
Diagnosis of Fabry Disease in a Patient with a Surgically Repaired Congenital Heart Defect: When Clinical History and Genetics Make the Difference

 ,
,  , , , ,
, , , ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
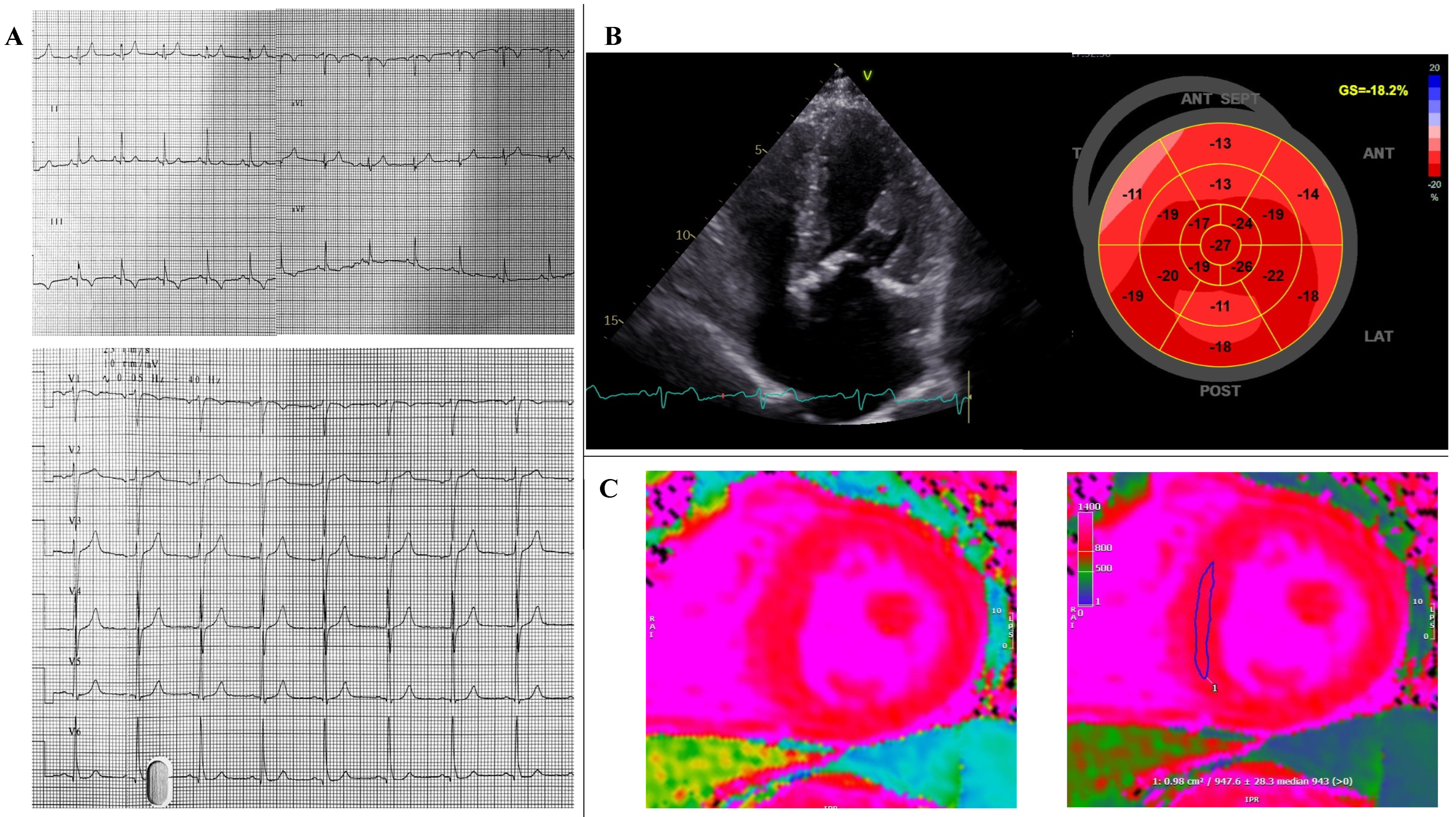
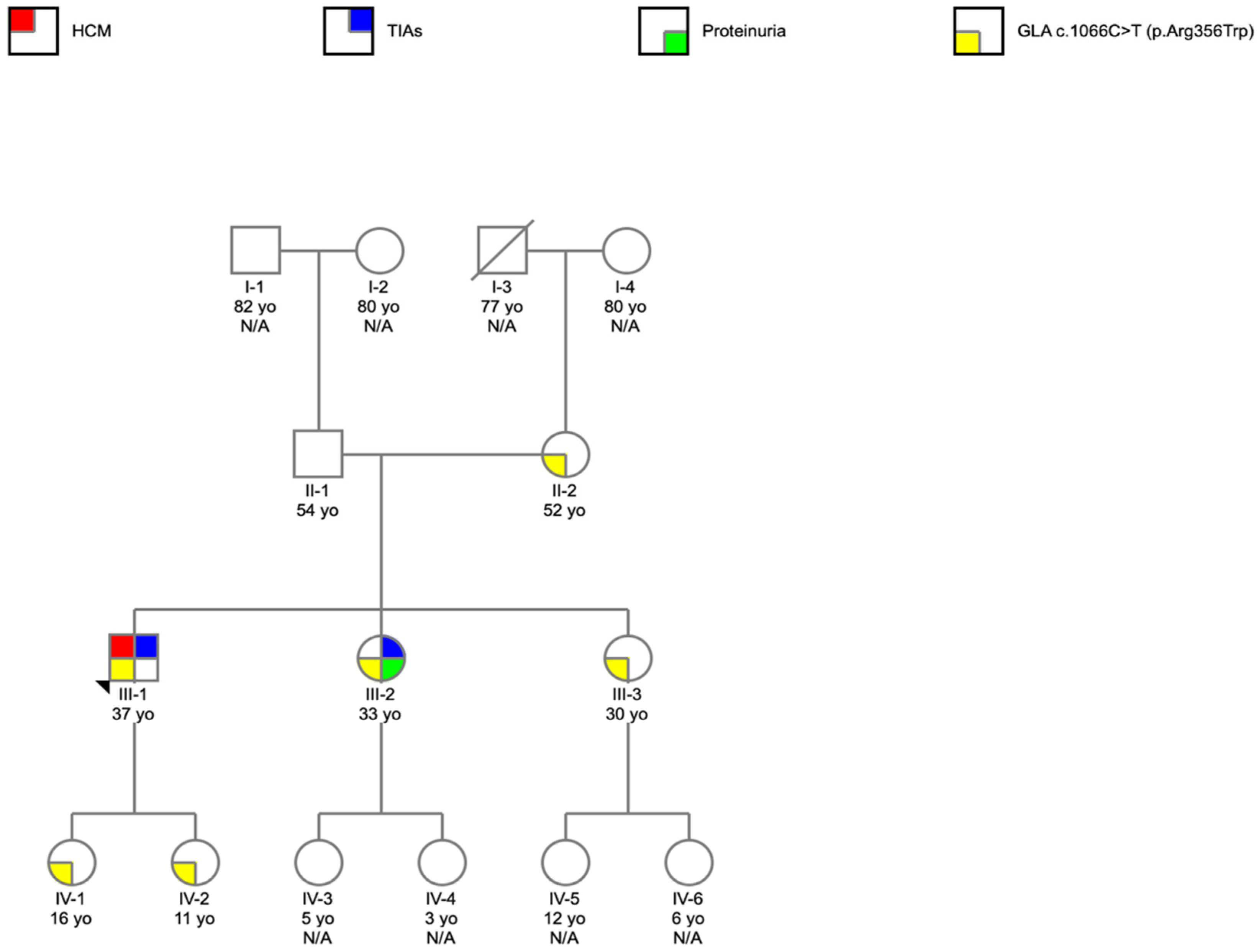
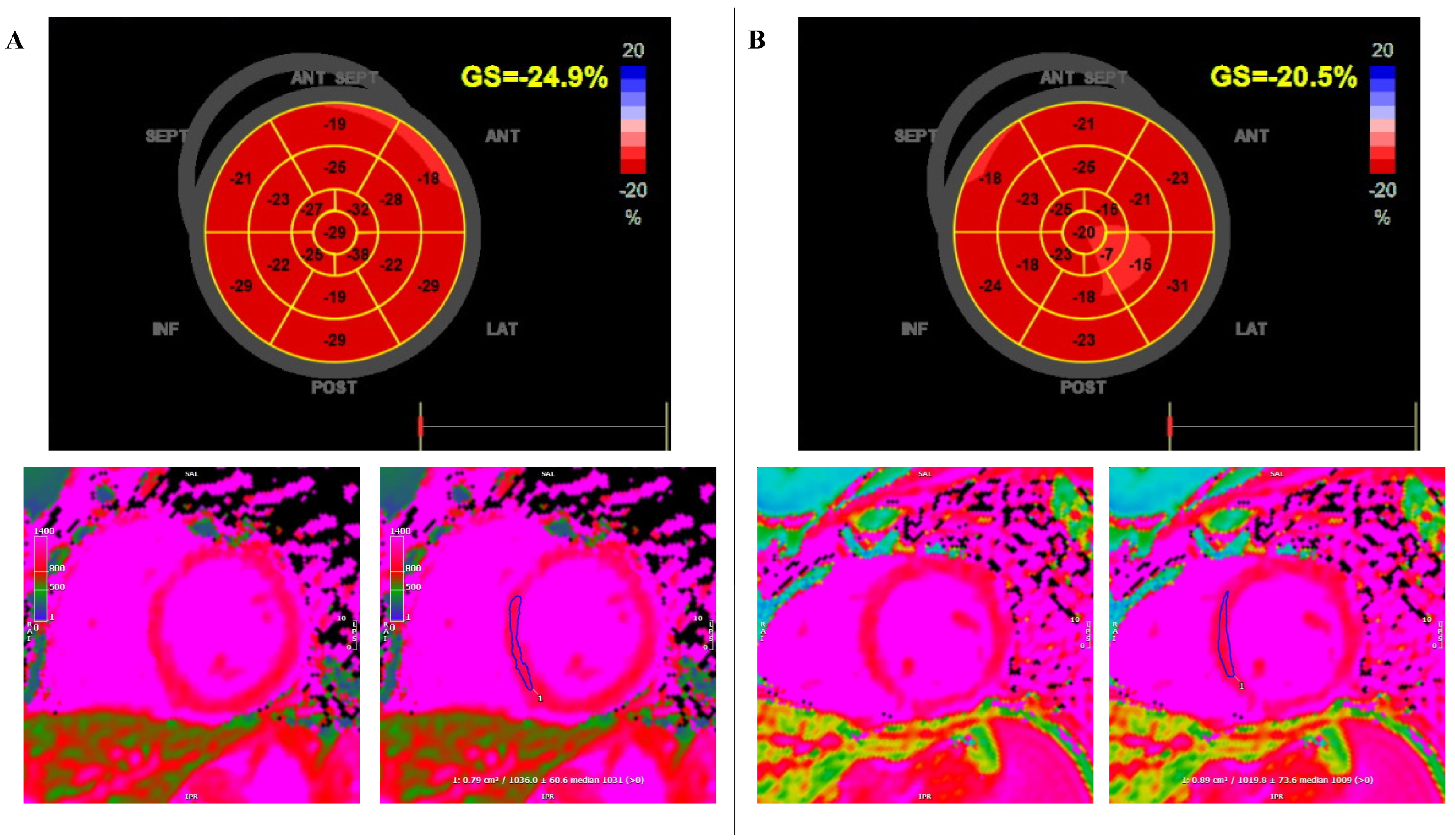
2. Case Report
3. Discussion
- The diagnostic delay that generally characterizes rare diseases;
- The potential coexistence with other diseases (i.e., congenital heart defect) and the importance of identifying specific red flags that raise the possibility of the disease;
- The importance of the cascade program screening to identify family members at risk;
- The difficulty in the management of asymptomatic female patients or with mild clinical manifestations.
3.1. Diagnostic Delay and Clinical Markers in FD
3.2. Genetic Diagnosis and Cascade Program Screening in FD, and Treatment in Females with FD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rubino, M.; Monda, E.; Lioncino, M.; Caiazza, M.; Palmiero, G.; Dongiglio, F.; Fusco, A.; Cirillo, A.; Cesaro, A.; Capodicasa, L.; et al. Diagnosis and Management of Cardiovascular Involvement in Fabry Disease. Heart Fail. Clin. 2022, 18, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry Registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- Rapezzi, C.; Arbustini, E.; Caforio, A.L.; Charron, P.; Gimeno-Blanes, J.; Helio, T.; Linhart, A.; Mogensen, J.; Pinto, Y.; Ristic, A.; et al. Diagnostic work-up in cardiomyopathies: Bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2013, 34, 1448–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2017, 19, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef]
- Lidove, O.; Kaminsky, P.; Hachulla, E.; Leguy-Seguin, V.; Lavigne, C.; Marie, I.; Maillot, F.; Serratrice, C.; Masseau, A.; Chérin, P.; et al. Fabry disease ‘The New Great Imposter’: Results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin. Genet. 2012, 81, 571–577. [Google Scholar] [CrossRef]
- Reisin, R.; Perrin, A.; García-Pavía, P. Time delays in the diagnosis and treatment of Fabry disease. Int. J. Clin. Pract. 2017, 71, e12914. [Google Scholar] [CrossRef]
- Linhart, A.; Elliott, P.M. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [PubMed]
- Militaru, S.; Ginghina, C.; Popescu, B.A.; Saftoiu, A.; Linhart, A.; Jurcut, R. Multimodality imaging in Fabry cardiomyopathy: From early diagnosis to therapeutic targets. Eur. Heart J. Cardiovasc. Imaging 2018, 19, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.; Shah, R.; Saiedi, M.; Patil, S.; Ganesan, A.; Linhart, A.; Selvanayagam, J.B. The Role of Cardiac Imaging in the Diagnosis and Management of Anderson-Fabry Disease. JACC Cardiovasc. Imaging 2019, 12 Pt 1, 1230–1242. [Google Scholar] [CrossRef]
- Esposito, A.; Monda, E.; Gragnano, F.; Simone, F.; Cesaro, A.; Natale, F.; Concilio, C.; Moscarella, E.; Caiazza, M.; Pazzanese, V.; et al. Prevalence and clinical implications of hyperhomocysteinaemia in patients with hypertrophic cardiomyopathy and MTHFR C6777T polymorphism. Eur. J. Prev. Cardiol. 2020, 27, 1906–1908. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. [Google Scholar] [CrossRef]
- Barretta, F.; Mirra, B.; Monda, E.; Caiazza, M.; Lombardo, B.; Tinto, N.; Scudiero, O.; Frisso, G.; Mazzaccara, C. The Hidden Fragility in the Heart of the Athletes: A Review of Genetic Biomarkers. Int. J. Mol. Sci. 2020, 21, 6682. [Google Scholar] [CrossRef]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020, 28, 1134–1137. [Google Scholar] [CrossRef]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020, 28, 1081–1090. [Google Scholar] [CrossRef]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alström syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Monda, E.; Fusco, A.; Melis, D.; Caiazza, M.; Gragnano, F.; Mauriello, A.; Cirillo, A.; Rubino, M.; Esposito, A.; Grammegna, A.; et al. Clinical significance of family history and bicuspid aortic valve in children and young adult patients with Marfan syndrome. Cardiol. Young 2020, 30, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, M.; Rubino, M.; Monda, E.; Passariello, A.; Fusco, A.; Cirillo, A.; Esposito, A.; Pierno, A.; De Fazio, F.; Pacileo, R.; et al. Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy. Genes 2020, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; DArgenio, V.; Esposito, M.V.; Monda, E.; Maggio, F.D.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes 2020, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Frisso, G.; Rubino, M.; Caiazza, M.; Esposito, A.; Cirillo, A.; Fusco, A.; Palmiero, G.; Mazzaccara, C.; Pacileo, R.; et al. Potential role of imaging markers in predicting future disease expression of arrhythmogenic cardiomyopathy. Future Cardiol. 2020, 17, 647–654. [Google Scholar] [CrossRef]
- Germain, D.P.; Arad, M.; Burlina, A.; Elliott, P.M.; Falissard, B.; Feldt-Rasmussen, U.; Hilz, M.J.; Hughes, D.A.; Ortiz, A.; Wanner, C.; et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease—A systematic literature review by a European panel of experts. Mol. Genet. Metab. 2019, 126, 224–235. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rubino, M.; Monda, E.; Caiazza, M.; Palmiero, G.; Lioncino, M.; Cirillo, A.; Fusco, A.; Verrillo, F.; Perna, A.; Diana, G.; et al. Diagnosis of Fabry Disease in a Patient with a Surgically Repaired Congenital Heart Defect: When Clinical History and Genetics Make the Difference. Cardiogenetics 2022, 12, 102-108. https://doi.org/10.3390/cardiogenetics12010010
Rubino M, Monda E, Caiazza M, Palmiero G, Lioncino M, Cirillo A, Fusco A, Verrillo F, Perna A, Diana G, et al. Diagnosis of Fabry Disease in a Patient with a Surgically Repaired Congenital Heart Defect: When Clinical History and Genetics Make the Difference. Cardiogenetics. 2022; 12(1):102-108. https://doi.org/10.3390/cardiogenetics12010010
Chicago/Turabian StyleRubino, Marta, Emanuele Monda, Martina Caiazza, Giuseppe Palmiero, Michele Lioncino, Annapaola Cirillo, Adelaide Fusco, Federica Verrillo, Alessia Perna, Gaetano Diana, and et al. 2022. "Diagnosis of Fabry Disease in a Patient with a Surgically Repaired Congenital Heart Defect: When Clinical History and Genetics Make the Difference" Cardiogenetics 12, no. 1: 102-108. https://doi.org/10.3390/cardiogenetics12010010
APA StyleRubino, M., Monda, E., Caiazza, M., Palmiero, G., Lioncino, M., Cirillo, A., Fusco, A., Verrillo, F., Perna, A., Diana, G., Amodio, F., Cesaro, A., Duro, G., Sarubbi, B., Russo, M. G., Calabrò, P., & Limongelli, G. (2022). Diagnosis of Fabry Disease in a Patient with a Surgically Repaired Congenital Heart Defect: When Clinical History and Genetics Make the Difference. Cardiogenetics, 12(1), 102-108. https://doi.org/10.3390/cardiogenetics12010010