
Relevance of Fluorodopa PET Scan in Dopamine Responsive Dystonia and Juvenile Parkinsonism: A Systematic Review
 , ,
, ,  ,
, 
Abstract
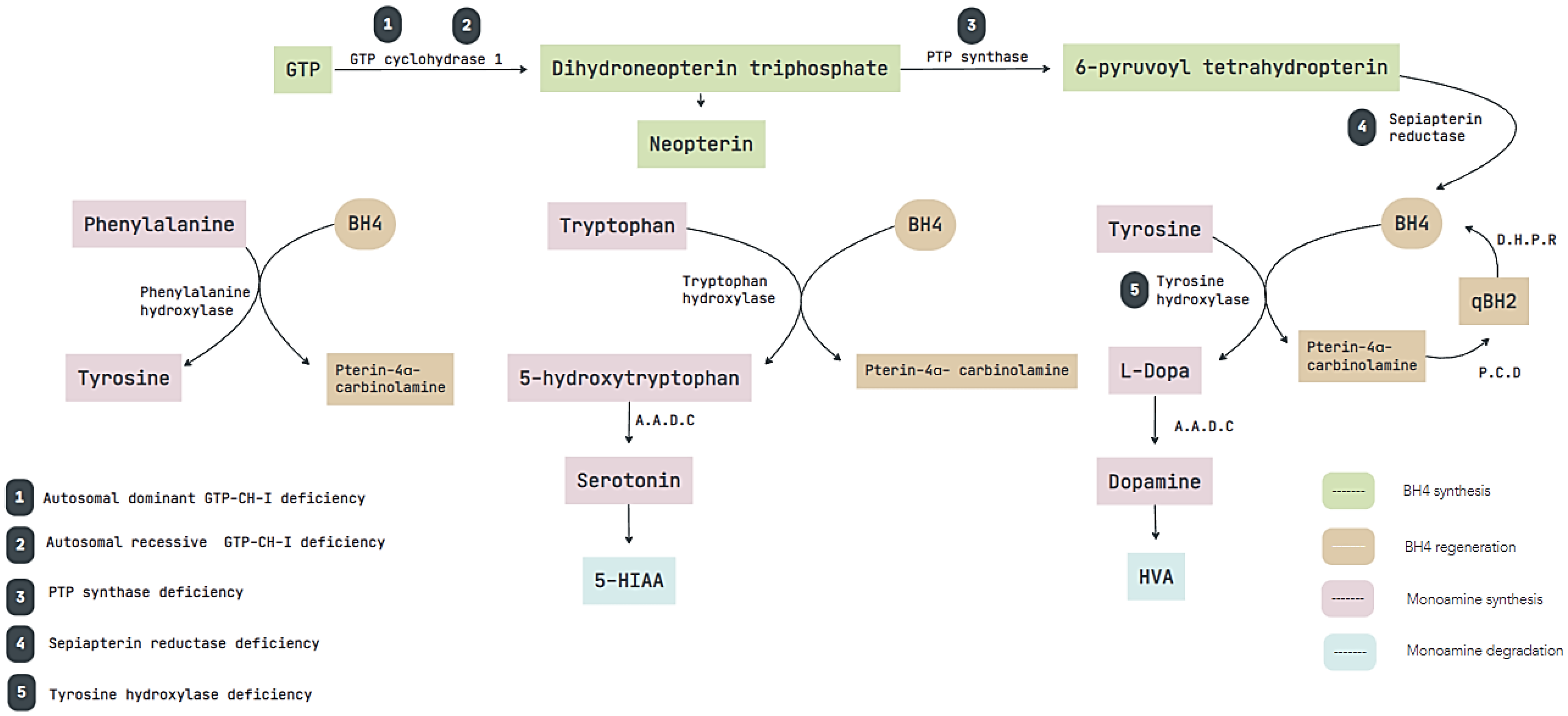
:1. Introduction
2. Materials and Methods
2.1. Protocol
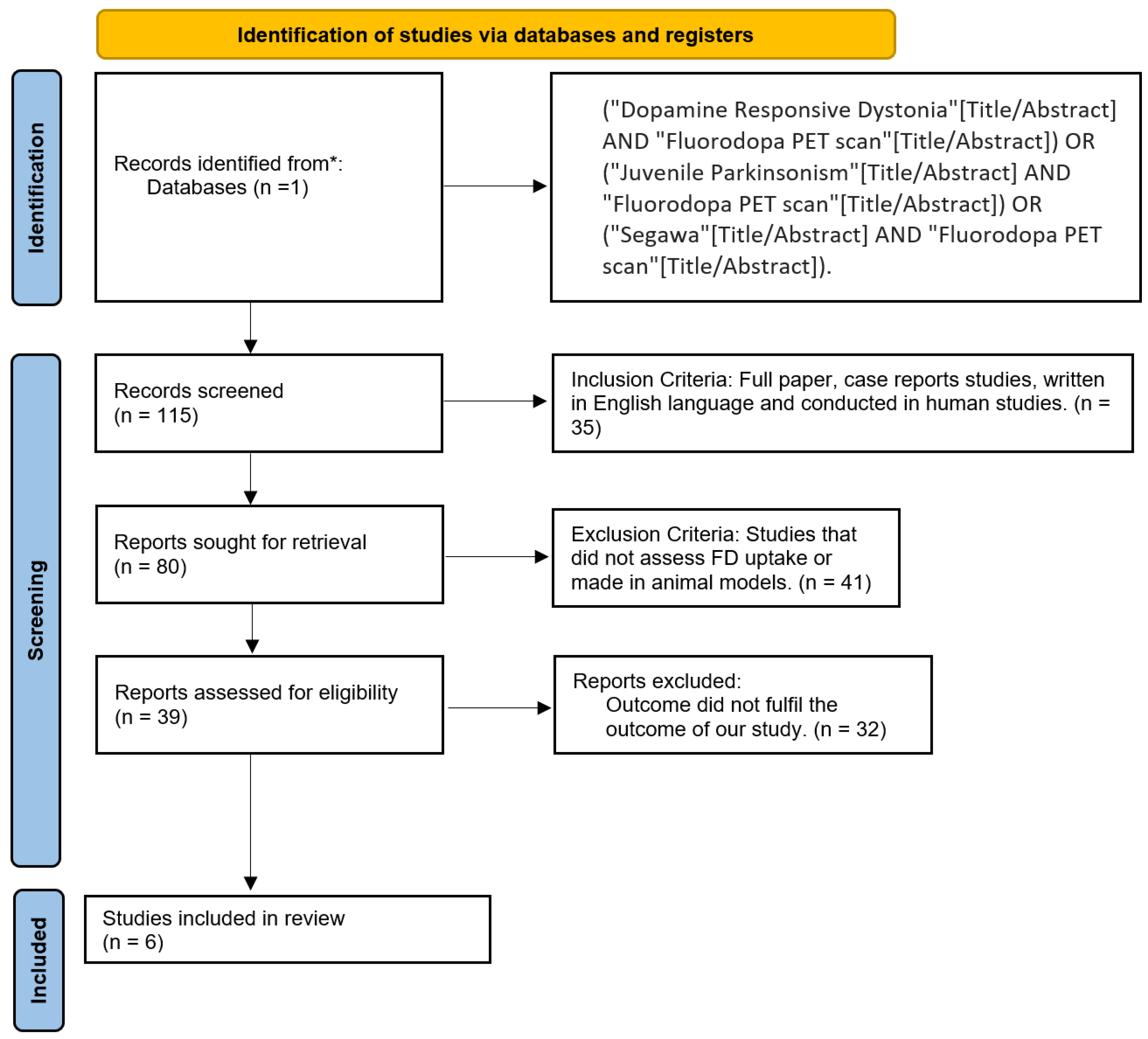
2.2. Eligibility Criteria and Study Selection
2.3. Database and Search Strategy
2.4. Data Extraction and Analysis
2.5. Bias Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FD | Fluorodopa |
| DRD | Dopamine Responsive Dystonia |
| PET | Positron Emission Tomography |
| PD | Parkinson’s Disease |
| JP | Juvenile Parkinsonism |
| LD | Levodopa |
| TDH | Tyrosine Hydroxylase Deficiency |
| TD | Tyrosine Hydroxylase |
| SNpc | Substantia Nigra Pars Compacta |
| EOPD | Early-Onset Parkinson’s Disease |
| iPD | Idiopathic Parkinson’s Disease |
| GTP-CH-I | Guanosine Triphosphate-Cyclohydrolase 1 |
| PTP | Pyruvoyltetrahydropterin |
| PRISMA | Preferred reporting items for systematic reviews and meta-analyses |
| MOOSE | Meta-analysis of observational studies in epidemiology |
References
- Weng, Y.C.; Wang, C.C.; Wu, Y.R. Atypical presentation of dopa-responsive dystonia in Taiwan. Brain Behav. 2018, 8, e00906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijemanne, S.; Jankovic, J. Dopa-responsive dystonia--clinical and genetic heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Fletcher, N.; Newman, E. Diagnosing dopamine-responsive dystonias. Pract. Neurol. 2015, 15, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Himmelreich, N.; Blau, N.; Thöny, B. Molecular and metabolic bases of tetrahydrobiopterin (BH4) deficiencies. Mol. Genet. Metab. 2021, 133, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Horvath, G.A.; Stockler-Ipsiroglu, S.G.; Salvarinova-Zivkovic, R.; Lillquist, Y.P.; Connolly, M.; Hyland, K.; Blau, N.; Rupar, T.; Waters, P.J. Autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia: Evidence of a phenotypic continuum between dominant and recessive forms. Mol. Genet. Metab. 2008, 94, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Zhang, Y.; Ye, J.; Han, L.; Qiu, W.; Gu, X.; Zhang, H. Tyrosine hydroxylase deficiency: A case of autosomal recessive dopa-responsive dystonia. Zhonghua Er Ke Za Zhi Chin. J. Pediatr. 2014, 52, 616–619. [Google Scholar]
- Willemsen, M.A.; Verbeek, M.M.; Kamsteeg, E.-J.; Andel, J.F.D.R.-V.; Aeby, A.; Blau, N.; Burlina, A.; Donati, M.A.; Geurtz, B.; Grattan-Smith, P.J.; et al. Tyrosine hydroxylase deficiency: A treatable disorder of brain catecholamine biosynthesis. Brain J. Neurol. 2010, 133, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.-W.; Jeon, B.S. Clinical Spectrum of Dopa-Responsive Dystonia and Related Disorders. Curr. Neurol. Neurosci. Rep. 2014, 14, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koht, J.; Rengmark, A.; Opladen, T.; Bjørnarå, K.A.; Selberg, T.; Tallaksen, C.M.E.; Blau, N.; Toft, M. Clinical and genetic studies in a family with a novel mutation in the sepiapterin reductase gene. Acta Neurol. Scand Suppl. 2014, 129, 7–12. [Google Scholar] [CrossRef]
- Verbeek, M.M.; Willemsen, M.A.; Wevers, R.A.; Lagerwerf, A.J.; Abeling, N.G.; Blau, N.; Thöny, B.; Vargiami, E.; Zafeiriou, D.I. Two Greek siblings with sepiapterin reductase deficiency. Mol. Genet. Metab. 2008, 94, 403–409. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Incidence and Distribution of Parkinsonism in Olmsted County Minnesota, 1991–2005 (S42.007)|Neurology. Available online: https://n.neurology.org/content/78/1_Supplement/S42.007 (accessed on 20 June 2022).
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.X.; Rosado, L.; Ross, B.M.; Verbitsky, M.; Kisselev, S.; Louis, E.D.; Comella, C.L.; et al. Frequency of known mutations in early-onset Parkinson disease: Implication for genetic counseling: The consortium on risk for early onset Parkinson disease study. Arch. Neurol. 2010, 67, 1116–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehanna, R.; Jankovic, J. Young-onset Parkinson’s disease: Its unique features and their impact on quality of life. Parkinsonism Relat. Disord. 2019, 65, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A.; Schott, J.M. Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol. 2006, 5, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, T.R.; Rodnitzky, R.L. Juvenile parkinsonism: Epidemiology, diagnosis and treatment. CNS Drugs 2010, 24, 467–477. [Google Scholar] [CrossRef]
- Quinn, N.; Critchley, P.; Marsden, C.D. Young onset Parkinson’s disease. Mov. Disord. 1987, 2, 73–91. [Google Scholar] [CrossRef]
- Schrag, A.; Ben-Shlomo, Y.; Brown, R.; Marsden, C.D.; Quinn, N. Young-onset Parkinson’s disease revisited--clinical features, natural history, and mortality. Mov. Disord. Off. J. Mov. Disord. Soc. 1998, 13, 885–894. [Google Scholar] [CrossRef]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B.; Brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; et al. Point Mutations (Thr240Arg and Ala311Stop) in theParkinGene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Brownstein, M.J.; Jonnalagada, S.; Chernova, T.; et al. The ubiquitin pathway in Parkinson’s disease. Nature 1998, 395, 451–452. [Google Scholar] [CrossRef]
- Morett, E.; Bork, P. A novel transactivation domain in parkin. Trends Biochem. Sci. 1999, 24, 229–231. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. New Genes Causing Hereditary Parkinson’s Disease or Parkinsonism. Curr. Neurol. Neurosci. Rep. 2017, 17, 66. [Google Scholar]
- Niemann, N.; Jankovic, J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Defazio, G.; Jinnah, H.A.; Berardelli, A.; Perlmutter, J.S.; Berkmen, G.K.; Berman, B.D.; Jankovic, J.; Bäumer, T.; Comella, C.; Cotton, A.C.; et al. Diagnostic criteria for blepharospasm: A multicenter international study. Parkinsonism Relat. Disord. 2021, 91, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, G.; Participants, T.I.D.R.; Conte, A.; Ferrazzano, G.; Esposito, M.; Albanese, A.; Pellicciari, R.; Di Biasio, F.; Bono, F.; Eleopra, R.; et al. Neuroimaging in idiopathic adult-onset focal dystonia. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2021, 42, 2947–2950. [Google Scholar] [CrossRef] [PubMed]
- Lidstone, S.C.; Costa-Parke, M.; Robinson, E.J.; Ercoli, T.; Stone, J.; FMD GAP Study Group. Functional movement disorder gender, age and phenotype study: A systematic review and individual patient meta-analysis of 4905 cases. J. Neurol. Neurosurg. Psychiatry 2022, 93, 609–616. [Google Scholar] [CrossRef]
- Takahashi, H.; Levine, R.A.; Galloway, M.P.; Snow, B.J.; Calne, D.B.; Nygaard, T.G. Biochemical and fluorodopa positron emission tomographic findings in an asymptomatic carrier of the gene for dopa-responsive dystonia. Ann. Neurol. 1994, 35, 354–356. [Google Scholar] [CrossRef]
- Snow, B.J.; Nygaard, T.G.; Takahashi, H.; Calne, D.B. Positron emission tomographic studies of dopa-responsive dystonia and early-onset idiopathic parkinsonism. Ann. Neurol. 1993, 34, 733–738. [Google Scholar] [CrossRef]
- Sawle, G.V.; Leenders, K.L.; Brooks, D.J.; Harwood, G.; Lees, A.J.; Frackowiak, R.S.; Marsden, C.D. Dopa-responsive dystonia: [18F]dopa positron emission tomography. Ann Neurol. 1991, 30, 24–30. [Google Scholar] [CrossRef]
- Tanji, H.; Nagasawa, H.; Araki, T.; Onodera, J.; Takase, S.; Itoh, M.; Itoyama, Y. PET study of striatal fluorodopa uptake and dopamine D2 receptor binding in a patient with juvenile parkinsonism. Eur. J. Neurol. 1998, 5, 243–248. [Google Scholar] [CrossRef]
- Hanakawa, T.; Fukuyama, H.; Akiguchi, I.; Kato, M.; Kimura, J.; Shibasaki, H. A case of familial juvenile dystonia-parkinsonism: 18F-6-fluorodopa and 18F-fluoro-2-deoxyglucose PET study. Rinsho Shinkeigaku 1996, 36, 655–660. [Google Scholar]
- Pal, P.K.; Leung, J.; Hedrich, K.; Samii, A.; Lieberman, A.; Nausieda, P.A.; Dm, D.B.C.; Breakefield, X.O.; Klein, C.; Stoessl, A.J. [18F]-Dopa positron emission tomography imaging in early-stage, non-parkin juvenile parkinsonism. Mov. Disord. Off. J. Mov. Disord. Soc. 2002, 17, 789–794. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Typical Juvenile Parkinsonism | Subtype | Clinical Symptoms |
|---|---|---|
| Autosomal Dominant | PARK SNCA (alpha synuclein) | These three subtypes are clinically indistinguishable from idiopathic Parkinson Disease. However, patients with the PARK-SNCA may have a higher rate of non-motor symptoms, atypical signs, and cognitive decline compared to the other two [24]. |
| PARK-LRRK2 (Leucine Rich Repeat Kinase 2) | ||
| PARK VPS35 (Vacuolar protein sorting-associated protein 35) | ||
| Autosomal Recessive | PARK-parkin (Parkin—E3 ubiquitin ligase) | These three subtypes are clinically indistinguishable between them.The most common reported symptoms are bradykinesia, tremor, rigidity, dystonia, and postural instability [24].Non-motor symptoms usually are more frequently reported in PARK-DJ1 (57%) and PARK-PINK1 (42%), compared to PARK-parkin (13%) [24]. |
| PARK-PINK1 (PTEN-induced putative kinase 1) | ||
| PARK-DJ1 (DJ1 oncogene) |
| Study | Newcastle-Ottawa Scale | Overall Risk of Bias | ||
|---|---|---|---|---|
| Selection (Total 4) | Comparability (Total 2) | Outcome/Exposure (Total 3) | (Total Score) | |
| Takahashi et al., 1993, USA. [28] | *** | - | *** | Moderate (6) |
| Snow et al., 1993, USA. [29] | *** | * | **** | Low (8) |
| Sawle et al., 1991, USA. [30] | ** | ** | **** | Low (8) |
| Tanji et al., 1998, Japan. [31] | * | * | *** | Moderate (5) |
| Hanawaka et al., 1996, Japan. [32] | - | * | ** | High (3) |
| Pal et al., 2002, USA. [33] | * | * | **** | Moderate (6) |
| Author, Year, Country | Number of Cases Described | Family History | Age When Scanned |
|---|---|---|---|
| Takahashi et al., 1993, USA. [28] | DRD
|
|
|
| Snow et al., 1993, USA. [29] | DRD:
| DRD:
| DRD:
|
| Sawle et al., 1991, USA. [30] | DRD:
|
|
|
| Tanji et al., 1998, Japan. [31] | JP:
|
|
|
| Hanawaka et al., 1996, Japan. [32] | JP:
|
|
|
| Pal et al., 2002, USA. [33] | JP:
|
|
|
| Author, Year, Country | LD Response * | FD PET Scan Uptake | Genetic Testing |
|---|---|---|---|
| Takahashi et al., 1993, USA. [28] | DRD:
| DRD: FD uptake was within the acceptable range compared to healthy control subjects, but tyrosine hydroxylase activity was reduced by 40% in the striatum. | Not described |
| Snow et al., 1993, USA. [29] | DRD:
| DRD: Normal striatal FD uptake compared to EOPD where the structural integrity of the nigrostriatal dopaminergic systems was affected, revealing a decrease in FD uptake. | Not described |
| Sawle et al., 1991, USA. [30] | DRD:
| DRD: Difference in the uptake of the familial cases where the uptake of FD was modest and decreased uptake in caudate and putamen as opposed to the idiopathic case where the uptake was severely decreased and showed a dramatic reduction in tracer activity than in JP. | Not described |
| Tanji et al., 1998, Japan. [31] | JP:
| JP: FD accumulation and its uptake rate constant into the putamen were markedly decreased bilateral. FD uptake was preserved only in the bilateral lower parts of the caudate nucleus. | Not described |
| Hanawaka et al., 1996, Japan. [32] | JP:
| JP: A marked decrease in uptake and metabolites was observed, especially in the striatum at the level of the putamen. | Not described |
| Pal et al., 2002, USA. [33] | JP:
| JP: Severe decrease in FD uptake in the caudate nucleus and in the putamen. FD uptake in the caudate had decreased by almost 50% of values considered normal, while uptake in the putamen was only 28% of that expected in a normal subject. | The parkin gene was screened extensively. Sequencing of the 12 exons from genomic DNA did not show any mutations. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moncayo, J.A.; Vargas, M.; Ortiz, J.F.; Granda, P.; Aguirre, A.S.; Argudo, J.; Tambo, W.; Garofalo, G.; Capirig, C.J.; German-Montenegro, M.; et al. Relevance of Fluorodopa PET Scan in Dopamine Responsive Dystonia and Juvenile Parkinsonism: A Systematic Review. Neurol. Int. 2022, 14, 997-1006. https://doi.org/10.3390/neurolint14040079
Moncayo JA, Vargas M, Ortiz JF, Granda P, Aguirre AS, Argudo J, Tambo W, Garofalo G, Capirig CJ, German-Montenegro M, et al. Relevance of Fluorodopa PET Scan in Dopamine Responsive Dystonia and Juvenile Parkinsonism: A Systematic Review. Neurology International. 2022; 14(4):997-1006. https://doi.org/10.3390/neurolint14040079
Chicago/Turabian StyleMoncayo, Juan A., Maite Vargas, Juan F. Ortiz, Pablo Granda, Alex S. Aguirre, Jennifer Argudo, Willians Tambo, Gabriela Garofalo, Christian John Capirig, Melisa German-Montenegro, and et al. 2022. "Relevance of Fluorodopa PET Scan in Dopamine Responsive Dystonia and Juvenile Parkinsonism: A Systematic Review" Neurology International 14, no. 4: 997-1006. https://doi.org/10.3390/neurolint14040079
APA StyleMoncayo, J. A., Vargas, M., Ortiz, J. F., Granda, P., Aguirre, A. S., Argudo, J., Tambo, W., Garofalo, G., Capirig, C. J., German-Montenegro, M., & Rueda, L. G. (2022). Relevance of Fluorodopa PET Scan in Dopamine Responsive Dystonia and Juvenile Parkinsonism: A Systematic Review. Neurology International, 14(4), 997-1006. https://doi.org/10.3390/neurolint14040079