Interaction Potential of the Multitargeted Receptor Tyrosine Kinase Inhibitor Dovitinib with Drug Transporters and Drug Metabolising Enzymes Assessed in Vitro



Abstract
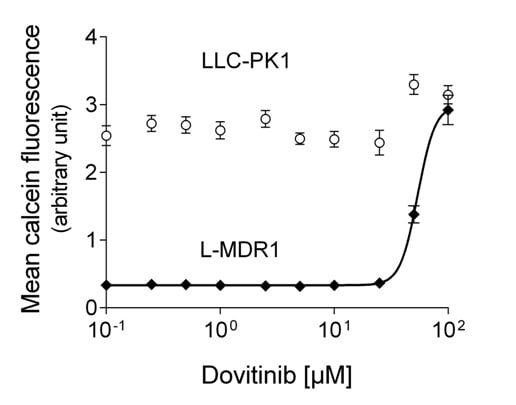
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Stock Solutions
2.3. Cell Lines
2.3.1. LS180 Cells
2.3.2. MDCKII Cells
2.3.3. LLC-PK1 and L-MDR1 Cells
2.3.4. HEK293 Cells
2.3.5. AZ-AHR Cells
2.4. Cytotoxicity Assay
2.5. P-gp Inhibition Assay (Calcein Uptake Assay)
2.6. BCRP Inhibition Assay (Pheophorbide A Flow Cytometry Efflux Assay)
2.7. OATP Inhibition Assay (8-FcA Flow Cytometry Uptake Assay)
2.8. Inhibition of CYP3A4, CYP2C19, and CYP2D6
2.9. Growth Inhibition Assay
2.10. Induction Assay
2.11. Quantification of mRNA Expression by Real-Time RT-PCR
2.12. Western Blot Analysis of P-gp Expression
2.13. AhR Reporter Gene Assay
2.14. Statistical Analysis
3. Results
3.1. Dovtinib Weakly Inhibits P-gp


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
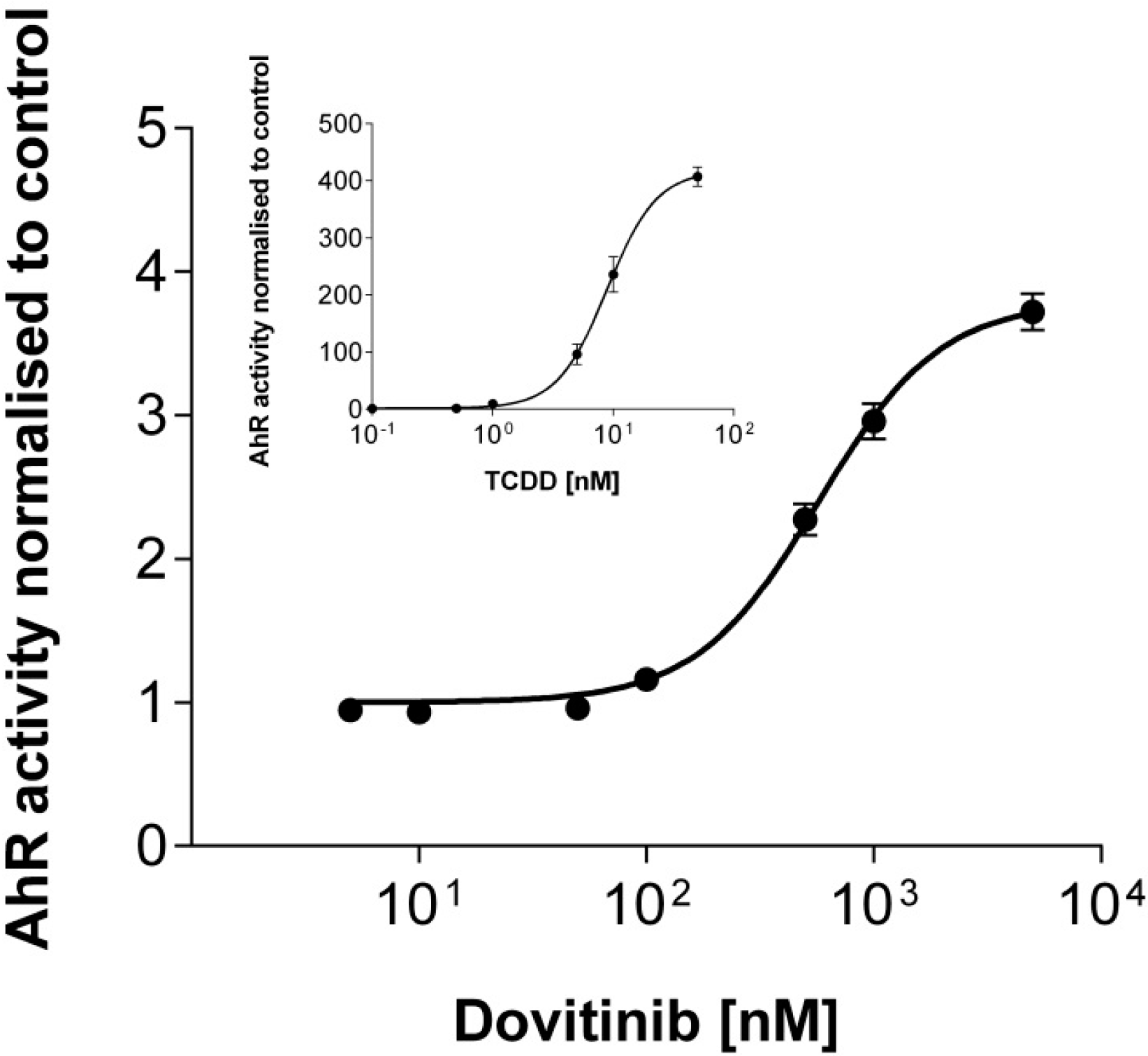{kind=link}
{kind=link}
| Protein Inhibited | Cell Model Used | Dovitinib f2 [µM] | Control Compound | f2 [µM] |
|---|---|---|---|---|
| P-gp | L-MDR1 | 48.4 ± 15.6 | Verapamil | 3.9 ± 1.9 [28] |
| IC50 [µM] | IC50 [µM] | |||
| BCRP | MDCKII-BCRP | 10.3 ± 4.5 | Fumitremorgin C | 0.7 ± 0.3 [37] |
| OATP1B1 | HEK-OATP1B1 | 76.5 ± 28.9 | Rifampicin | 2.4 ± 0.9 [13] |
| OATP1B3 | HEK-OATP1B3 | 105.2 ± 23.0 | Rifampicin | 2.1 ± 1.0 [13] |
3.2. Dovitinib Weakly Inhibits OATP1B1 and OATP1B3
3.3. Dovitinib Inhibits BCRP
3.4. Inhibition of CYPs
3.5. Influence of Dovitinib on the mRNA Expression of Drug Transporters, Drug Metabolising Enzymes, and PXR



3.6. Activation of AhR Activity

3.7. Efficacy in MDR Cell Lines

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, K.B.; Saro, J.; Moschos, S.S.; Hwu, P.; Tarhini, A.A.; Hwu, W.; Jones, G.; Wang, Y.; Rupani, H.; Kirkwood, J.W. A phase I dose finding and biomarker study of TKI258 (dovitinib lactate) in patients with advanced melanoma. J. Clin. Oncol. 2008, 26, 9026. [Google Scholar]
- André, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; PHurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with Dovitinib (TKI258): Preclinical and Clinical Data in Breast Cancer. Clin. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Lopez-Martin, J.A.; Lin, C.C.; Gschwend, J.E.; Harzstark, A.; Castellano, D.; Soria, J.C.; Sen, P.; Chang, J.; Shi, M.; et al. Phase I study of dovitinib (TKI258); an oral FGFR; VEGFR; and PDGFR inhibitor; in advanced or metastatic renal cell carcinoma. Clin. Cancer Res. 2013, 19, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Yoo, C.; Ryoo, B.Y.; Lee, J.J.; Tan, E.; Park, I.; Park, J.H.; Choi, Y.J.; Jo, J.; Ryu, J.S.; et al. Phase II study of dovitinib in patients with metastatic and/or unresectable gastrointestinal stromal tumours after failure of imatinib and sunitinib. Br. J. Cancer 2013, 109, 2309–2315. [Google Scholar] [PubMed]
- Konecny, G.E.; Kolarova, T.; O’Brien, N.A.; Winterhoff, B.; Yang, G.; Qi, J.; Qi, Z.; Venkatesan, N.; Ayala, R.; Luo, T.; et al. Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol. Cancer Ther. 2013, 12, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Cheng, A.L.; Shiau, C.W.; Liu, C.Y.; Ko, C.H.; Lin, M.W.; Chen, P.J.; Chen, K.F. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol. Cancer Ther. 2012, 11, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Ryu, M.H.; Na, Y.S.; Ryoo, B.Y.; Park, S.R.; Kang, Y.K. Analysis of serum protein biomarkers, circulating tumor DNA, and dovitinib activity in patients with tyrosine kinase inhibitor-refractory gastrointestinal stromal tumors. Ann. Oncol. 2014, 25, 2272–7227. [Google Scholar] [PubMed]
- Galsky, M.D.; Posner, M.; Holcombe, R.F.; Lee, K.M.; Misiukiewicz, K.; Tsao, C.K.; Godbold, J.; Soto, R.; Gimpel-Tetra, K.; Lowe, N.; et al. Phase Ib study of dovitinib in combination with gemcitabine plus cisplatin or gemcitabine plus carboplatin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 465–471. [Google Scholar] [PubMed]
- Escudier, B.; Grünwald, V.; Ravaud, A.; Ou, Y.C.; Castellano, D.; Lin, C.C.; Gschwend, J.E.; Harzstark, A.; Beall, S.; Pirotta, N.; et al. Phase II results of Dovitinib (TKI258) in patients with metastatic renal cell cancer. Clin. Cancer Res. 2014, 20, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Porta, C.; Vogelzang, N.J.; Sternberg, C.N.; Szczylik, C.; Zolnierek, J.; Kollmannsberger, C.; Rha, S.Y.; Bjarnason, G.A.; Melichar, B.; et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 286–296. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kay, A.; Anak, O.; Angevin, E.; Escudier, B.; Zhou, W.; Feng, Y.; Dugan, M.; Schran, H. Population pharmacokinetic/pharmacodynamic modeling to assist dosing schedule selection for dovitinib. J. Clin. Pharmacol. 2013, 53, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Theile, D.; Spalwisz, A.; Burhenne, J.; Riedel, K.D.; Haefeli, W.E. Influence of sildenafil and tadalafil on the enzyme- and transporter-inducing effects of bosentan and ambrisentan in LS180 cells. Biochem. Pharmacol. 2013, 85, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Koster, A.S.; Beijnen, J.H.; Schellens, J.H.; Meijerman, I. Comparison of two immortalized human cell lines to study nuclear receptor-mediated CYP3A4 induction. Drug Metab. Dispos. 2008, 36, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mugundu, G.M.; Desai, P.B.; Thummel, K.E.; Unadkat, J.D. Intestinal human colon adenocarcinoma cell line LS180 is an excellent model to study pregnane X receptor; but not constitutive androstane receptor; mediated CYP3A4 and multidrug resistance transporter 1 induction: Studies with anti-human immunodeficiency virus protease inhibitors. Drug Metab. Dispos. 2008, 36, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Herzog, M.; Haefeli, W.E. Differential modulation of the expression of important drug metabolising enzymes and transporters by endothelin-1 receptor antagonists ambrisentan and bosentan in vitro. Eur. J. Pharmacol. 2011, 660, 298–304. [Google Scholar] [PubMed]
- Brandin, H.; Viitanen, E.; Myrberg, O.; Arvidsson, A.K. Effects of herbal medicinal products and food supplements on induction of CYP1A2; CYP3A4 and MDR1 in the human colon carcinoma cell line LS180. Phytother. Res. 2007, 21, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, D.; Nakamura, T.; Okamura, N.; Kokudai, M.; Inui, N.; Takeuchi, K.; Watanabe, H.; Hirai, M.; Okumura, K.; Sakaeda, T. Effects of acid and lactone forms of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors on the induction of MDR1 expression and function in LS180 cells. Eur. J. Pharm. Sci. 2009, 37, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Harper, P.A.; Tang, B.K.; Okey, A.B. Regulation of cytochrome P450 enzymes by aryl hydrocarbon receptor in human cells: CYP1A2 expression in the LS180 colon carcinoma cell line after treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 3-methylcholanthrene. Biochem. Pharmacol. 1998, 56, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Harper, P.A.; Prokipcak, R.D.; Bush, L.E.; Golas, C.L.; Okey, A.B. Detection and characterization of the Ah receptor for 2,3,7,8-tetrachlorodibenzo-p-dioxin in the human colon adenocarcinoma cell line LS180. Arch. Biochem. Biophys. 1991, 290, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; Borst, P. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J. Clin. Investig. 1995, 96, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Merino, G.; Wagenaar, E.; Bolscher, E.; Novotna, M.; Jonker, J.W.; Schinkel, A.H. Human breast cancer resistance protein: Interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J. Pharmacol. Exp. Ther. 2005, 312, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Wagenaar, E.; Mol, C.A.; van Deemter, L. P-Glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J. Clin. Investig. 1996, 97, 2517–2524. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Cui, Y.; Nies, A.T.; Keppler, D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J. Biol. Chem. 2000, 275, 23161–23168. [Google Scholar] [CrossRef] [PubMed]
- Köni, J.; Cui, Y.; Nies, A.T.; Keppler, D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G156–G164. [Google Scholar] [PubMed]
- Novotna, A.; Pavek, P.; Dvorak, Z. Novel stably transfected gene reporter human hepatoma cell line for assessment of aryl hydrocarbon receptor transcriptional activity: Construction and characterization. Environ. Sci. Technol. 2011, 45, 10133–10139. [Google Scholar] [CrossRef] [PubMed]
- Holló, Z.; Homolya, L.; Hegedüs, T.; Sarkadi, B. Transport properties of the multidrug resistance-associated protein (MRP) in human tumour cells. FEBS Lett. 1996, 383, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Dormann, S.M.; Martin-Facklam, M.; Kerpen, C.J.; Ketabi-Kiyanvash, N.; Haefeli, W.E. Inhibition of P-glycoprotein by newer antidepressants. J. Pharmacol. Exp. Ther. 2003, 305, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Haefeli, W.E. Evaluation of inhibitory potencies for compounds inhibiting P-glycoprotein but without maximum effects: f2 values. Drug Metab. Dispos. 2006, 34, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Rose, J.; Storch, C.H.; Ketabi-Kiyanvash, N.; Sauer, A.; Haefeli, W.E.; Efferth, T. Modulation of human BCRP (ABCG2) activity by anti-HIV drugs. J. Antimicrob. Chemother. 2007, 59, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.; Lindenmaier, H.; Haefeli, W.E.; Weiss, J. Interaction of the mitotic kinesin Eg5 inhibitor monastrol with P-glycoprotein. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 372, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Albermann, N.; Schmitz-Winnenthal, F.H.; Z’graggen, K.; Volk, C.; Hoffmann, M.M.; Haefeli, W.E.; Weiss, J. Expression of the drug transporters MDR1/ABCB1; MRP1/ABCC1; MRP2/ABCC2; BCRP/ABCG2; and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 2005, 70, 949–958. [Google Scholar] [CrossRef] [PubMed]
- König, S.J.; Herzog, M.; Theile, D.; Zembruski, N.; Haefeli, W.E.; Weiss, J. Impact of drug transporters for the cellular resistance towards saquinavir and darunavir. J. Antimicrob. Chemother. 2010, 65, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, Z.; Vrzal, R.; Henklova, P.; Jancova, P.; Anzenbacherova, E.; Maurel, P.; Svecova, L.; Pavek, P.; Ehrmann, J.; Havlik, R.; et al. JNK inhibitor SP600125 is a partial agonist of human aryl hydrocarbon receptor and induces CYP1A1 and CYP1A2 genes in primary human hepatocytes. Biochem. Pharmacol. 2008, 5, 580–588. [Google Scholar] [CrossRef]
- Ayed-Boussema, I.; Pascussi, J.M.; Maurel, P.; Bacha, H.; Hassen, W. Zearalenone activates pregnane X receptor; constitutive androstane receptor and aryl hydrocarbon receptor and corresponding phase I target genes mRNA in primary cultures of human hepatocytes. Environ. Toxicol. Pharmacol. 2011, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.; Sauer, A.; Divac, N.; Herzog, M.; Schwedhelm, E.; Böger, R.H.; Haefeli, W.E.; Benndorf, R.A. Interaction of angiotensin receptor type 1 blockers with ATP-binding cassette transporters. Biopharm. Drug Dispos. 2010, 31, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Mandery, K.; Glaeser, H.; Fromm, M.F. Interaction of innovative small molecule drugs used for cancer therapy with drug transporters. Br. J. Pharmacol. 2012, 165, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Molife, R.; Evans, T.R.; Hardie, M.; Marriott, C.; Butzberger-Zimmerli, P.; Morrison, R.; Fox, J.A.; Heise, C.; Louie, S.; et al. A phase I pharmacokinetic and pharmacodynamic study of TKI258; an oral; multitargeted receptor tyrosine kinase inhibitor in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.B.; Chesney, J.; Robinson, D.; Gardner, H.; Shi, M.M.; Kirkwood, J.M. Phase I/II and pharmacodynamic study of dovitinib (TKI258); an inhibitor of fibroblast growth factor receptors and VEGF receptors; in patients with advanced melanoma. Clin. Cancer Res. 2011, 17, 7451–7461. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Ding, N.; Xiao, G.; Wang, S.; Wu, Y.; Tang, L. Reversal of multidrug resistance by gefitinib via RAF1/ERK pathway in pancreatic cancer cell line. Anat. Rec. 2012, 295, 2122–2128. [Google Scholar] [CrossRef]
- Garcia, R.; Franklin, R.A.; McCubrey, J.A. EGF induces cell motility and multi-drug resistance gene expression in breast cancer cells. Cell Cycle 2006, 5, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Zhao, L.; Yang, Z.; Zhao, Q.; Qiang, L.; Ha, J.; Li, Z.Y.; You, Q.D.; Guo, Q.L. Oroxylin A reverses multi-drug resistance of human hepatoma BEL7402/5-FU cells via downregulation of P-glycoprotein expression by inhibiting NF-κB signaling pathway. Mol. Carcinog. 2012, 51, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Yueh, M.F.; Mellon, P.L.; Tukey, R.H. Inhibition of human UGT2B7 gene expression in transgenic mice by the constitutive androstane receptor. Mol. Pharmacol. 2011, 79, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, S.; Meijerman, I.; Maas-Bakker, R.F.; Beijnen, J.H.; Schellens, J.H. PXR-mediated P-glycoprotein induction by small molecule tyrosine kinase inhibitors. Eur. J. Pharm. Sci. 2013, 48, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.; Seidel, A.; Lampen, A. Identification of BCRP as transporter of benzo(a)pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005, 26, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y.D.; Strong, J.M.; Reynolds, K.S.; Huang, S.M. A regulatory viewpoint on transporter-based drug interactions. Xenobiotica 2008, 38, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Pfrunder, A.; Gutmann, H.; Beglinger, C.; Drewe, J. Gene expression of CYP3A4; ABC-transporters (MDR1 and MRP1-MRP5) and hPXR in three different human colon carcinoma cell lines. J. Pharm. Pharmacol. 2003, 55, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, B.L.; Tirona, R.G.; Kim, R.B. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J. Clin. Pharmacol. 2007, 47, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Jigorel, E.; Le Vee, M.; Boursier-Neyret, C.; Parmentier, Y.; Fardel, O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 2006, 34, 1756–1763. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.; Jekerle, V.; Piquette-Miller, M. Induction of ABCC3 (MRP3) by pregnane X receptor activators. Drug Metab. Dispos. 2003, 31, 1296–1299. [Google Scholar] [CrossRef] [PubMed]
- Kast, H.R.; Goodwin, B.; Tarr, P.T.; Jones, S.A.; Anisfeld, A.M.; Stoltz, C.M.; Tontonoz, P.; Kliewer, S.; Willson, T.M.; Edwards, P.A. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor; farnesoid X-activated receptor; and constitutive androstane receptor. J. Biol. Chem. 2002, 277, 2908–2915. [Google Scholar] [CrossRef] [PubMed]
- Geick, A.; Eichelbaum, M.; Burk, O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J. Biol. Chem. 2001, 276, 14581–14587. [Google Scholar] [CrossRef] [PubMed]
- Hariparsad, N.; Nallani, S.C.; Sane, R.S.; Buckley, D.J.; Buckley, A.R.; Desai, P.B. Induction of CYP3A4 by efavirenz in primary human hepatocytes: Comparison with rifampin and phenobarbital. J. Clin. Pharmacol. 2004, 44, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, J.; Theile, D.; Dvorak, Z.; Haefeli, W.E. Interaction Potential of the Multitargeted Receptor Tyrosine Kinase Inhibitor Dovitinib with Drug Transporters and Drug Metabolising Enzymes Assessed in Vitro. Pharmaceutics 2014, 6, 632-650. https://doi.org/10.3390/pharmaceutics6040632
Weiss J, Theile D, Dvorak Z, Haefeli WE. Interaction Potential of the Multitargeted Receptor Tyrosine Kinase Inhibitor Dovitinib with Drug Transporters and Drug Metabolising Enzymes Assessed in Vitro. Pharmaceutics. 2014; 6(4):632-650. https://doi.org/10.3390/pharmaceutics6040632
Chicago/Turabian StyleWeiss, Johanna, Dirk Theile, Zdenek Dvorak, and Walter Emil Haefeli. 2014. "Interaction Potential of the Multitargeted Receptor Tyrosine Kinase Inhibitor Dovitinib with Drug Transporters and Drug Metabolising Enzymes Assessed in Vitro" Pharmaceutics 6, no. 4: 632-650. https://doi.org/10.3390/pharmaceutics6040632
APA StyleWeiss, J., Theile, D., Dvorak, Z., & Haefeli, W. E. (2014). Interaction Potential of the Multitargeted Receptor Tyrosine Kinase Inhibitor Dovitinib with Drug Transporters and Drug Metabolising Enzymes Assessed in Vitro. Pharmaceutics, 6(4), 632-650. https://doi.org/10.3390/pharmaceutics6040632