Application of Pharmacokinetics Modelling to Predict Human Exposure of a Cationic Liposomal Subunit Antigen Vaccine System




Abstract
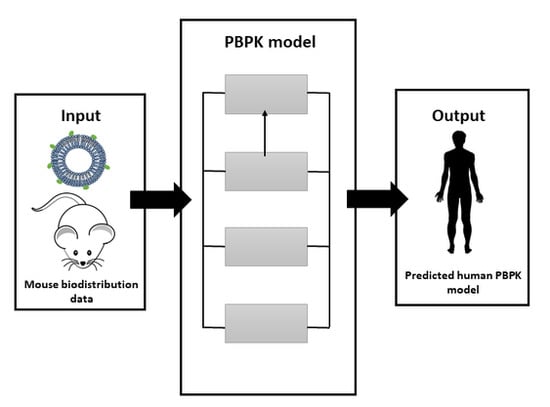
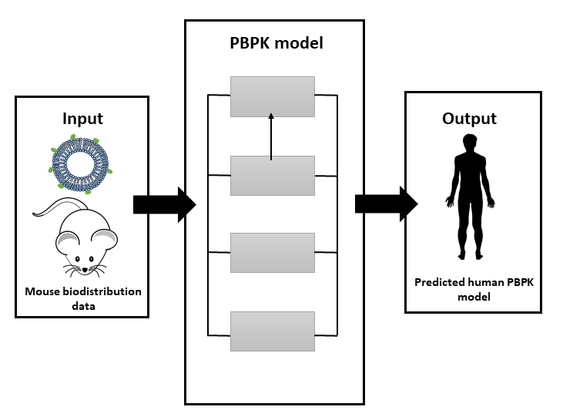
:
1. Introduction
2. Materials and Methods
2.1. Data Collection
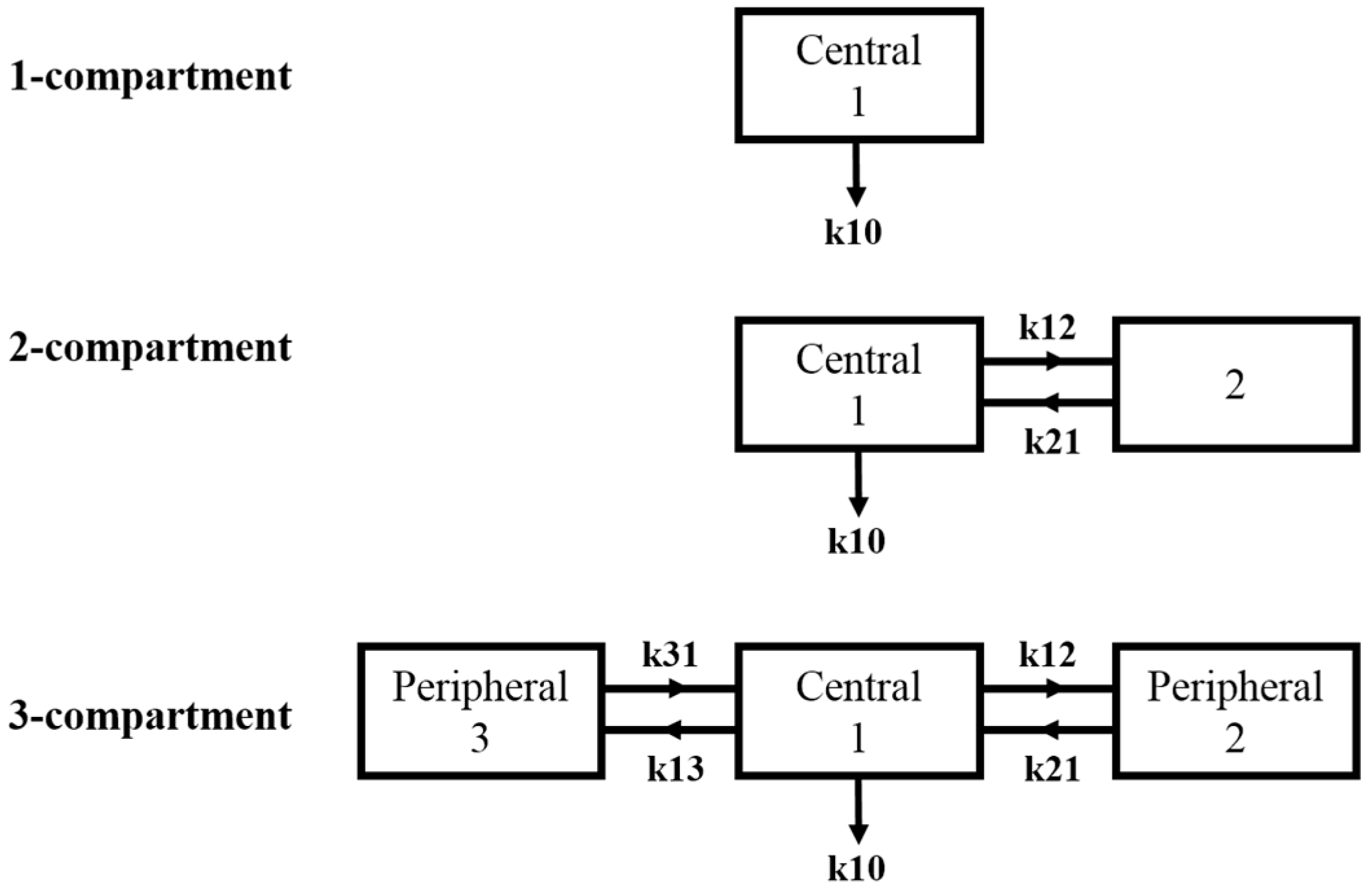
2.2. Compartmental Modelling of Data
2.3. Mechanistic Modelling of Data
- All tissues were modelled based on the total tissue volume (derived from tissue mass), and, where necessary, assuming a density of 1.
- The ‘muscle’ compartment was modeled solely by the quadriceps tissue component in mice and the deltoid tissue in humans.
- Plasma flow to and drainage from the PLN were assumed to be 0.012% cardiac output [27].
- The fraction escaping (fe) the quadriceps and being drained into the PLN was fixed at 3 × 10−5 for liposome and 3.6 × 10−6 for antigen, to reflect fraction escaping the muscle based upon the average ratio of percent accumulation for all time points in the target tissues compared to the dose administered. The small-pore theory of molecular translocation across a membrane would preclude molecules below 60 nm in size from undergoing transvascular flow across a capillary wall [29,30,31,32].
- Dosing was modelled as a rapid first-order dose into the quadriceps (approximating a bolus dose) with a ka = 10 day−1. The human model focused on simulating antigen only and therefore a human dose of 50 µg was modelled.
- In the absence of plasma concentration of both liposome and antigen and the limited muscle and PLN biodistribution data, an attempt to estimate an appropriate tissue partition coefficient was not conducted and transfer of liposome/antigen out of the site of administration was assumed to occur only through exiting via the muscle blood flow (accounting for the fraction escaping) and the transfer via lymphatics. Transvascular flow was therefore modelled as a rate constant (when accounting for tissue volume):
2.4. Parameter Sensitivity Analysis
3. Results
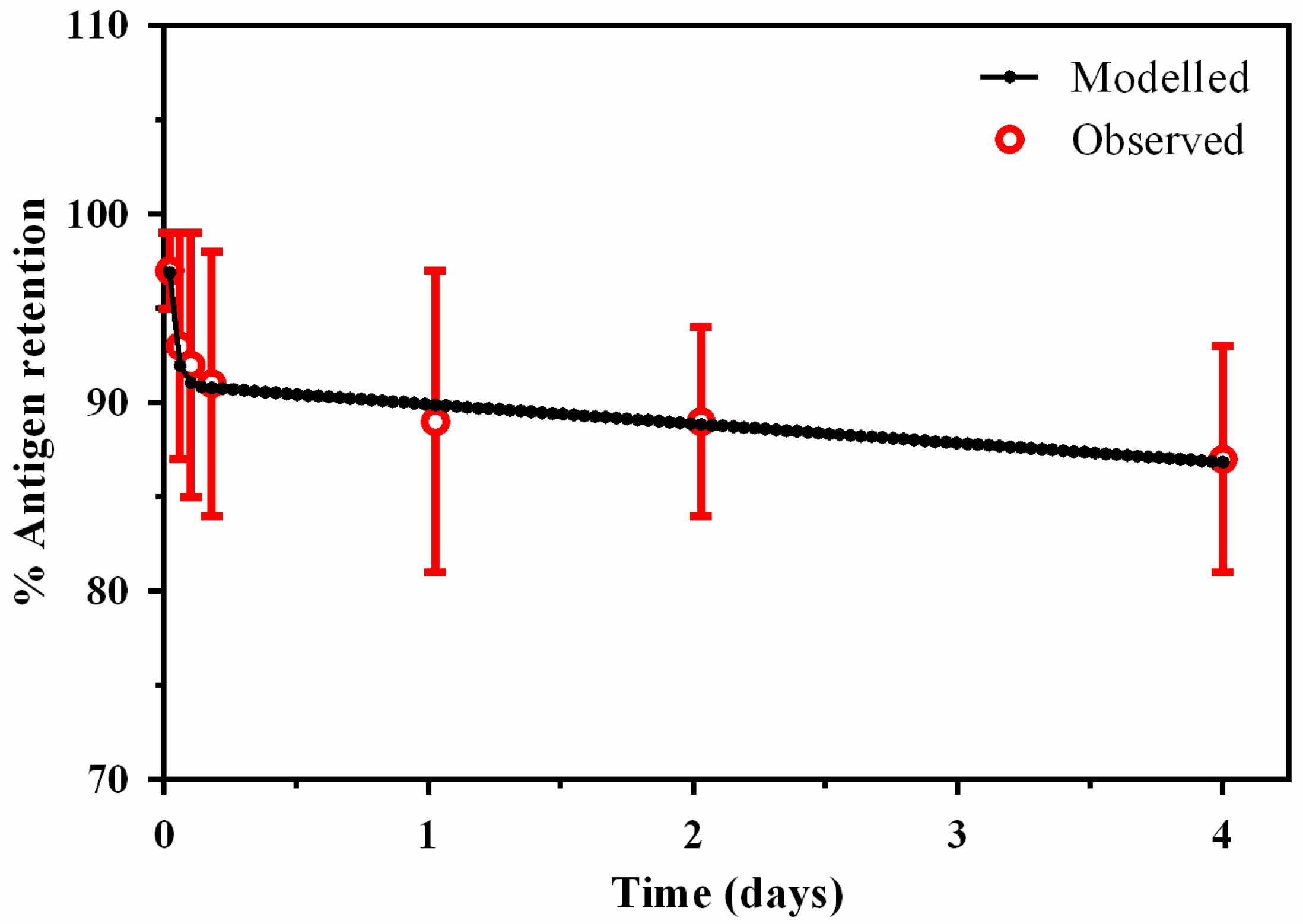
3.1. Compartment Modelling
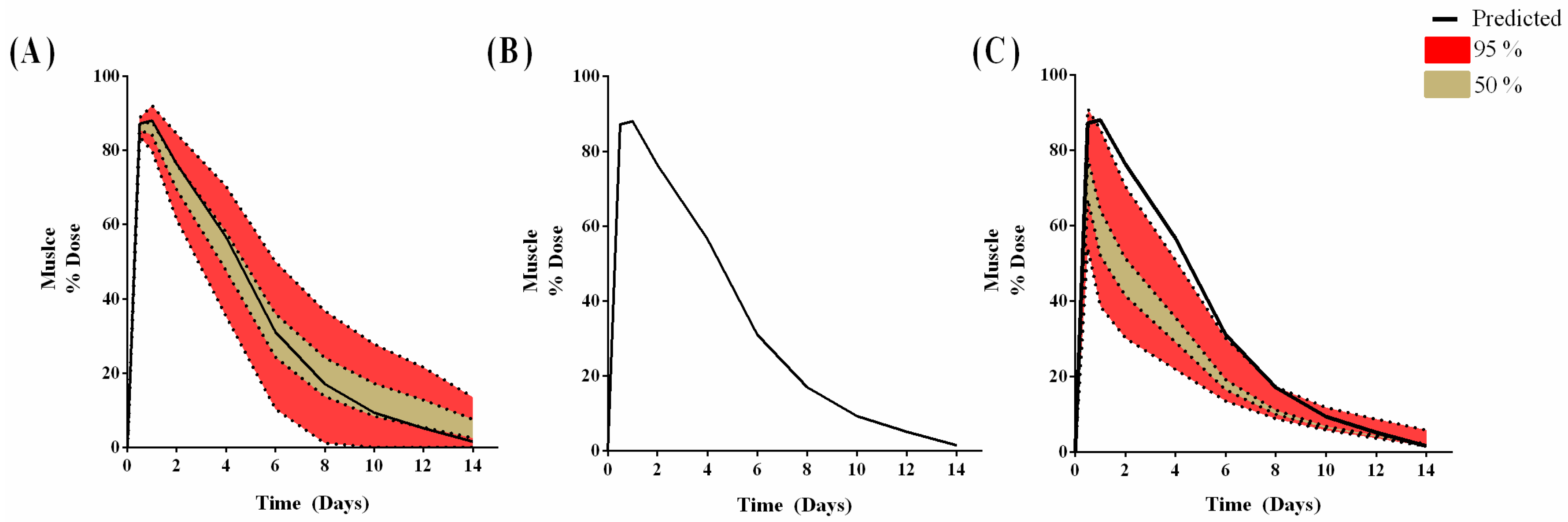
3.2. Minimal-PBPK Models
3.3. Sensitivity Analysis
3.4. Human Model
4. Discussion
4.1. Compartmental Modelling
4.2. Physiological Modelling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Agger, E.M.; Rosenkrands, I.; Hansen, J.; Brahimi, K.; Vandahl, B.S.; Aagaard, C.; Werninghaus, K.; Kirschning, C.; Lang, R.; Christensen, D.; et al. Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): A versatile adjuvant for vaccines with different immunological requirements. PLoS ONE 2008, 3, e3116. [Google Scholar] [CrossRef] [PubMed]
- Holten-Andersen, L.; Doherty, T.M.; Korsholm, K.S.; Andersen, P. Combination of the Cationic Surfactant Dimethyl Dioctadecyl Ammonium Bromide and Synthetic Mycobacterial Cord Factor as an Efficient Adjuvant for Tuberculosis Subunit Vaccines. Infect. Immun. 2004, 72, 1608–1617. [Google Scholar] [CrossRef] [PubMed]
- Korsholm, K.S.; Agger, E.M.; Foged, C.; Christensen, D.; Dietrich, J.; Andersen, C.S.; Geisler, C.; Andersen, P. The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology 2007, 121, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Werninghaus, K.; Babiak, A.; Groß, O.; Hölscher, C.; Dietrich, H.; Agger, E.M.; Mages, J.; Mocsai, A.; Schoenen, H.; Finger, K.; et al. Adjuvanticity of a synthetic cord factor analogue for subunit Mycobacterium tuberculosis vaccination requires FcRgamma-Syk-Card9-dependent innate immune activation. J. Exp. Med. 2009, 206, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Bramwell, V.W.; Christensen, D.; Agger, E.-M.; Andersen, P.; Perrie, Y. Liposomes based on dimethyldioctadecylammonium promote a depot effect and enhance immunogenicity of soluble antigen. J. Control. Release 2010, 142, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Bramwell, V.; Perrie, Y. Radiolabelling of Antigen and Liposomes for Vaccine Biodistribution Studies. Pharmaceutics 2010, 2, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Christensen, D.; Bramwell, V.W.; Lindenstrøm, T.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J. Control. Release 2010, 145, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Devitt, A.; Perrie, Y. The vesicle size of DDA:TDB liposomal adjuvants plays a role in the cell-mediated immune response but has no significant effect on antibody production. J. Control. Release 2011, 154, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Bramwell, V.W.; Kirby, D.J.; Perrie, Y. Manipulation of the surface pegylation in combination with reduced vesicle size of cationic liposomal adjuvants modifies their clearance kinetics from the injection site, and the rate and type of T cell response. J. Control. Release 2012, 164, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Bramwell, V.W.; Kirby, D.J.; Perrie, Y. Pegylation of DDA:TDB liposomal adjuvants reduces the vaccine depot effect and alters the Th1/Th2 immune responses. J. Control. Release 2012, 158, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Baxter, L.T.; Zhu, H.; Mackensen, D.G.; Jain, R.K. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994, 54, 1517–1528. [Google Scholar] [PubMed]
- Chetty, M.; Li, L.; Rose, R.; Machavaram, K.; Jamei, M.; Rostami-Hodjegan, A.; Gardner, I. Prediction of the Pharmacokinetics, Pharmacodynamics, and Efficacy of a Monoclonal Antibody, Using a Physiologically Based Pharmacokinetic FcRn Model. Front. Immunol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Balthasar, J.P. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J. Pharmacokinet. Pharmacodyn. 2007, 34, 687–709. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Chen, Y.; Balthasar, J.P. Scale-up of a physiologically-based pharmacokinetic model to predict the disposition of monoclonal antibodies in monkeys. J. Pharmacokinet. Pharmacodyn. 2015, 42, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gardner, I.; Dostalek, M.; Jamei, M. Simulation of monoclonal antibody pharmacokinetics in humans using a minimal physiologically based model. AAPS J. 2014, 16, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Mould, D.R.; Sweeney, K.R. The pharmacokinetics and pharmacodynamics of monoclonal antibodies—Mechanistic modeling applied to drug development. Curr. Opin. Drug Discov. Dev. 2007, 10, 84–96. [Google Scholar]
- Wang, J.; Iyer, S.; Fielder, P.J.; Davis, J.D.; Deng, R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: Comparative evaluation of prediction approaches in early drug development. Biopharm. Drug Dispos. 2015, 37, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Van Dissel, J.T.; Arend, S.M.; Prins, C.; Bang, P.; Tingskov, P.N.; Lingnau, K.; Nouta, J.; Klein, M.R.; Rosenkrands, I.; Ottenhoff, T.H.M.; et al. Ag85B–ESAT-6 adjuvanted with IC31® promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naïve human volunteers. Vaccine 2010, 28, 3571–3581. [Google Scholar] [CrossRef] [PubMed]
- Reither, K.; Katsoulis, L.; Beattie, T.; Gardiner, N.; Lenz, N.; Said, K.; Mfinanga, E.; Pohl, C.; Fielding, K.L.; Jeffery, H.; et al. Safety and immunogenicity of H1/IC31(R), an adjuvanted TB subunit vaccine, in HIV-infected adults with CD4+ lymphocyte counts greater than 350 cells/mm3: A phase II, multi-centre, double-blind, randomized, placebo-controlled trial. PLoS ONE 2014, 9, e114602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Henriksen-Lacey, M.; Wilkhu, J.; Devitt, A.; Christensen, D.; Perrie, Y. Effect of Incorporating Cholesterol into DDA:TDB Liposomal Adjuvants on Bilayer Properties, Biodistribution, and Immune Responses. Mol. Pharm. 2014, 11, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milicic, A.; Kaur, R.; Reyes-Sandoval, A.; Tang, C.-K.; Honeycutt, J.; Perrie, Y.; Hill, A.V.S. Small Cationic DDA:TDB Liposomes as Protein Vaccine Adjuvants Obviate the Need for TLR Agonists in Inducing Cellular and Humoral Responses. PLoS ONE 2012, 7, e34255. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Kastner, E.; Kaur, R.; Wilkinson, A.; Ingham, A.J. A case-study investigating the physicochemical characteristics that dictate the function of a liposomal adjuvant. Hum. Vaccines Immunother. 2013, 9, 1374–1381. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.P.; Delp, M.D.; Lindstedt, S.L.; Rhomberg, L.R.; Beliles, R.P. Physiological parameter values for physiologically based pharmacokinetic models. Toxicol. Ind. Health 1997, 13, 407–484. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Betts, A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J. Pharmacokinet. Pharmacodyn. 2011, 39, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Tegenge, M.A.; Mitkus, R.J. A physiologically-based pharmacokinetic (PBPK) model of squalene-containing adjuvant in human vaccines. J. Pharmacokinet. Pharmacodyn. 2013, 40, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Leamy, V.L.; Martin, T.; Mahajan, R.; Vilalta, A.; Rusalov, D.; Hartikka, J.; Bozoukova, V.; Hall, K.D.; Morrow, J.; Rolland, A.P.; et al. Comparison of rabbit and mouse models for persistence analysis of plasmid-based vaccines. Hum. Vaccines 2006, 2, 113–118. [Google Scholar] [CrossRef]
- Hay, J.B.; Hobbs, B.B. The flow of blood to lymph nodes and its relation to lymphocyte traffic and the immune response. J. Exp. Med. 1977, 145, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, Y.; Proulx, S.T.; Guo, R.; Yao, Z.; Schwarz, E.M.; Boyce, B.F.; Xing, L. Increased lymphangiogenesis in joints of mice with inflammatory arthritis. Arthritis Res. Ther. 2007, 9, R118. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.C. The investigation of capillary permeability in single vessels. Acta Physiol. Scand. Suppl. 1979, 463, 67–74. [Google Scholar] [PubMed]
- Pappenheimer, J.R. Passage of molecules through capillary walls. Physiol. Rev. 1953, 33, 387–423. [Google Scholar] [PubMed]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes. Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, K.; Mayerson, H.S. Dynamics of Lymph and Plasma Protein Exchange. Cardiologia 1952, 21, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2007, 96, 3151–3152. [Google Scholar] [CrossRef]
- Rodgers, T.; Rowland, M. Physiologically-based Pharmacokinetic Modeling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compartment | Flow (L/Day) Symbol | Mouse | Human | Volume (L) Symbol | Mouse | Human |
|---|---|---|---|---|---|---|
| Plasma | Qplasma | - | - | Vplasma | 9.44 × 10−4 [24] | 3.13 |
| Quadriceps | Qmuscle | 2.60 [24] | 32.04 [25] | Vmuscle | 1.6 × 10−4 [26] | 0.19 [25] |
| PLN | QPLN | 2 × 10−3 [27] | 0.52 [27] | VPLN | 5 × 10−6 [26] | 3.5 × 10−3 [27] |
| Rest of Body | Qrest | 18.10 | 3.02 × 104 | Vrest | 2.6 × 10−2 | 67.68 |
| Liposome | |
| K10 | 0.0306 day−1 |
| t1/2 | 22.6 days |
| MRT | 32.6 days |
| AIC | 49.54 |
| Antigen | |
| k10 | 0.34 day−1 |
| k12 | 22.26 day−1 |
| k21 | 77.58 day−1 |
| t1/2α | 0.0069 day |
| t1/2β | 2.62 days |
| MRT | 3.78 days |
| AIC | 37.4 |
| Time (Days) | % Precision Error | |||
|---|---|---|---|---|
| Muscle | PLN | |||
| Liposome | Antigen | Liposome | Antigen | |
| 0.25 | 16.4 | 13.5 | - | - |
| 1 | 32.7 | 5.3 | 28.4 | 32.7 |
| 4 | 3.9 | 13.8 | 13.90 | 41.5 |
| 14 | 3.8 | 65.9 | 66.3 | 74.0 |
| Compartment | Degradation Constant (Day−1 ± SD) | ||
|---|---|---|---|
| Symbol | Liposome | Antigen | |
| Plasma | - | - | - |
| Quadriceps | kdeg,muscle | 0.051 ± 0.008 | 0.320 ± 0.028 |
| PLN | kdeg,pln | 0.132 ± 0.004 | 0.280 ± 0.013 |
| Rest of Body | kdeg,rest | 0.091 ± 0.040 | 0.110 ± 0.030 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badhan, R.K.S.; Khadke, S.; Perrie, Y. Application of Pharmacokinetics Modelling to Predict Human Exposure of a Cationic Liposomal Subunit Antigen Vaccine System. Pharmaceutics 2017, 9, 57. https://doi.org/10.3390/pharmaceutics9040057
Badhan RKS, Khadke S, Perrie Y. Application of Pharmacokinetics Modelling to Predict Human Exposure of a Cationic Liposomal Subunit Antigen Vaccine System. Pharmaceutics. 2017; 9(4):57. https://doi.org/10.3390/pharmaceutics9040057
Chicago/Turabian StyleBadhan, Raj K. S., Swapnil Khadke, and Yvonne Perrie. 2017. "Application of Pharmacokinetics Modelling to Predict Human Exposure of a Cationic Liposomal Subunit Antigen Vaccine System" Pharmaceutics 9, no. 4: 57. https://doi.org/10.3390/pharmaceutics9040057
APA StyleBadhan, R. K. S., Khadke, S., & Perrie, Y. (2017). Application of Pharmacokinetics Modelling to Predict Human Exposure of a Cationic Liposomal Subunit Antigen Vaccine System. Pharmaceutics, 9(4), 57. https://doi.org/10.3390/pharmaceutics9040057