
Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy


Abstract
:1. Introduction
2. Pathophysiology
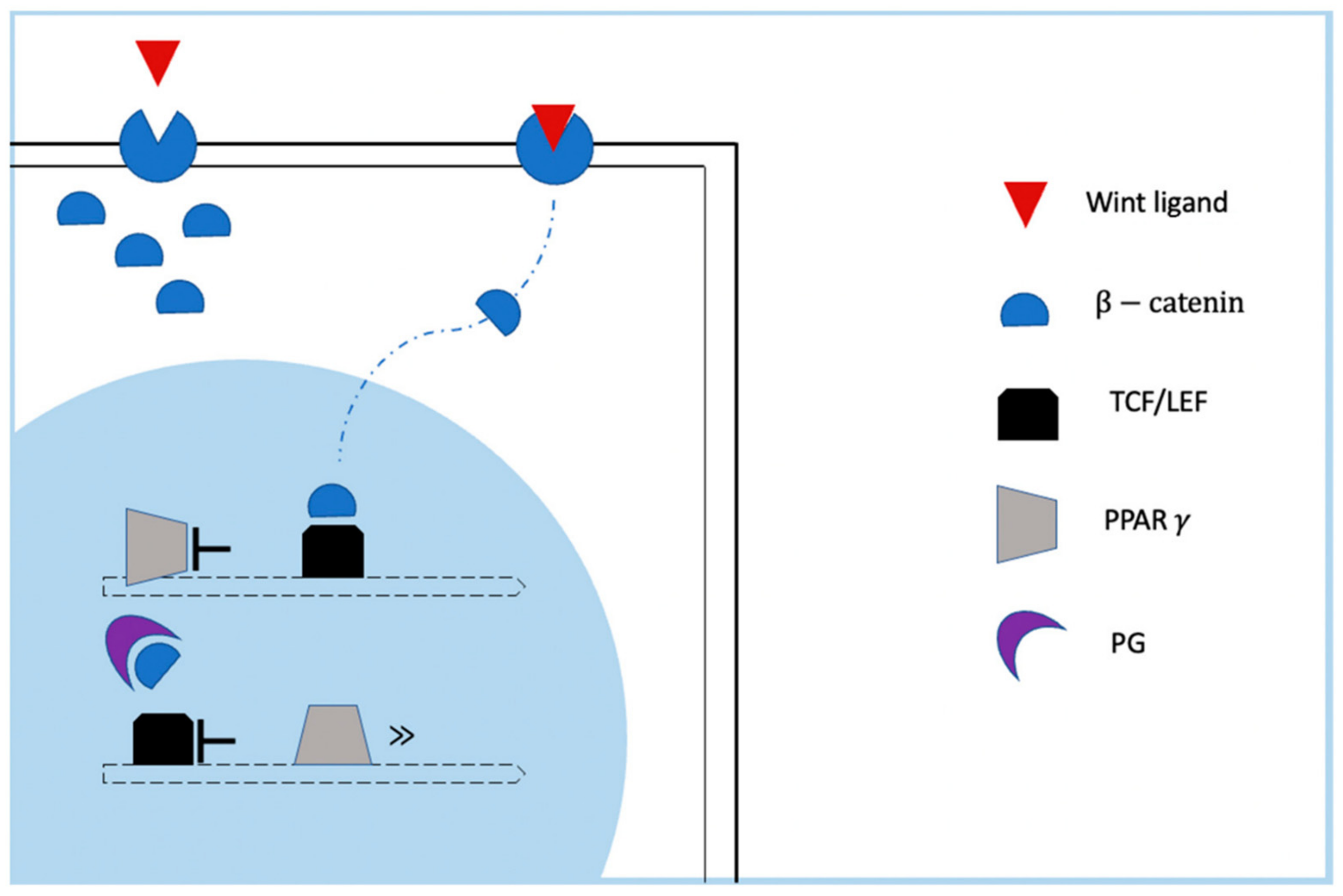
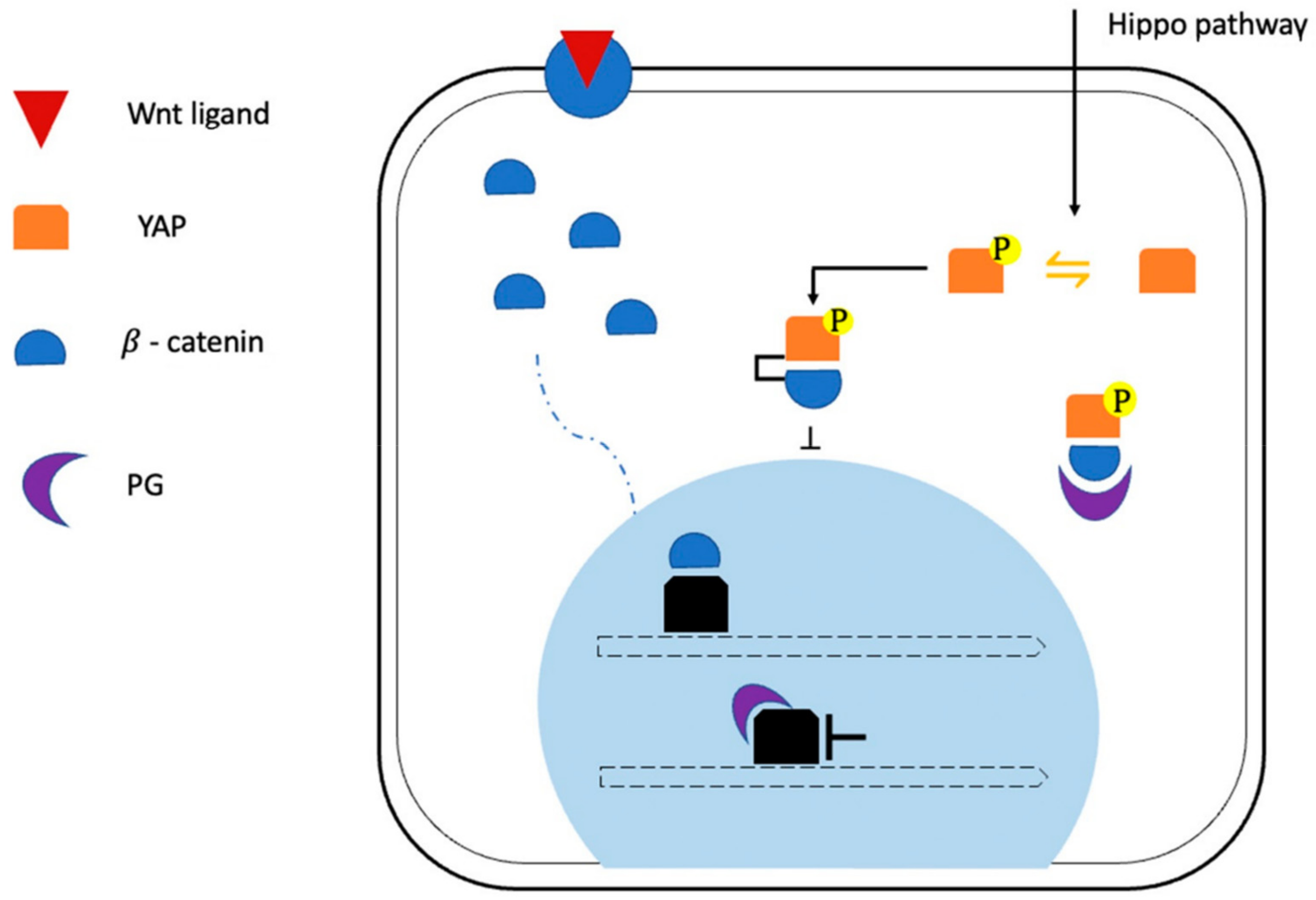
2.1. Role of Altered Signaling Pathways in ACM Pathogenesis: Desmosomal and Junctional Gene Mutations
2.2. Role of Altered Signaling Pathways in ACM Pathogenesis: Non-Desmosomal Gene Mutations
2.3. Role of Large Genomic Rearrangements of Desmosomal Genes and Whole-Exome Sequencing in ACM
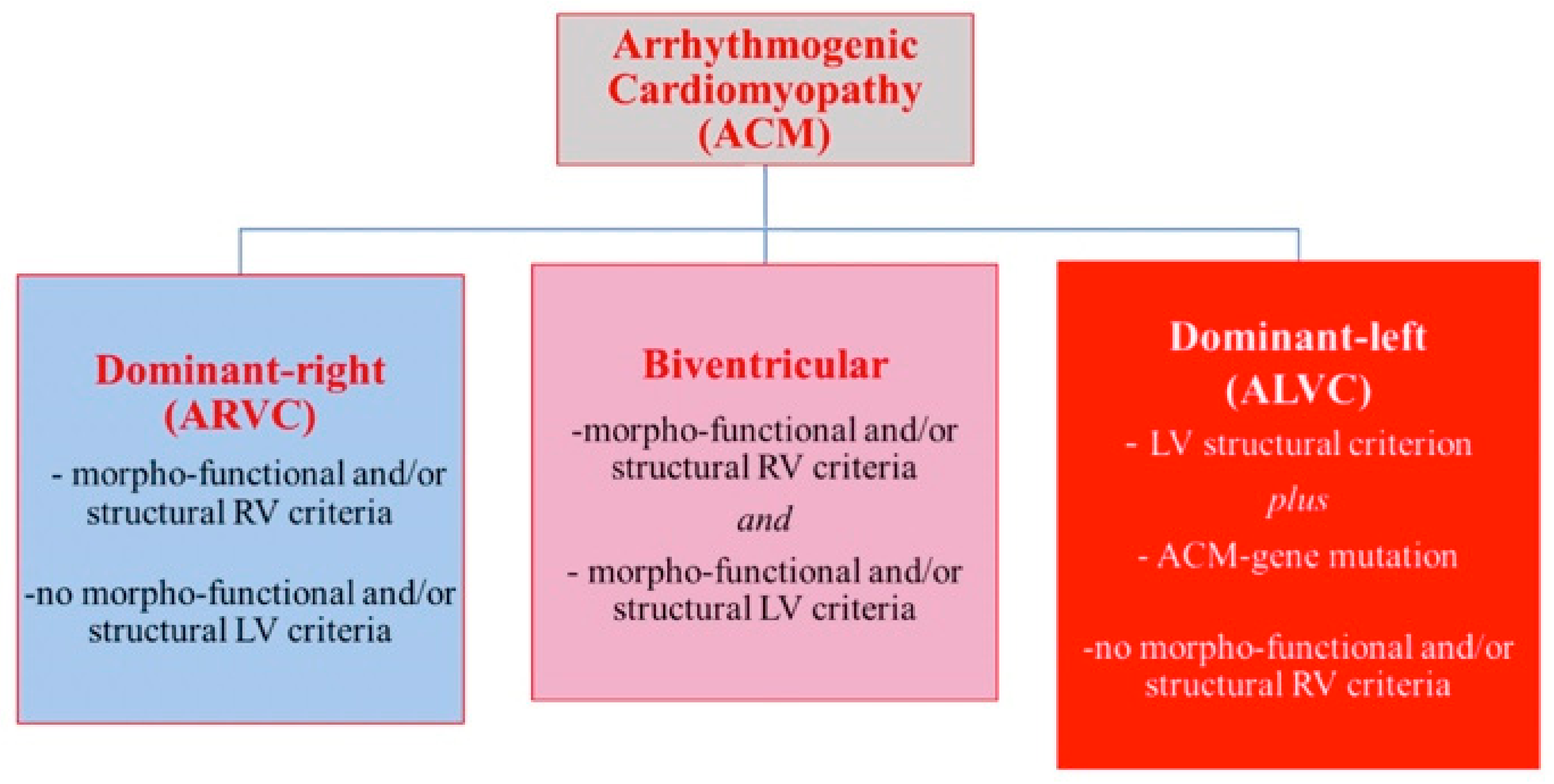
2.4. Arrhythmogenic Cardiomyopathy Is Not a Right-Ventricular Disease
2.4.1. Arrhythmogenic Phenotypes
2.4.2. The Arrhythmogenic Profile in Left-Side Dilated Hearts: Role of Genetics
2.4.3. Desmoplakin Cardiomyopathy
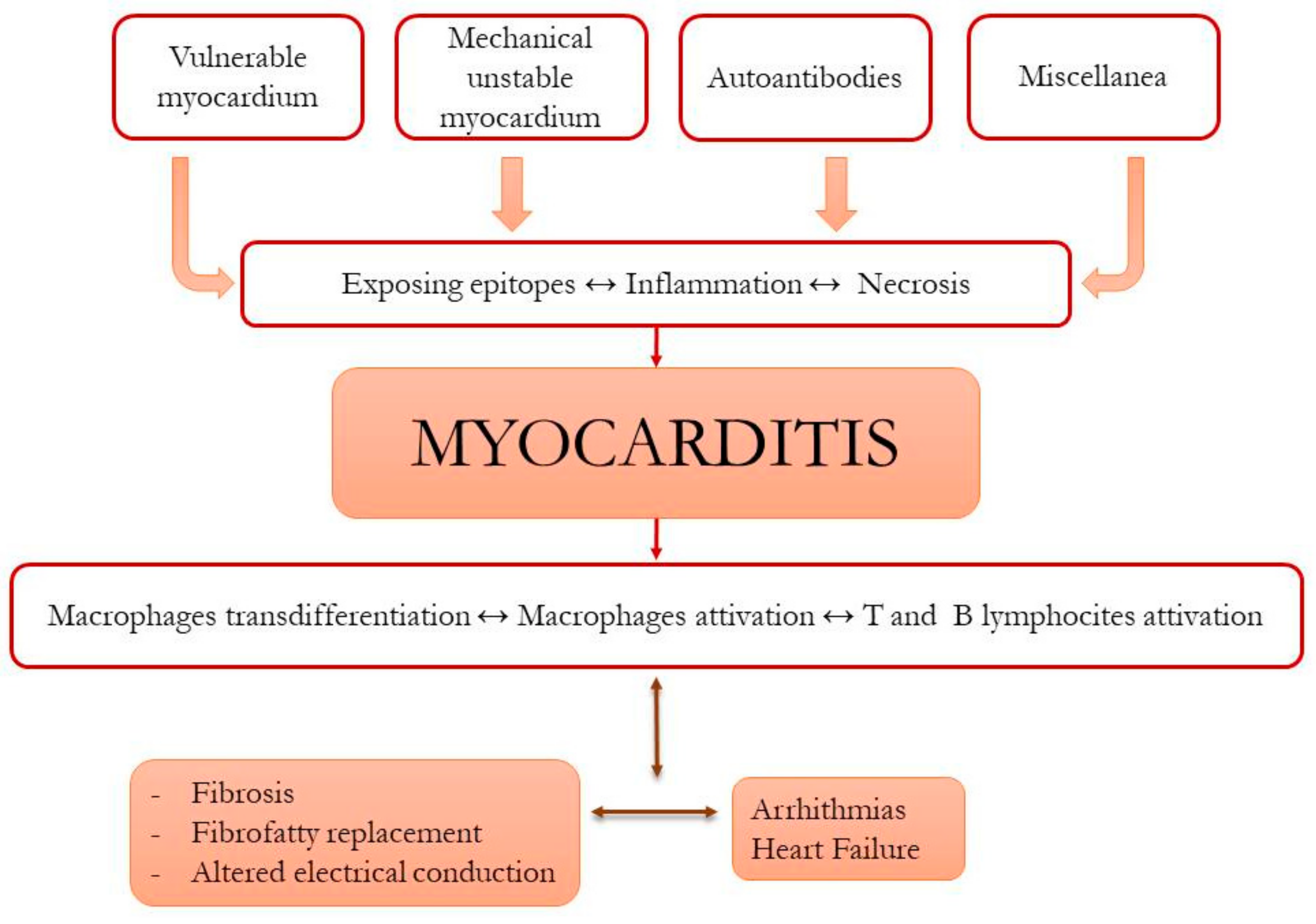
2.5. Role of Immunity in ACM Pathophysiology
3. Diagnosis
3.1. Electrocardiographic Signs
3.2. First-Line Echocardiography
3.3. The Power of Cardiac Magnetic Resonance
3.4. The Role of Computed Tomography
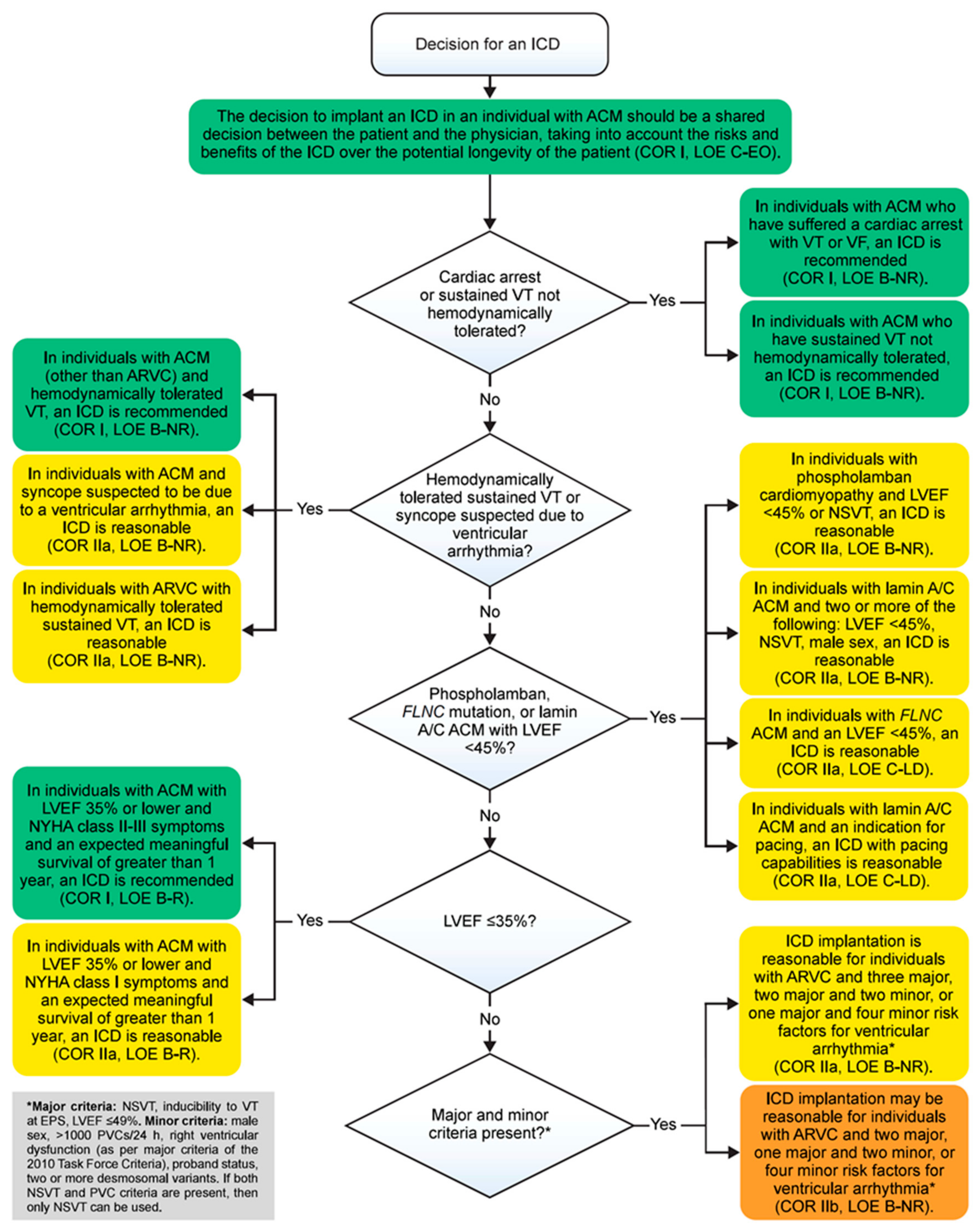
4. Risk Stratification
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malargue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical Diagnosis, Imaging, and Genetics of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; De Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int. J. Cardiol. 2020, 319, 106–114. [Google Scholar] [CrossRef] [PubMed]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J.; et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Gao, S.; Puthenvedu, D.; Lombardi, R.; Chen, S.N. Established and Emerging Mechanisms in the Pathogenesis of Arrhythmogenic Cardiomyopathy: A Multifaceted Disease. Int. J. Mol. Sci. 2020, 21, 6320. [Google Scholar] [CrossRef]
- Costa, S.; Cerrone, M.; Saguner, A.M.; Brunckhorst, C.; Delmar, M.; Duru, F. Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends Cardiovasc. Med. 2021, 31, 395–402. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit. Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.A.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfled, A.C.P.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Chen, L.; Rao, M.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Zhao, Q.; Yu, W.; Yuan, B.; Song, J.; et al. A founder homozygous DSG2 variant in East Asia results in ARVC with full penetrance and heart failure phenotype. Int. J. Cardiol. 2019, 274, 263–270. [Google Scholar] [CrossRef] [PubMed]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Wong, J.; Wen, J.; Wang, S.; Wang, C.; Spiering, S.; Kan, N.G.; Forcales, S.; Puri, P.L.; Leone, T.C.; et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 2013, 494, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Pu, W.T. Hippo activation in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Paige, S.L.; Osugi, T.; Afanasiev, O.K.; Pabon, L.; Reinecke, H.; Murry, C.E. Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS ONE 2010, 5, e11134. [Google Scholar] [CrossRef]
- Van Gijn, M.E.; Blankesteijn, W.M.; Smits, J.F.; Hierck, B.; Gittenberger-de Groot, A.C. Frizzled 2 is transiently expressed in neural crest-containing areas during development of the heart and great arteries in the mouse. Anat. Embryol. 2001, 203, 185–192. [Google Scholar] [CrossRef]
- Van de Schans, V.A.; Smits, J.F.; Blankesteijn, W.M. The Wnt/frizzled pathway in cardiovascular development and disease: Friend or foe? Eur. J. Pharmacol. 2008, 585, 338–345. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Imajo, M.; Miyatake, K.; Iimura, A.; Miyamoto, A.; Nishida, E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. 2012, 31, 1109–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1, e85923. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, Y.; Maruyama, M.; Zhu, W.; Chen, H.; Zhang, W.; Reuter, S.; Lin, S.F.; Haneline, L.S.; Field, L.J.; et al. Restrictive loss of plakoglobin in cardiomyocytes leads to arrhythmogenic cardiomyopathy. Hum. Mol. Genet. 2011, 20, 4582–4596. [Google Scholar] [CrossRef] [Green Version]
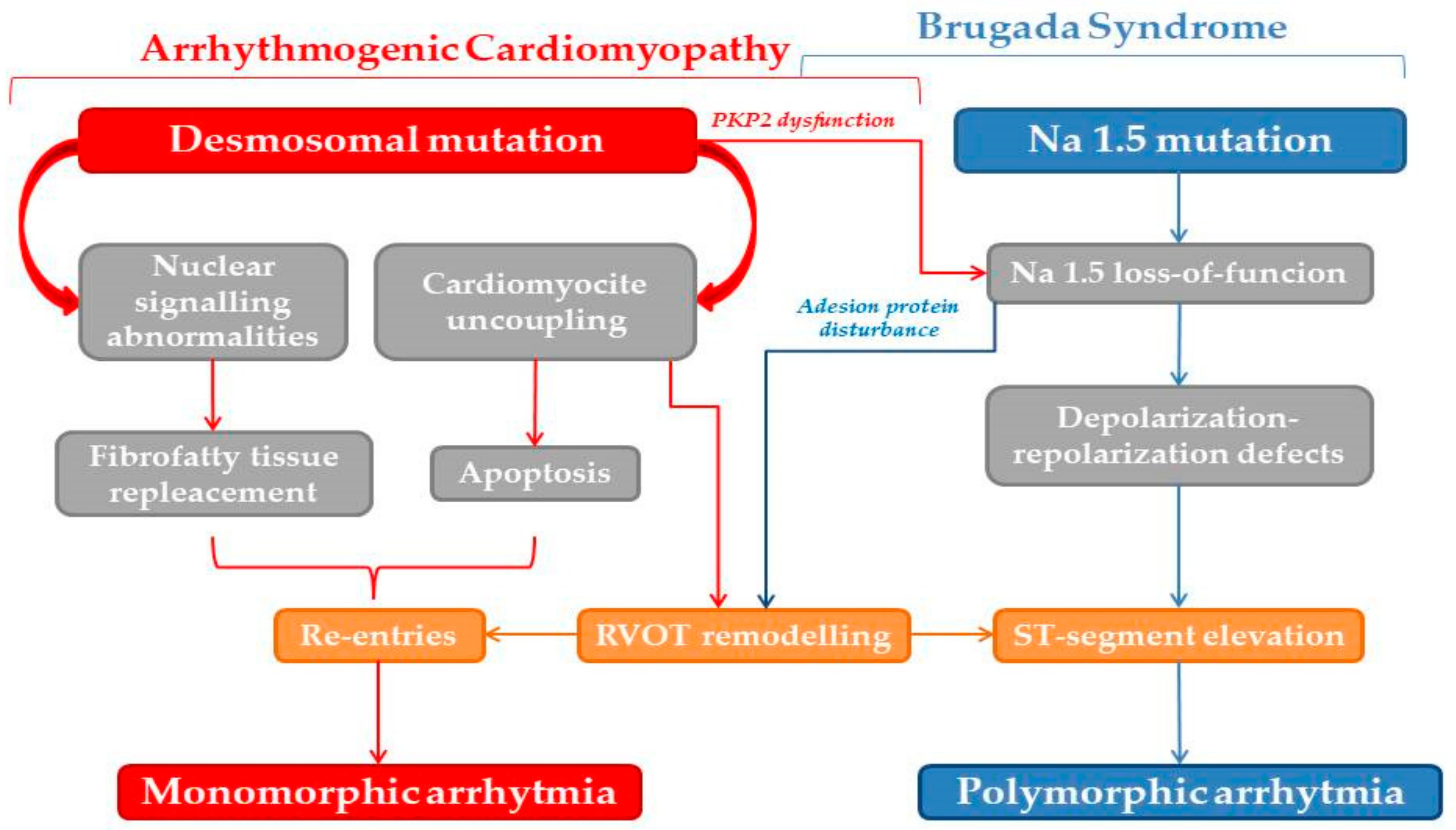
- BBen-Haim, Y.; Asimaki, A.; Behr, E.R. Brugada syndrome and arrhythmogenic cardiomyopathy: Overlapping disorders of the connexome? EP Europace 2021, 23, 653–664. [Google Scholar] [CrossRef]
- Zaklyazminskaya, E.; Dzemeshkevich, S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim. Biophys. Acta 2016, 1863, 1799–1805. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Leo-Macias, A.; Agullo-Pascual, E.; Sanchez-Alonso, J.L.; Keegan, S.; Lin, X.; Arcos, T.; Liang, F.X.; Korchev, Y.E.; Gorelik, J.; Fenyo, D.; et al. Erratum: Nanoscale visualization of functional adhesion/excitability nodes at the intercalated disc. Nat. Commun. 2016, 7, 1. [Google Scholar] [CrossRef]
- Beyder, A.; Rae, J.L.; Bernard, C.; Strege, P.R.; Sachs, F.; Farrugia, G. Mechanosensitivity of Nav1.5, a voltage-sensitive sodium channel. J. Physiol. 2010, 588 Pt 24, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Gusky, H.C.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.J.; Isom, L.L.; Taffet, S.M.; Delmar, M. Interactions between andkyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ. Ris. 2011, 109, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.; Asimaki, A.; van Rijen, H.V.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.; et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm 2013, 10, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Catalano, O.; Antonaci, S.; Moro, G.; Mussida, M.; Frascaroli, M.; Baldi, M.; Cobelli, F.; Baiardi, P.; Nastoli, J.; Bloise, R.; et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 2009, 30, 2241–2248. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.; Finlay, M.; Ahmed, A.K.; Ciaccio, E.J.; Asimaki, A.; Saffitz, J.E.; Quarta, G.; Nobles, M.; Syrris, P.; Chaubey, S.; et al. Electrophysiological abnormalities precede overt structural changes in arrhythmogenic right ventricular cardiomyopathy due to mutations in desmoplakin-A combined murine and human study. Eur. Heart J. 2012, 33, 1942–1953. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Liu, N.; Priori, S.G. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 2009, 6, 337–348. [Google Scholar] [CrossRef]
- FFranke, W.W.; Borrmann, C.M.; Grund, C.; Pieperhoff, S. The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur. J. Cell Biol. 2006, 85, 69–82. [Google Scholar] [CrossRef]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Basso, C.; Bauce, B.; Corrado, D.; Thiene, G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2011, 9, 223–233. [Google Scholar] [CrossRef]
- Moncayo-Arlandi, J.; Brugada, R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat. Rev. Cardiol. 2017, 14, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Pilichou, K.; Lazzarini, E.; Rigato, I.; Celeghin, R.; De Bortoli, M.; Perazzolo Marra, M.; Cason, M.; Jongbloed, J.; Calore, M.; Rizzo, S.; et al. Large Genomic Rearrangements of Desmosomal Genes in Italian Arrhythmogenic Cardiomyopathy Patients. Circ. Arrhythm. Electrophysiol. 2017, 10, e005324. [Google Scholar] [CrossRef] [PubMed]
- Fedida, J.; Fressart, V.; Charron, P.; Surget, E.; Hery, T.; Richard, P.; Donal, E.; Keren, B.; Duthoit, G.; Hidden-Lucet, F.; et al. Contribution of exome sequencing for genetic diagnostic in arrhythmogenic right ventricular cardiomyopathy/dysplasia. PLoS ONE 2017, 12, e0181840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [Green Version]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegkos, T.; Panagiotidis, T.; Parcharidou, D.; Efthimiadis, G. Emerging concepts in arrhythmogenic dilated cardiomyopathy. Heart Fail. Rev. 2021, 26, 1219–1229. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, W.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Køber, L.; Thune, J.J.; Nielsen, J.C.; Haarbo, J.; Videbaek, L.; Korup, E.; Jensen, G.; Hildebrandt, P.; Steffensen, F.H.; Bruun, N.E.; et al. Defibrillator Implantation in Patients with Nonischemic Systolic Heart Failure. N. Engl. J. Med. 2016, 375, 1221–1230. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 Linee guida ESC per la gestione dei pazienti con aritmie ventricolari e la prevenzione della morte cardiaca improvvisa: La task force per la gestione dei pazienti con aritmie ventricolari e la prevenzione della morte cardiaca improvvisa della European Society of Cardiology (ESC) Approvato da: Association for European Pediatric and Congenital Cardiology (AEPC). G. Eur. Del Cuore 2015, 36, 2793–2867. [Google Scholar] [CrossRef] [Green Version]
- Gigli, M.; Merlo, M.; Graw, S.L.; Barbati, G.; Rowland, T.J.; Slavov, D.B.; Stolfo, D.; Haywood, M.E.; Dal Ferro, M.; Altinier, A.; et al. Genetic Risk of Arrhythmic Phenotypes in Patients with Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 1480–1490. [Google Scholar] [CrossRef] [PubMed]
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signalling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jimenez-Jaimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell-Cell Adhesion Structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Alvarez Abril, B.; Alvarez, M.; Lopez-Fernandez, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfled, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Hodgkinson, K.A.; Connors, S.P.; Merner, N.; Haywood, A.; Young, T.L.; McKenna, W.J.; Gallagher, B.; Curtis, F.; Bassett, A.S.; Parfreu, P.S.; et al. The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin. Genet. 2013, 83, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Rivera Sorensen, C.; Sheppard, M.N.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322. [Google Scholar] [CrossRef]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.; Murray, B.; Te Riele, A.S.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef]
- Price, A.J.; Cost, A.L.; Ungewiß, H.; Waschke, J.; Dunn, A.R.; Grashoff, C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat. Commun. 2018, 9, 5284. [Google Scholar] [CrossRef] [PubMed]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heliö, K.; Kangas-Kontio, T.; Weckström, S.; Vanninen, S.U.; Aalto-Setälä, K.; Alastalo, T.P.; Myllykangas, S.; Heliö, T.M.; Koskenvuo, J.W. DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC Med. Genet. 2020, 21, 19. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.; Asatryan, B.; Siripanthong, B.; Munroe, P.B.; Tiku-Owens, A.; Lopes, L.R.; Khanji, M.Y.; Protonotarios, A.; Santangeli, P.; Muser, D.; et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6615. [Google Scholar] [CrossRef]
- Sabel, K.G.; Blomström-Lundqvist, C.; Olsson, S.B.; Eneström, S. Arrhythmogenic right ventricular dysplasia in brother and sister: Is it related to myocarditis? Pediatr. Cardiol. 1990, 11, 113–116. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G.; Corrado, D.; Angelini, A.; Nava, A.; Valente, M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996, 94, 983–991. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm 2015, 12, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Patrianakos, A.P.; Protonotarios, N.; Nyktari, E.; Pagonidis, K.; Tsatsopoulou, A.; Parthenakis, F.I.; Vardas, P.E. Arrhythmogenic right ventricular cardiomyopathy/dysplasia and troponin release. Myocarditis or the “hot phase” of the disease? Int. J. Cardiol. 2012, 157, e26–e28. [Google Scholar] [CrossRef] [PubMed]
- Kostis, W.J.; Tedford, R.J.; Miller, D.L.; Schulman, S.P.; Tomaselli, G.F. Troponin-I elevation in a young man with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Interv. Card. Electrophysiol. 2008, 22, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, A.; Elliott, P.M. Arrhythmogenic Cardiomyopathy: A Disease or Merely a Phenotype? Eur. Cardiol. 2020, 15, 1–5. [Google Scholar] [CrossRef]
- Bowles, N.E.; Ni, J.; Marcus, F.; Towbin, J.A. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 892–895. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, F.; Basso, C.; Carturan, E.; Valente, M.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Is there a role for viruses? Cardiovasc. Pathol. 2006, 15, 11–17. [Google Scholar] [CrossRef]
- Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Sanchez, O.; Cesar, S.; Mademont, I.; Allegue, C.; Mates, J.; Perez-Serra, A.; Coll, M.; et al. A Genetically Vulnerable Myocardium May Predispose to Myocarditis. J. Am. Coll. Cardiol. 2015, 66, 2913–2914. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Lubos, N.; van der Gaag, S.; Gerçek, M.; Kant, S.; Leube, R.E.; Krusche, C.A. Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Res. Cardiol. 2020, 115, 42. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Ama-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies With Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albayda, J.; Khan, A.; Casciola-Rosen, L.; Corse, A.M.; Paik, J.J.; Christopher-Stine, L. Inflammatory myopathy associated with anti-mitochondrial antibodies: A distinct phenotype with cardiac involvement. Semin. Arthritis Rheum. 2018, 47, 552–556. [Google Scholar] [CrossRef]
- Koyama, M.; Yano, T.; Kikuchi, K.; Nagahara, D.; Ishibashi-Ueda, H.; Miura, T. Lethal heart failure with anti-mitochondrial antibody: An arrhythmogenic right ventricular cardiomyopathy mimetic. Eur. Heart J. 2017, 38, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poller, W.; Haas, J.; Klingel, K.; Kuhnisch, J.; Gast, M.; Kaya, Z.; Escher, F.; Kayvanpour, E.; Degener, F.; Opgen-Rhein, B.; et al. Familial Recurrent Myocarditis Triggered by Exercise in Patients with a Truncating Variant of the Desmoplakin Gene. J. Am. Heart Assoc. 2020, 9, e015289. [Google Scholar] [CrossRef]
- Campbell, J.P.; Turner, J.E. Debunking the Myth of Exercise-Induced Immune Suppression: Redefining the Impact of Exercise on Immunological Health across the Lifespan. Front. Immunol. 2018, 9, 648. [Google Scholar] [CrossRef]
- Beffagna, G.; Sommariva, E.; Bellin, M. Mechanotransduction and Adrenergic Stimulation in Arrhythmogenic Cardiomyopathy: An Overview of in vitro and in vivo Models. Front. Physiol. 2020, 11, 568535. [Google Scholar] [CrossRef]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.A.; van Moerkerken, A.F.; van Eck-Smit, B.L.F.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef] [Green Version]
- Mavrogeni, S.; Protonotarios, N.; Tsatsopoulou, A.; Papachristou, P.; Sfendouraki, E.; Papadopoulos, G. Naxos disease evolution mimicking acute myocarditis: The role of cardiovascular magnetic resonance imaging. Int. J. Cardiol. 2013, 166, e14–e15. [Google Scholar] [CrossRef]
- Piriou, N.; Marteau, L.; Kyndt, F.; Serfaty, J.M.; Toquet, C.; Le Gloan, L.; Warin-Fresse, K.; Guijarro, D.; Le Tourneaur, T.; Conan, E.; et al. Familial screening in case of acute myocarditis reveals inherited arrhythmogenic left ventricular cardiomyopathies. ESC Heart Fail. 2020, 7, 1520–1533. [Google Scholar] [CrossRef]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2019, 284, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, D.; Basso, C.; Thiene, G.; McKenna, W.J.; Davies, M.J.; Fontaliran, F.; Nava, A.; Silvestri, F.; Blomstrom-Lundqvist, C.; Wlodarska, E.K.; et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J. Am. Coll. Cardiol. 1997, 30, 1512–1520. [Google Scholar] [CrossRef] [Green Version]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomsrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.; Singh, R.; Yamini, S.; Das, M.K. Fragmented ECG as a risk marker in cardiovascular diseases. Curr. Cardiol. Rev. 2014, 10, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Marcus, F.I.; Fontaine, G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: A review. Pacing Clin. Electrophysiol. 1995, 18, 1298–1314. [Google Scholar] [CrossRef]
- Peters, S.; Trümmel, M.; Koehler, B. QRS fragmentation in standard ECG as a diagnostic marker of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Heart Rhythm 2008, 5, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Peters, S. Electrocardiographic morphology in right precordial T waves in arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2016, 214, 228. [Google Scholar]
- Park, Y.; Cho, Y.; Lee, D.Y.; Jang, G.L.; Lee, H.; Yang, D.H.; Park, H.S.; Chae, S.C.; Jun, J.E.; Park, W.H. Correlation between the parameters of signal-averaged ECG and two-dimensional echocardiography in patients with arrhythmogenic right ventricular cardiomyopathy. Ann. Noninvasive Electrocardiol. 2009, 14, 50–56. [Google Scholar] [CrossRef]
- Kamath, G.S.; Zareba, W.; Delaney, J.; Koneru, J.N.; McKenna, W.J.; Gear, K.; Polonsky, S.; Sherrill, D.; Bluemke, D.; Marcus, F.; et al. Value of the signal-averaged electrocardiogram in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2011, 8, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Prakasa, K.R.; Dalal, D.; Wang, J.; Bomma, C.; Tandri, H.; Dong, J.; James, C.; Tichnell, C.; Russell, S.D.; Spevak, P.; et al. Feasibility and variability of three dimensional echocardiography in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am. J. Cardiol. 2006, 97, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.H.; Basso, C.; Badano, L.P.; Bucciarelli-Ducci, C.; Cardim, N.; Gaemperli, O.; Galderisi, M.; Habib, G.; Knuuti, J.; Lancellotti, P.; et al. Comprehensive multi-modality imaging approach in arrhythmogenic cardiomyopathy-an expert consensus document of the European Association of Cardiovascular Imaging. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 237–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, N.; Win, S.; James, C.A.; Kutty, S.; Mukherjee, M.; Gilotra, N.A.; Tichnell, C.; Murray, B.; Agafonova, J.; Tandri, H.; et al. Right Ventricular Strain Predicts Structural Disease Progression in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e015016. [Google Scholar] [CrossRef]
- Mast, T.P.; James, C.A.; Calkins, H.; Teske, A.J.; Tichnell, C.; Murray, B.; Loh, O.; Russell, S.D.; Velthuis, B.K.; Judge, D.P.; et al. Evaluation of Structural Progression in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. JAMA Cardiol. 2017, 2, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Saguner, A.M.; Vecchiati, A.; Baldinger, S.H.; Rüeger, S.; Medeiros-Domingo, A.; Mueller-Burri, A.S.; Haegeli, L.M.; Biaggi, P.; Manka, R.; Lüscher, T.F.; et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ. Cardiovasc. Imaging 2014, 7, 230–239. [Google Scholar] [CrossRef]
- Li, K.H.C.; Bazoukis, G.; Liu, T.; Li, G.; Wu, W.K.K.; Wong, S.H.; Wong, W.T.; Chan, Y.S.; Wong, M.C.S.; Wassilew, K.; et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) in clinical practice. J. Arrhythm. 2017, 34, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Raman, S.V.; Cook, S.C.; McCarthy, B.; Ferketich, A.K. Usefulness of multidetector row computed tomography to quantify right ventricular size and function in adults with either tetralogy of Fallot or transposition of the great arteries. Am. J. Cardiol. 2005, 95, 683–686. [Google Scholar] [CrossRef]
- Cochet, H.; Denis, A.; Komatsu, Y.; Jadidi, A.S.; Ali, T.A.; Sacher, F.; Derval, N.; Relan, J.; Sermesant, M.; Corneloup, O.; et al. Automated Quantification of Right Ventricular Fat at Contrast-enhanced Cardiac Multidetector CT in Arrhythmogenic Right Ventricular Cardiomyopathy. Radiology 2015, 275, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, Y.; Jadidi, A.; Sacher, F.; Denis, A.; Daly, M.; Derval, N.; Shah, A.; Lehrmann, H.; Park, C.I.; Weber, R.; et al. Relationship between MDCT-imaged myocardial fat and ventricular tachycardia substrate in arrhythmogenic right ventricular cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etoom, Y.; Govindapillai, S.; Hamilton, R.; Manlhiot, C.; Yoo, S.J.; Farhan, M.; Sarikouch, S.; Peters, B.; McCrindle, B.W.; Grosse-Wortmann, L. Importance of CMR within the Task Force Criteria for the diagnosis of ARVC in children and adolescents. J. Am. Coll. Cardiol. 2015, 65, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquaro, G.D.; Barison, A.; Todiere, G.; Grigoratos, C.; Ait Ali, L.; Di Bella, G.; Emdin, M.; Festa, P. Usefulness of combined functional assessment by cardiac magnetic resonance and tissue characterization versus task force criteria for diagnosis of Arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2016, 118, 1730–1736. [Google Scholar] [CrossRef]
- Segura-Rodríguez, D.; Bermúdez-Jiménez, F.J.; Carriel, V.; Lopez-Fernandez, S.; Gonzalez-Molina, M.; Oyonarte Ramirez, J.M.; Fernandez-Navarro, L.; Garcia-Roa, M.D.; Cabrerizo, E.M.; Durand-Herrera, D.; et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: A genotype-phenotype correlation study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, A.; Bauce, B.; De Lazzari, M.; Rigato, I.; Bariani, R.; Meneghin, S.; Pilichou, K.; Motta, R.; Aliberti, C.; Thiene, G.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy: Characterization of Left Ventricular Phenotype and Differential Diagnosis with Dilated Cardiomyopathy. J. Am. Heart Assoc. 2020, 9, e014628. [Google Scholar] [CrossRef]
- Boulé, S.; Fressart, V.; Laux, D.; Mallet, A.; Simon, F.; de Groote, P.; Bonnet, D.; Klug, D.; Charron, P. Expanding the phenotype associated with a desmoplakin dominant mutation: Carvajal/Naxos syndrome associated with leukonychia and oligodontia. Int. J. Cardiol. 2012, 161, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Prati, G.; Vitrella, G.; Allocca, G.; Muser, D.; Buttignoni, S.C.; Piccoli, G.; Morocutti, G.; Delise, P.; Pinamonti, B.; Proclemer, A.; et al. Right Ventricular Strain and Dyssynchrony Assessment in Arrhythmogenic Right Ventricular Cardiomyopathy: Cardiac Magnetic Resonance Feature-Tracking Study. Circ. Cardiovasc. Imaging 2015, 8, e003647. [Google Scholar] [CrossRef] [Green Version]
- Zghaib, T.; Ghasabeh, M.A.; Assis, F.R.; Chrispin, J.; Keramati, A.; Misra, S.; Berger, R.; Calkins, H.; Kamel, I.; Nazarian, S.; et al. Regional Strain by Cardiac Magnetic Resonance Imaging Improves Detection of Right Ventricular Scar Compared With Late Gadolinium Enhancement on a Multimodality Scar Evaluation in Patients With Arrhythmogenic Right Ventricular Cardiomyopathy. Circ. Cardiovasc. Imaging 2018, 11, e007546. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.T.; Yang, Z.G.; Diao, K.Y.; Jiang, L.; Zhang, Y.; Liu, X.; Gao, Y.; Hu, B.Y.; Huang, S.; Guo, Y.K. Left Ventricular Involvement in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Predicts Adverse Clinical Outcomes: A Cardiovascular Magnetic Resonance Feature Tracking Study. Sci. Rep. 2019, 9, 14235. [Google Scholar] [CrossRef]
- Tansey, D.K.; Aly, Z.; Sheppard, M.N. Fat in the right ventricle of the normal heart. Histopathology 2005, 46, 98–104. [Google Scholar] [CrossRef]
- Bluemke, D.A.; Krupinski, E.A.; Ovitt, T.; Gear, K.; Unger, E.; Axel, L.; Boxt, L.M.; Casolo, G.; Ferrari, V.A.; Funaki, B.; et al. MR Imaging of arrhythmogenic right ventricular cardiomyopathy: Morphologic findings and interobserver reliability. Cardiology 2003, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Tandri, H.; Castillo, E.; Ferrari, V.A.; Nasir, K.; Dalal, D.; Bomma, C.; Calkins, H.; Bluemke, D.A. Magnetic resonance imaging of arrhythmogenic right ventricular dysplasia: Sensitivity, specificity, and observer variability of fat detection versus functional analysis of the right ventricle. J. Am. Coll. Cardiol. 2006, 48, 2277–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandri, H.; Calkins, H.; Marcus, F.I. Controversial role of magnetic resonance imaging in the diagnosis of arrhythmogenic right ventricular dysplasia. Am. J. Cardiol. 2003, 92, 649. [Google Scholar] [CrossRef]
- Corrado, D.; Leoni, L.; Link, M.S.; Della Bella, P.; Gaita, F.; Curnis, A.; Salerno, J.U.; Igidbashian, D.; Raviele, A.; Disertori, M.; et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation 2003, 108, 3084–3091. [Google Scholar] [CrossRef] [Green Version]
- Schinkel, A.F. Implantable cardioverter defibrillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ. Arrhythm. Electrophysiol. 2013, 6, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Wichter, T.; Paul, M.; Wollmann, C.; Acil, T.; Gerdesm, O.; Ashraf, O.; Tjan, T.D.T.; Soeparwata, R.; Block, M.; Borggrefe, M.; et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: Single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004, 109, 1503–1508. [Google Scholar] [CrossRef]
- Piccini, J.P.; Dalal, D.; Roguin, A.; Bomma, C.; Cheng, A.; Prakasa, K.; Dong, J.; Tichnell, C.; James, C.; Russell, S.; et al. Predictors of appropriate implantable defibrillator therapies in patients with arrhythmogenic right ventricular dysplasia. Heart Rhythm 2005, 2, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Calkins, H.; Link, M.S.; Leoni, L.; Favale, S.; Bevilacqua, M.; Basso, C.; Ward, D.; Boriani, G.; Ricci, R.; et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation 2010, 122, 1144–1152. [Google Scholar] [CrossRef]
- Bhonsale, A.; James, C.A.; Tichnell, C.; Murray, B.; Gagarin, D.; Philips, B.; Dalal, D.; Tedford, R.; Russell, S.D.; Abraham, T.; et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J. Am. Coll. Cardiol. 2011, 58, 1485–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orgeron, G.M.; James, C.A.; Te Riele, A.; Tichnell, C.; Murray, B.; Bhonsale, A.; Kamel, I.R.; Zimmerman, S.L.; Judge, D.P.; Crosson, J.; et al. Implantable Cardioverter-Defibrillator Therapy in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: Predictors of Appropriate Therapy, Outcomes, and Complications. J. Am. Heart Assoc. 2017, 6, e006242. [Google Scholar] [CrossRef]
- Ellenbogen, K.A.; Levine, J.H.; Berger, R.D.; Daubert, J.P.; Winters, S.L.; Greenstein, E.; Shalaby, A.; Schaechter, A.; Subacius, H.; Kadish, A. Are implantable cardioverter defibrillator shocks a surrogate for sudden cardiac death in patients with nonischemic cardiomyopathy? Circulation 2006, 113, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Brun, F.; Groeneweg, J.A.; Gear, K.; Sinagra, G.; van der Heijden, J.; Mestroni, L.; Hauer, R.N.; Borgstrom, M.; Marcus, F.I.; Hughes, T. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy Without Implantable Cardioverter-Defibrillators. JACC Clin. Elettrofisiolo. 2016, 2, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Corrado, D.; Wichter, T.; Link, M.S.; Hauer, R.N.W.; Marchlinski, F.E.; Anastasakis, A.; Bauce, B.; Basso, C.; Brunckhorst, C.; Tsatsopoulou, A.; et al. Treatment of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: An International Task Force Consensus Statement. Circulation 2015, 132, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Anselme, F.; Moubarak, G.; Savouré, A.; Godin, B.; Borz, B.; Drouin-Garraud, V.; Gay, A. Implantable cardioverter-defibrillators in lamin A/C mutation carriers with cardiac conduction disorders. Heart Rhythm 2013, 10, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Maupain, C.; Badenco, N.; Pousset, F.; Waintraub, X.; Duthoit, G.; Chastre, T.; Himbert, C.; Hebert, J.L.; Franck, R.; Hidden-Lucet, F.; et al. Risk Stratification in Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia Without an Implantable Cardioverter-Defibrillator. JACC Clin. Electrophysiol. 2018, 4, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Cadrin-Tourigny, J.; Bosman, L.P.; Nozza, A.; Wang, W.; Tadros, R.; Bhonsale, A.; Bourfiss, M.; Fortier, A.; Lie, O.H.; Saguner, A.M.; et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2019, 40, 1850–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquaro, G.D.; De Luca, A.; Cappelletto, C.; Raimondi, F.; Bianco, F.; Botto, N.; Barison, A.; Romani, S.; Lesizza, P.; Fabris, E.; et al. Comparison of different prediction models for the indication of implanted cardioverter defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy. ESC Heart Fail. 2020, 7, 4080–4088. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Right Ventricle (Upgraded 2010 ITF Diagnostic Criteria) | Left Ventricle (New Diagnostic “Padua” Criteria) |
|---|---|---|
| Morpho-functional ventricular abnormalities by cardiovascular imaging (echocardiography, CMR) or angiography | Major
| Minor
|
| Structural myocardial abnormalities by CE-CMR | Major
| Major
|
| Structural myocardial abnormalities by EMB (limited indications) | Major
| |
| Repolarization abnormalities | Major
| Minor
|
| Ventricular arrhythmias | Major
| Minor
|
| Family history/genetics | Major
| |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Florio, M.T.; Boccia, F.; Vetrano, E.; Borrelli, M.; Gossios, T.; Palmiero, G. Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy. Cardiogenetics 2021, 11, 263-289. https://doi.org/10.3390/cardiogenetics11040025
Florio MT, Boccia F, Vetrano E, Borrelli M, Gossios T, Palmiero G. Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy. Cardiogenetics. 2021; 11(4):263-289. https://doi.org/10.3390/cardiogenetics11040025
Chicago/Turabian StyleFlorio, Maria Teresa, Filomena Boccia, Erica Vetrano, Marco Borrelli, Thomas Gossios, and Giuseppe Palmiero. 2021. "Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy" Cardiogenetics 11, no. 4: 263-289. https://doi.org/10.3390/cardiogenetics11040025
APA StyleFlorio, M. T., Boccia, F., Vetrano, E., Borrelli, M., Gossios, T., & Palmiero, G. (2021). Pathogenesis, Diagnosis and Risk Stratification in Arrhythmogenic Cardiomyopathy. Cardiogenetics, 11(4), 263-289. https://doi.org/10.3390/cardiogenetics11040025