In Vitro Phase I Metabolism of CRV431, a Novel Oral Drug Candidate for Chronic Hepatitis B

Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs and Reagents
2.2. Metabolic Stability of CRV431 in Human Liver Microsomes
2.3. Cytochrome P450 Metabolism of CRV431 Using Recombinant Human CYP Enzymes
2.4. Cytochrome P450 Metabolism Using Chemical Inhibition in Human Liver Microsomes
2.5. LC-MS/MS Analysis of CRV431 for Cytochrome P450 Studies
2.6. Microsome Incubation Procedure for Generation of CRV431 Metabolites
2.7. CRV431 and Metabolite Extraction from Rat, Monkey and Human Microsomes
2.8. LC-MS Analysis of CRV431 Metabolites
2.9. Verapamil LC-MS Analysis for Microsome Viability
3. Results
3.1. CRV431 Metabolic Stability in Human Liver Microsomes
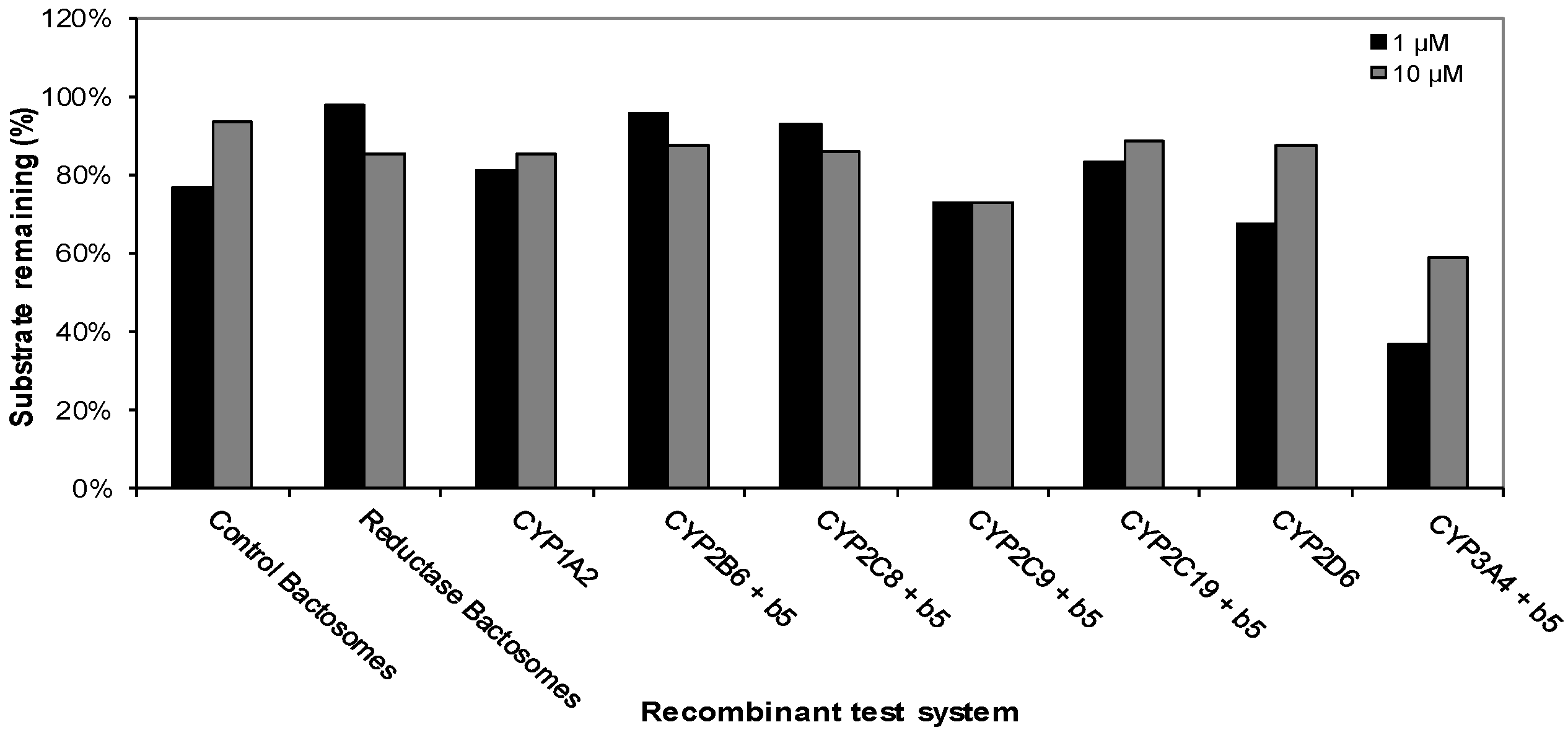
3.2. Cytochrome P450 Metabolism of CRV431 Using Recombinant Human CYP Enzymes
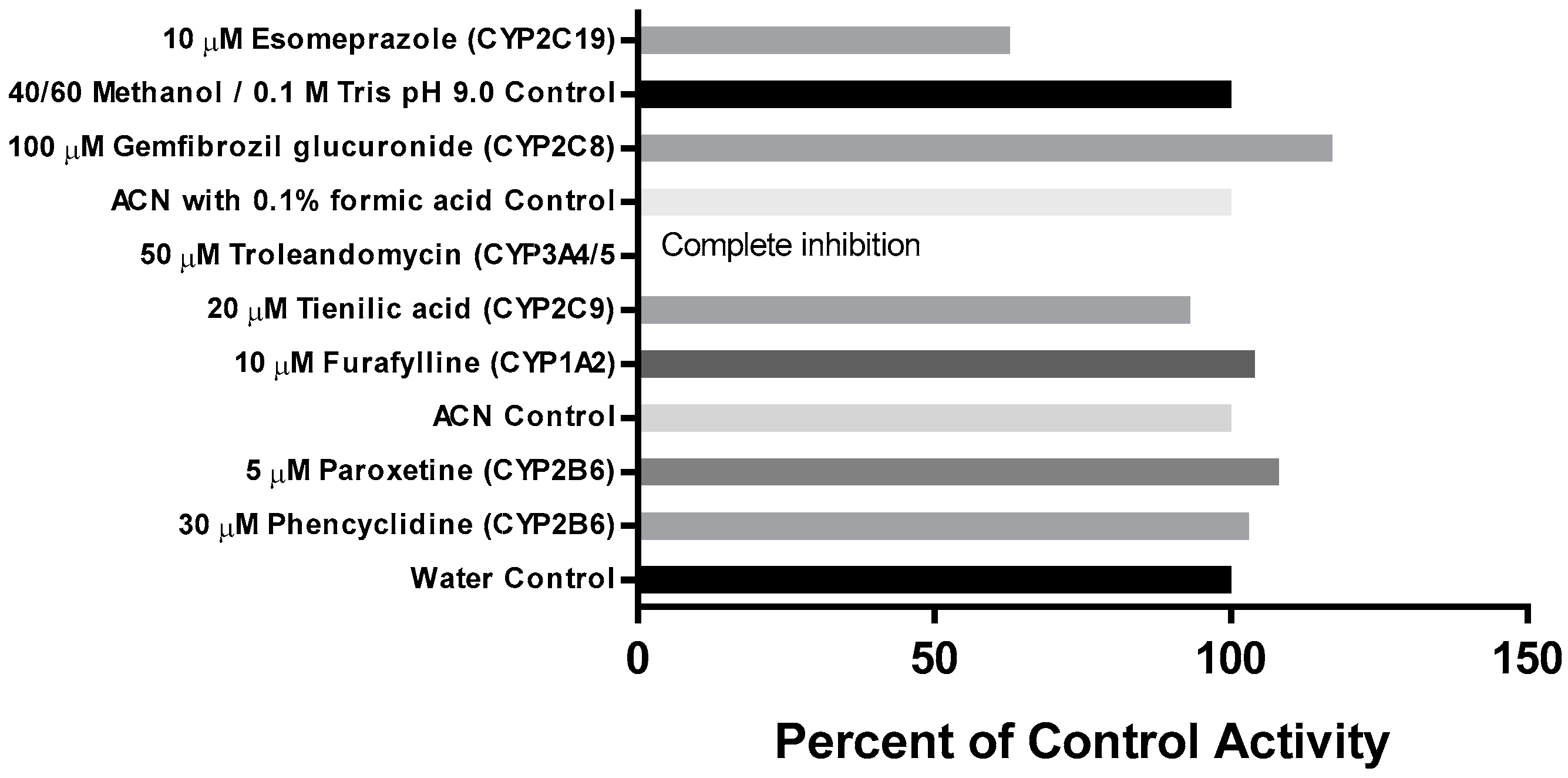
3.3. Cytochrome P450 Metabolism of CRV431 Using Chemical Inhibitors
3.4. Liver Microsome Viability for Metabolic Profiling Studies
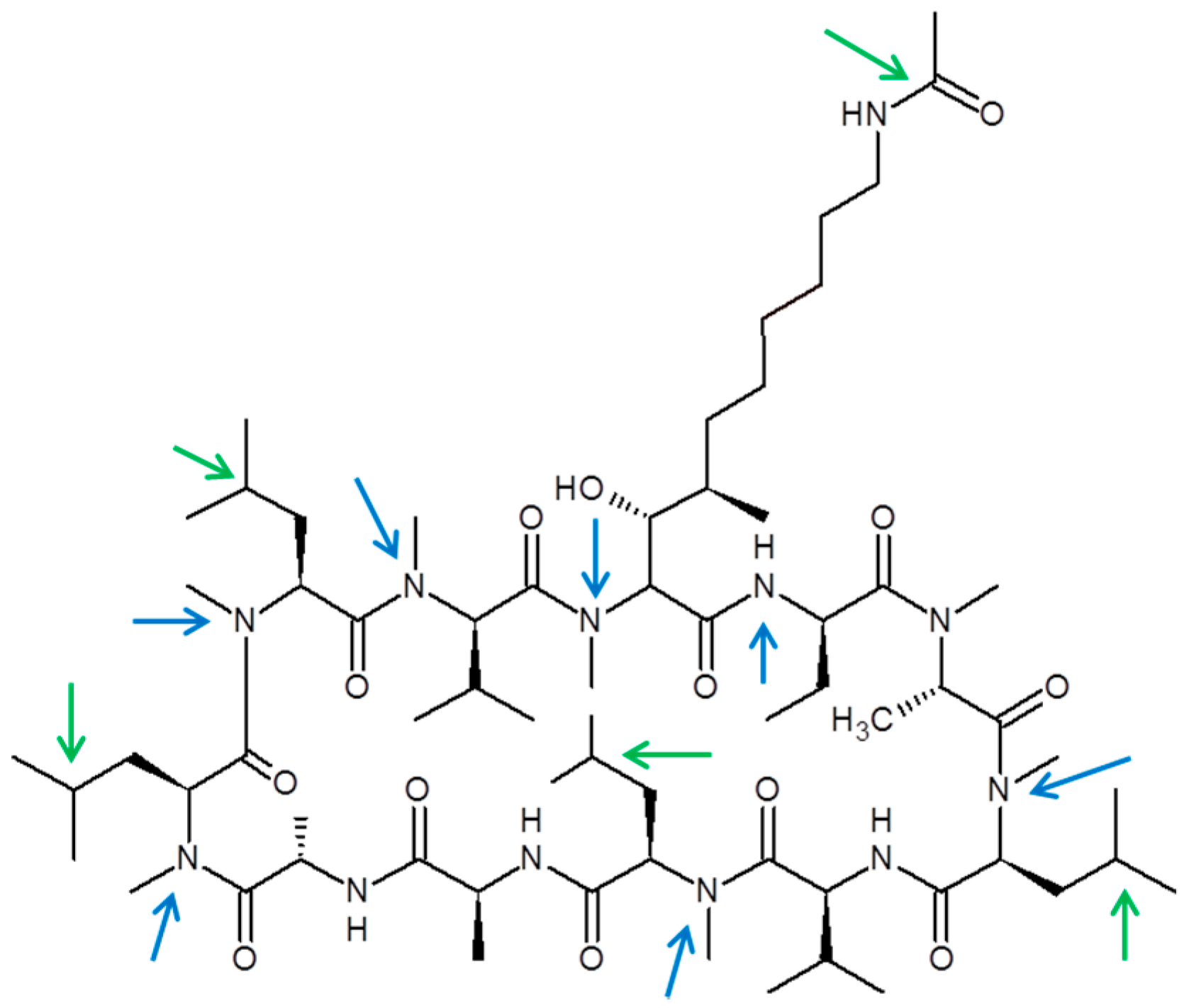
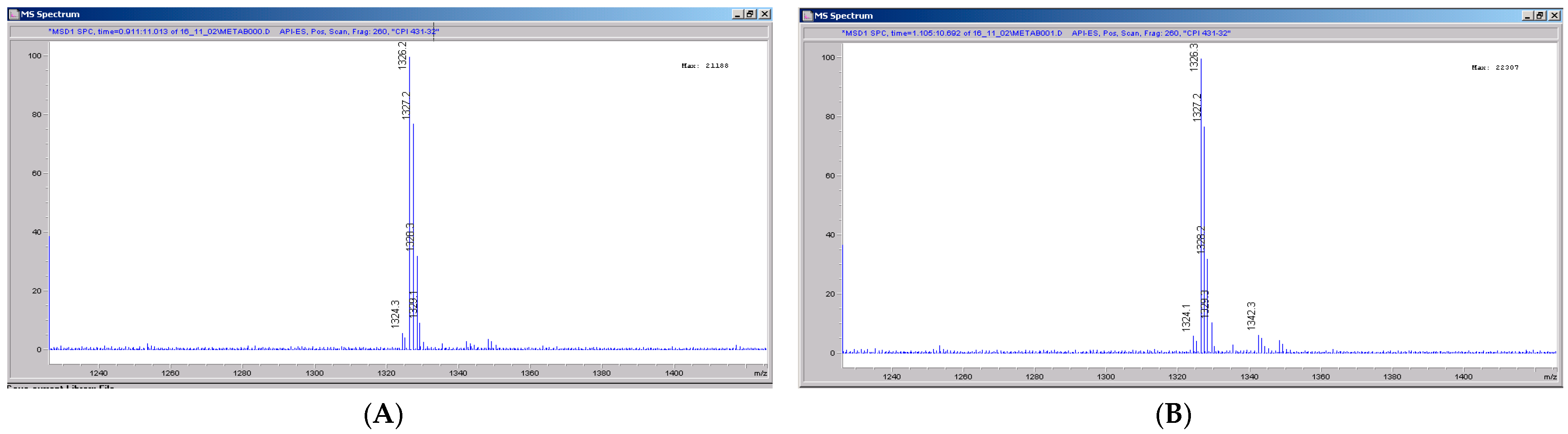
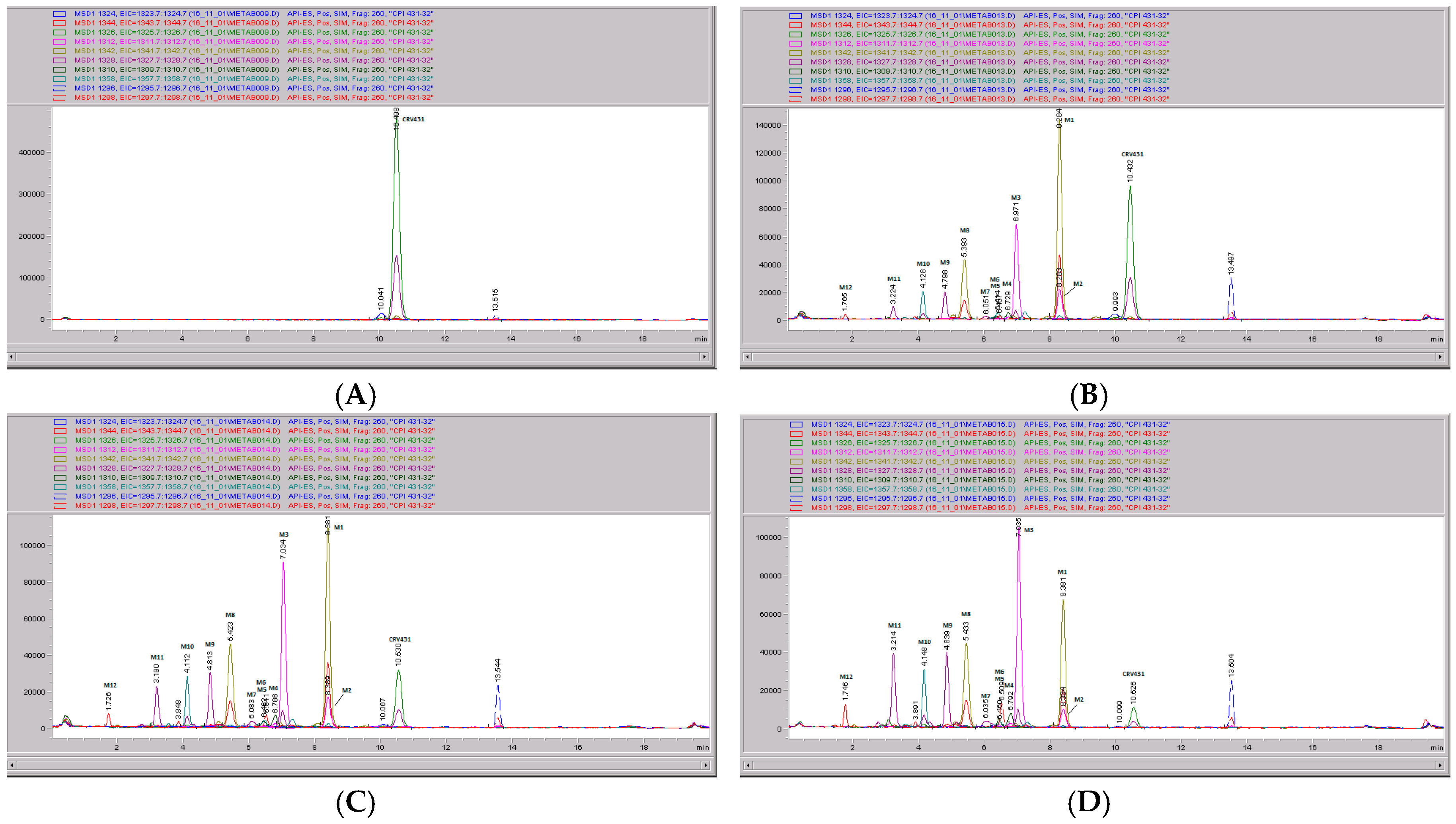
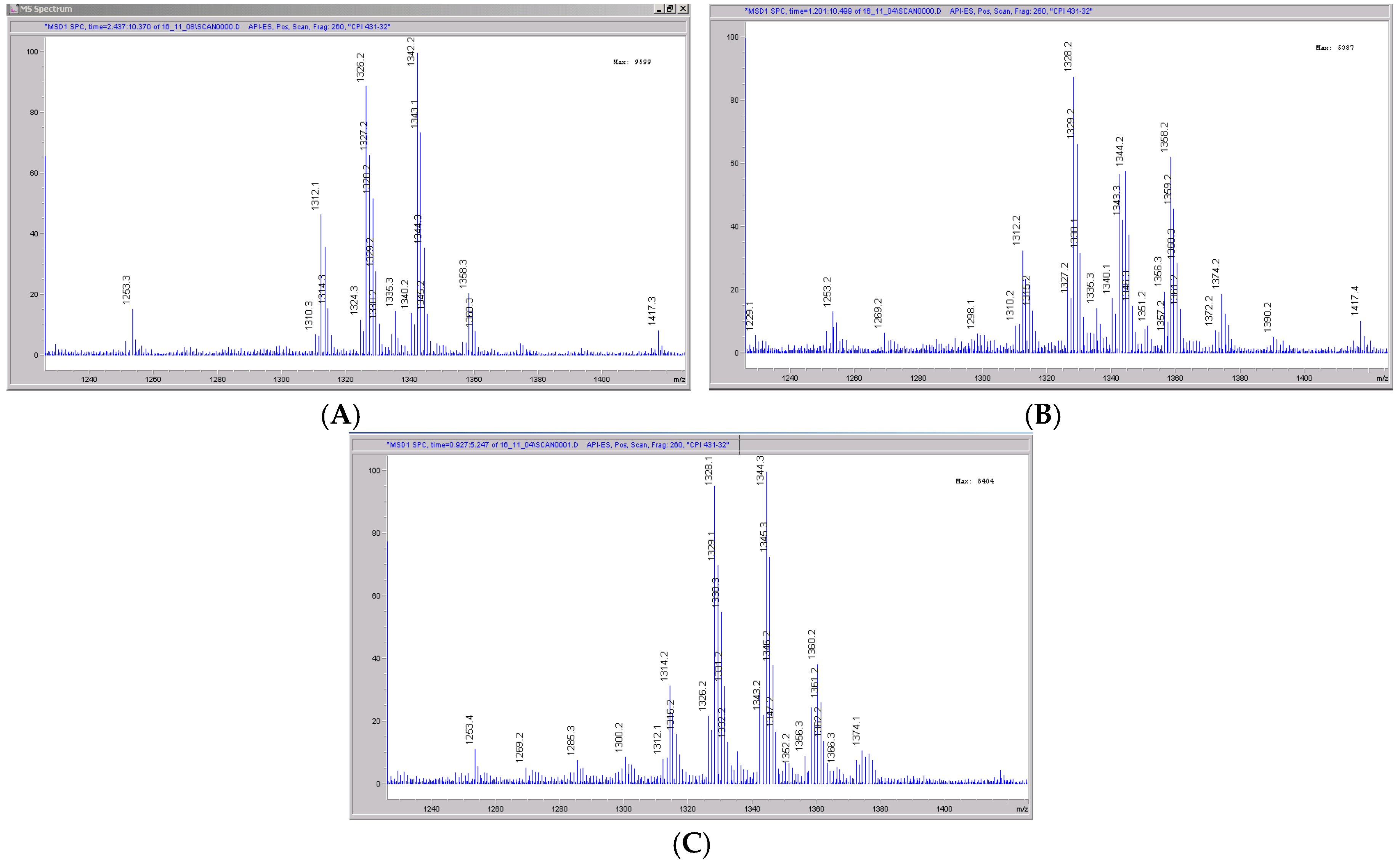
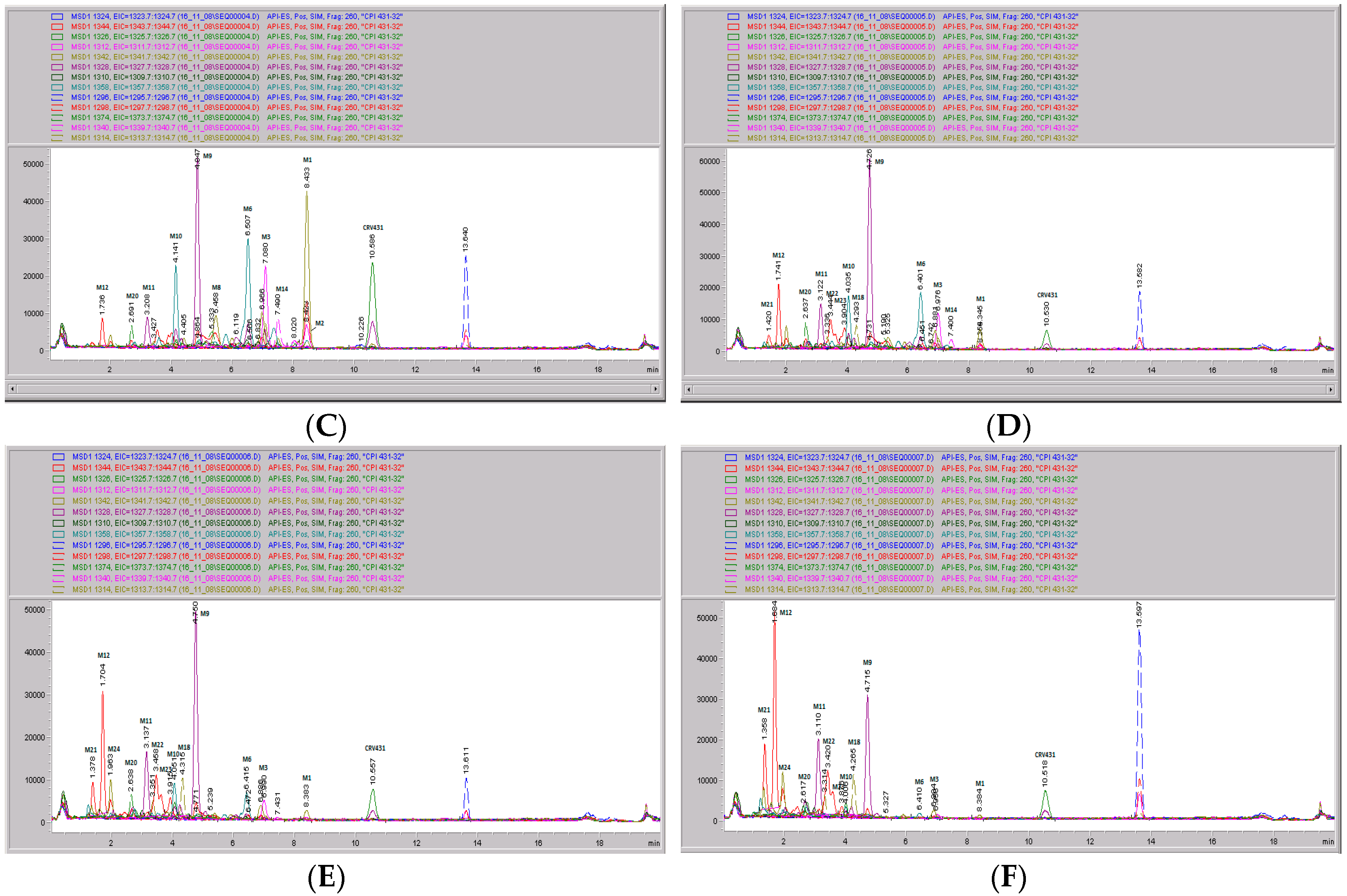
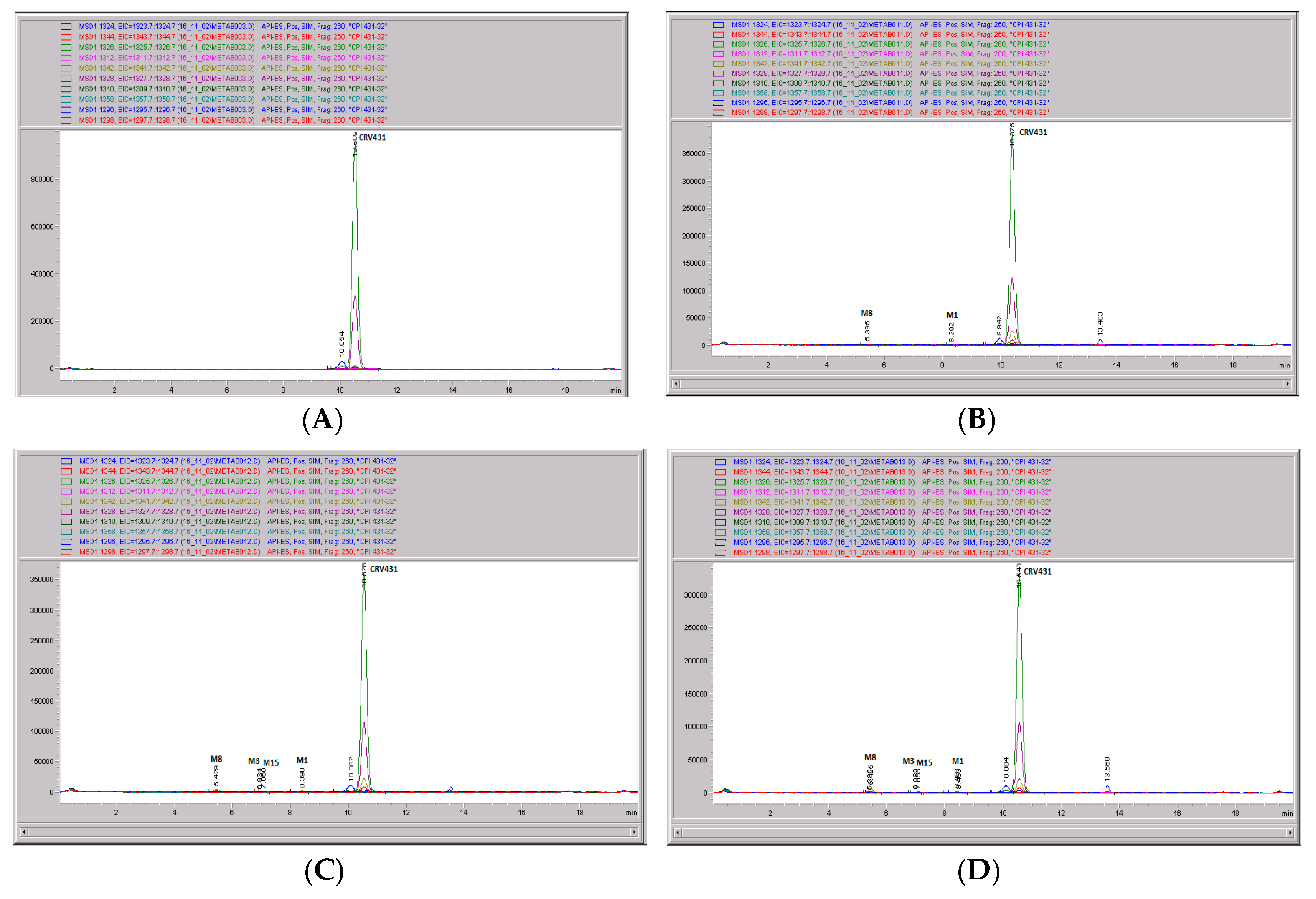
3.5. CRV431 Metabolite Identification in Human Liver Microsomes
3.6. CRV431 Metabolite Identification in Monkey Liver Microsomes
3.7. CRV431 Metabolite Identification in Rat Liver Microsomes
4. Conclusions and Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Borel, J.F.; Kis, Z.L. The Discovery and Development of Cyclosporine (Sandimmune). Transpl. Proc. 1991, 23, 1867–1874. [Google Scholar]
- Wiederrecht, G.; Lam, E.; Hung, S.; Martin, M.; Sigal, N. The Mechanism of Action of FK-506 and Cyclosporin A. Ann. N. Y. Acad. Sci. 1993, 696, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.A.; Bobardt, M.D.; Chatterji, U.; Trepanier, D.J.; Ure, D.; Ordonez, C.; Foster, R. The Novel Cyclophilin Inhibitor CRV431 Concurrently Blocks HCV and HIV-1 Infections via a Similar Mechanism of Action. PLoS ONE 2015, 10, e0134707. [Google Scholar] [CrossRef] [PubMed]
- Ure, D.R.; Bobardt, M.D.; Chatterji, U.; Trepanier, D.J.; Gallay, P.A.; Foster, R.T. The Cyclophilin Inhibitor, CPI-431-32, is a Hepatitis B Oral Drug Candidate with Antiviral and Antifibrotic Actions. In Proceedings of the HEP DART 2015 Conference, Wailea, HI, USA, 5 October 2015. [Google Scholar]
- Gallay, P.; Chatterji, U.; Bobardt, M.D.; Ure, D.; Trepanier, D.; Foster, R.; Ordonez, C. Novel Cyclophilin Inhibitor CPI-431-32 Shows Broad Spectrum Antiviral Action by Blocking Replication of HCV, HBV, and HIV-1. J. Hepatol. 2015, 62, S677. [Google Scholar] [CrossRef]
- Maurer, G.; Loosli, H.R.; Schreier, E.; Keller, B. Disposition of Cyclosporine in Several Animal Species and Man. I. Structural elucidation of its metabolites. Drug Metab. Dispos. 1984, 12, 120–127. [Google Scholar] [PubMed]
- Kronbach, T.; Fischer, V.; Meyer, U.A. Cyclosporine Metabolism in Human Liver: Identification of a Cytochrome P450 III Gene Family as the Major Cyclosporine-Metabolizing Enzyme Explains Interactions of Cyclosporine with other Drugs. Clin. Pharmacol. Ther. 1988, 43, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Combalbert, J.; Fabre, I.; Fabre, G.; Dalet, I.; Derancourt, J.; Cano, J.P.; Maurel, P. Metabolism of Cyclosporine A. IV. Purification and Identification of Rifampicin-Inducible Human Liver Cytochrome P-450 (cyclosporine A oxidase) as a Product of P-450 IIIA Gene Subfamily. Drug Metab. Dispos. 1989, 17, 197–207. [Google Scholar] [PubMed]
- Grbac, R.T.; Stanley, F.A.; Ambo, T.; Barbara, J.E.; Haupt, L.J. High Content Automated Metabolic Stability and CYP Inhibition Cocktail Screening Assays for Early Drug Development. Personal communication, 2014. [Google Scholar]
- Kelly, P.A.; Wang, H.; Napoli, K.L.; Kahan, B.D.; Strobel, H.W. Metabolism of Cyclosporine by Cytochromes P450 3A9 and 3A4. Eur. J. Drug Metab. Pharm. 1999, 24, 321–328. [Google Scholar] [CrossRef]
- Kuo, A.; Gish, R. Chronic Hepatitis B Infection. Clin. Liver Dis. 2012, 16, 347–369. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | 95:5 v/v Water: Methanol * (%) | Methanol * | Flow Rate (mL/min) |
|---|---|---|---|
| 0.00 | 55 | 45 | 0.5 |
| 0.2 | 55 | 45 | 0.5 |
| 3.0 | 5 | 95 | 0.5 |
| 3.5 | 5 | 95 | 0.5 |
| 3.51 | 55 | 45 | 0.5 |
| 4.2 | Stop | Stop | 0.5 |
| Time (min) | dH20 * (%) | ACN * | Flow Rate (mL/min) |
|---|---|---|---|
| 0.00 | 55 | 45 | 1.0 |
| 16.0 | 25 | 75 | 1.0 |
| 16.1 | 0 | 100 | 1.0 |
| 18.1 | 0 | 100 | 1.0 |
| 18.2 | 55 | 45 | 1.0 |
| Time (min) | dH20 * (%) | ACN * | Flow Rate (mL/min) |
|---|---|---|---|
| 0.00 | 70 | 30 | 1.0 |
| 8.0 | 45 | 55 | 1.0 |
| 8.1 | 0 | 100 | 1.0 |
| 10.1 | 0 | 100 | 1.0 |
| 10.2 | 70 | 30 | 1.0 |
| Component | Proposed Biotransformation | Relative LC-MS Retention | m/z | Δ m/z | % of Total Drug-Related Mass Versus Time (minutes) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 min | 10 min | 20 min | 40 min | 80 min | |||||
| CRV431 | NA | 1.0 | 1326 | 0 | 96.4 | 57.0 | 27.5 | 10.4 | 4.0 |
| CRV431 unsaturated impurity | NA | 0.96 | 1324 | NA | 3.6 | 2.9 | 1.8 | 0.7 | 0.2 |
| M1 | Hydroxylation | 0.8 | 1342 | +16 | 0 | 21.2 | 28.3 | 27.4 | 18.3 |
| M2 | Demethylation | 0.8 | 1312 | −14 | 0 | 3.6 | 4.9 | 4.6 | 3.1 |
| M3 | Demethylation | 0.67 | 1312 | −14 | 0 | 5.3 | 13.8 | 21.9 | 27.4 |
| M4 | Demethylation of unsaturated CRV431 | 0.65 | 1310 | −16 | 0 | 0.3 | 1.1 | 1.7 | 2.0 |
| M5 | Didemethylation | 0.62 | 1298 | −28 | 0 | 0 | 0.4 | 1.3 | 2.5 |
| M6 | Dihydroxylation | 0.62 | 1358 | +32 | 0 | 0 | 1.3 | 1.4 | 1.1 |
| M7 | Demethylation + Hydroxylation | 0.58 | 1328 | +2 | 0 | 0.1 | 0.6 | 1.2 | 1.7 |
| M8 | Hydroxylation | 0.52 | 1342 | +16 | 0 | 5.9 | 10.3 | 13.0 | 13.5 |
| M9 | Demethylation + Hydroxylation | 0.47 | 1328 | +2 | 0 | 1.1 | 3.2 | 5.6 | 8.6 |
| M10 | Dihydroxylation | 0.39 | 1358 | +32 | 0 | 1.2 | 3.3 | 5.3 | 6.6 |
| M11 | Demethylation + Hydroxylation | 0.30 | 1328 | +32 | 0 | 0.4 | 1.6 | 4.4 | 8.9 |
| M12 | Dihydroxylation + demethylation | 0.17 | 1344 | +18 | 0 | 0.1 | 0.5 | 1.0 | 1.9 |
| Component | Proposed Biotransformation | Relative LC-MS Retention | m/z | Δ m/z | % of Total Drug-Related Mass Versus Time (minutes) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 min | 2.5 min | 5 min | 10 min | 20 min | 40 min | 80 min | |||||
| CRV431 | NA | 1.0 | 1326 | 0 | 96.3 | 59.4 | 21.5 | 12.3 | 4.3 | 6.6 | 6.7 |
| CRV431 unsaturated impurity | NA | 0.96 | 1324 | −2 | 3.7 | 3.8 | 1.7 | 0.8 | 0 | 0 | 0 |
| M1 | Hydroxylation | 0.8 | 1342 | +16 | 0 | 18.1 | 24.2 | 16.1 | 3.2 | 1.7 | 0.9 |
| M2 | Demethylation | 0.8 | 1312 | −14 | 0 | 3.1 | 6.7 | 2.7 | 0.7 | 0 | 0 |
| M3 | Demethylation | 0.67 | 1312 | −14 | 0 | 3.8 | 7.6 | 8.3 | 5.4 | 3.3 | 0.7 |
| M4 | Demethylation of unsaturated CRV431 | 0.65 | 1310 | −16 | 0 | 0.3 | 0.5 | 0.6 | 0.6 | 0 | 0 |
| M5 | Didemethylation | 0.62 | 1298 | −28 | 0 | 0 | 0.2 | 0.4 | 0.7 | 0.6 | 0 |
| M6 | Dihydroxylation | 0.62 | 1358 | +32 | 0 | 2.4 | 8.2 | 12.6 | 10.9 | 4.8 | 1.3 |
| M7 | Demethylation + Hydroxylation | 0.58 | 1328 | +2 | 0 | 0 | 0 | 1.1 | 0 | 0 | 0 |
| M8 | Hydroxylation | 0.52 | 1342 | +16 | 0 | 1.9 | 3.7 | 3.7 | 2.0 | 0 | 0.4 |
| M9 | Demethylation + Hydroxylation | 0.47 | 1328 | +2 | 0 | 1.9 | 8.6 | 15.6 | 24.8 | 25.2 | 17.6 |
| M10 | Dihydroxylation | 0.39 | 1358 | +32 | 0 | 0.8 | 4.4 | 6.9 | 6.7 | 4.2 | 1.4 |
| M11 | Demethylation + Hydroxylation | 0.30 | 1328 | +32 | 0 | 0.2 | 1.3 | 2.8 | 6.5 | 8.7 | 11.7 |
| M12 | Dihydroxylation + demethylation | 0.17 | 1344 | + 18 | 0 | 0.1 | 0.7 | 1.8 | 6.2 | 11.7 | 21.6 |
| Additional Metabolites Not Detected in Human Liver Microsome Experiments | |||||||||||
| M13 | 0.75 | 1340 | +14 | 0 | 1.1 | 1.6 | 1.1 | 0 | 0 | 0 | |
| M14 | 0.70 | 1340 | +14 | 0 | 0.9 | 2.4 | 3.2 | 1.7 | 0.6 | 0 | |
| M15 | Hydroxylation | 0.65 | 1342 | +16 | 0 | 1.6 | 3.3 | 3.9 | 3.3 | 2.4 | 1.7 |
| M16 | Trihydroxylation | 0.50 | 1374 | +48 | 0 | 0.4 | 1.8 | 2.6 | 2.2 | 1.4 | 0 |
| M17 | Trihydroxylation | 0.46 | 1374 | +48 | 0 | 0.4 | 0.8 | 0.9 | 1.1 | 0 | |
| M18 | Didemethylation + hydroxylation | 0.41 | 1314 | −12 | 0 | 0 | 0.2 | 0.6 | 2.3 | 4.1 | 5.4 |
| M19 | Didemethylation + hydroxylation | 0.31 | 1314 | −12 | 0 | 0 | 0.2 | 0.4 | 1.3 | 1.8 | 2.8 |
| M20 | Trihydroxylation | 0.25 | 1374 | +48 | 0 | 0.1 | 0.8 | 1.6 | 3.3 | 2.9 | 1.8 |
| M21 | Dihydroxylation + demethylation | 0.13 | 1344 | +18 | 0 | 0 | 0 | 0 | 1.2 | 3.7 | 8.0 |
| M22 | Dihydroxylation + demethylation | 0.32 | 1344 | +18 | 0 | 0 | 0 | 0 | 5.6 | 8.4 | 11.1 |
| M23 | Dihydroxylation + demethylation | 0.35 | 1344 | +18 | 0 | 0 | 0 | 0 | 3.6 | 2.6 | 1.4 |
| M24 | Hydroxylation | 0.18 | 1342 | +16 | 0 | 0 | 0 | 0 | 2.3 | 4.0 | 5.7 |
| Component | Proposed Biotransformation | Relative LC-MS Retention | m/z | Δ m/z | % of Total Drug-Related Mass Versus Time (minutes) | ||||
|---|---|---|---|---|---|---|---|---|---|
| 0 min | 10 min | 20 min | 40 min | 80 min | |||||
| CRV431 | NA | 1.0 | 1326 | 0 | 96.2 | 95.4 | 94.6 | 93.6 | 92.3 |
| CRV431 unsaturated impurity | NA | 0.96 | 1324 | NA | 3.8 | 3.9 | 3.8 | 3.9 | 4.6 |
| M1 | Hydroxylation | 0.8 | 1342 | +16 | 0 | 0.1 | 0.3 | 0.5 | 0.5 |
| M3 | Demethylation | 0.67 | 1312 | −14 | 0 | 0 | 0.07 | 0.19 | 0.4 |
| M8 | Hydroxylation | 0.52 | 1342 | +16 | 0 | 0.4 | 0.8 | 1.3 | 1.7 |
| M15 | Hydroxylation | 0.65 | 1342 | +16 | 0 | 0.08 | 0.1 | 0.2 | 0.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trepanier, D.J.; Ure, D.R.; Foster, R.T. In Vitro Phase I Metabolism of CRV431, a Novel Oral Drug Candidate for Chronic Hepatitis B. Pharmaceutics 2017, 9, 51. https://doi.org/10.3390/pharmaceutics9040051
Trepanier DJ, Ure DR, Foster RT. In Vitro Phase I Metabolism of CRV431, a Novel Oral Drug Candidate for Chronic Hepatitis B. Pharmaceutics. 2017; 9(4):51. https://doi.org/10.3390/pharmaceutics9040051
Chicago/Turabian StyleTrepanier, Daniel J., Daren R. Ure, and Robert T. Foster. 2017. "In Vitro Phase I Metabolism of CRV431, a Novel Oral Drug Candidate for Chronic Hepatitis B" Pharmaceutics 9, no. 4: 51. https://doi.org/10.3390/pharmaceutics9040051
APA StyleTrepanier, D. J., Ure, D. R., & Foster, R. T. (2017). In Vitro Phase I Metabolism of CRV431, a Novel Oral Drug Candidate for Chronic Hepatitis B. Pharmaceutics, 9(4), 51. https://doi.org/10.3390/pharmaceutics9040051