharmacokinetic study on a hesperetin cocrystal with piperine as conformer. Pharmaceutics 2022, 14, 94. [Google Scholar] [CrossRef]
- Kimoto, K.; Yamamoto, M.; Karashima, M.; Hohokabe, M.; Takeda, J.; Yamamoto, K.; Ikeda, Y. Pharmaceutical cocrystal development of TAK-020 with enhanced oral absorption. Crystals 2020, 10, 211. [Google Scholar] [CrossRef]
- Kataoka, M.; Minami, K.; Takagi, T.; Amidon, G.E.; Yamashita, S. In vitro-In vivo correlation in cocrystal dissolution: Consideration of drug release profiles based on coformer dissolution and absorption behavior. Mol. Pharm. 2021, 18, 4122–4130. [Google Scholar] [CrossRef] [PubMed]
- Mukoyoshi, M.; Nishimura, S.; Hoshide, S.; Umeda, S.; Kanou, M.; Taniguchi, K.; Muroga, H. In vitro drug-drug interaction studies with febuxostat, a novel non-purine selective inhibitor of xanthine oxidase: Plasma protein binding, identification of metabolic enzymes and cytochrome P450 inhibition. Xenobiotica 2008, 38, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Kamel, B.; Graham, G.G.; Williams, K.M.; Pile, K.D.; Day, R.O. Clinical pharmacokinetics and pharmacodynamics of febuxostat. Clin. Pharmacokinet. 2017, 56, 459–475. [Google Scholar] [CrossRef]
- Davies, N.M.; Takemoto, J.K.; Brocks, D.R.; Yáñez, J.A. Multiple peaking phenomena in pharmacokinetic disposition. Clin. Pharmacokinet. 2010, 49, 351–377. [Google Scholar] [CrossRef]
- Helmy, S.A. Therapeutic drug monitoring and pharmacokinetic compartmental analysis of sulpiride double-peak absorption profile after oral administration to human volunteers. Biopharm. Drug Dispos. 2013, 34, 288–301. [Google Scholar] [CrossRef]
- Ogungbenro, K.; Pertinez, H.; Aarons, L. Empirical and semi-mechanistic modelling of double-peaked pharmacokinetic profile phenomenon due to gastric emptying. AAPS J. 2015, 17, 227–236. [Google Scholar] [CrossRef]
- van Duijkeren, E.; Ensink, J.M.; Meijer, L.A. Distribution of orally administered trimethoprim and sulfadiazine into noninfected subcutaneous tissue chambers in adult ponies. J. Vet. Pharmacol. Ther. 2002, 25, 273–277. [Google Scholar] [CrossRef]
- You, B.H.; BasavanaGowda, M.K.; Lee, J.U.; Chin, Y.-W.; Choi, W.J.; Choi, Y.H. Pharmacokinetic properties of moracin C in mice. Planta Med. 2021, 87, 642–651. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | FBX (n = 12) | FBX-PG (n = 13) |
|---|---|---|
| Body weight (g) | 325 ± 37.4 | 330 ± 36.2 |
| Terminal t1/2 (min) | 434 ± 206 | 472 ± 207 |
| Cmax,1 (μg/mL) | 5.62 ± 2.86 | 14.6 ± 4.48 * |
| Tmax,1 (min) 1 | 30.0 (15–90) | 15.0 (15–30) * |
| Cmax,2 (μg/mL) | 5.63 ± 2.63 | 9.44 ± 3.27 * |
| Tmax,2 (min) 1 | 480 (120–960) | 600 (480–960) * |
| AUC0–1440min (μg min/mL) | 4144 ± 1784 | 8251 ± 2566 * |
| AUC0-inf (μg min/mL) | 4824 ± 1960 | 10,149 ± 3164 * |
| Vz/F (mL/kg) | 7502 ± 4709 | 3508 ± 1567 * |
| CL/F (mL/min/kg) | 12.5 ± 6.10 | 5.35 ± 1.58 * |
| Ae0–24h (% of dose) | 3.64 ± 1.25 | 3.69 ± 1.64 |
| GI24h (% of dose) | 51.7 ± 15.7 | 40.8 ± 9.68 * |
| Relative F0–1440min (%) | 199 | |
| Relative F (%) | 210 |
| Parameters | FBX (n = 22) | FBX-PG (n = 15) |
|---|---|---|
| Body weight (g) | 35.3 ± 3.94 | 37.5 ± 3.62 |
| Terminal t1/2 (min) | 880 | 883 |
| Cmax,1 (μg/mL) | 26.0 | 42.9 |
| Tmax,1 (min) 1 | 30.0 | 5.00 |
| Cmax,2 (μg/mL) | 15.3 | 17.6 |
| Tmax,2 (min) 1 | 480 | 720 |
| AUC0–1440min (μg min/mL) | 14,187 | 19,831 |
| AUC0-inf (μg min/mL) | 20,324 | 32,253 |
| Vz/F (mL/kg) | 3123 | 1974 |
| CL/F (mL/min/kg) | 2.46 | 1.55 |
| Relative F0–1440min (%) | 140 | |
| Relative F (%) | 159 |
| Tissue | FBX (n = 30) | FBX-PG (n = 15) |
|---|---|---|
| Liver | 17,234 ± 5813 | 54,403 ± 10,551 * |
| Kidney | 6961 ± 3954 | 9285 ± 619 |
| Stomach | 70,803 ± 19,964 | 114,976 ± 53,471 |
| Small intestine | 16,447 ± 8793 | 14,983 ± 2818 |
| Large intestine | 9434 ± 7230 | 18,978 ± 7719 |
| Lung | 48,314 ± 16,259 | 112,008 ± 19,080 * |
| Heart | 1971 ± 841 | 1746 ± 738 |
| Fat | 3356 ± 1392 | 2834 ± 1302 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.-E.; You, B.H.; Bae, M.; Han, S.Y.; Jung, K.; Choi, Y.H. Evaluation of Pharmacokinetic Feasibility of Febuxostat/L-pyroglutamic Acid Cocrystals in Rats and Mice. Pharmaceutics 2023, 15, 2167. https://doi.org/10.3390/pharmaceutics15082167
Yu J-E, You BH, Bae M, Han SY, Jung K, Choi YH. Evaluation of Pharmacokinetic Feasibility of Febuxostat/L-pyroglutamic Acid Cocrystals in Rats and Mice. Pharmaceutics. 2023; 15(8):2167. https://doi.org/10.3390/pharmaceutics15082167
Chicago/Turabian StyleYu, Jeong-Eun, Byoung Hoon You, Mingoo Bae, Seung Yon Han, Kiwon Jung, and Young Hee Choi. 2023. "Evaluation of Pharmacokinetic Feasibility of Febuxostat/L-pyroglutamic Acid Cocrystals in Rats and Mice" Pharmaceutics 15, no. 8: 2167. https://doi.org/10.3390/pharmaceutics15082167
APA StyleYu, J. -E., You, B. H., Bae, M., Han, S. Y., Jung, K., & Choi, Y. H. (2023). Evaluation of Pharmacokinetic Feasibility of Febuxostat/L-pyroglutamic Acid Cocrystals in Rats and Mice. Pharmaceutics, 15(8), 2167. https://doi.org/10.3390/pharmaceutics15082167